Introduction
Altered acetylation and deacetylation of the
histones is often associated with cancer since it causes changes in
the expression pattern of genes involved in the regulation of cell
proliferation and apoptosis (1,2). It
is well known that hyperacetylation of histones H3 and H4
correlates with gene activation whereas deacetylation mediates
chromatin condensation and gene transcription silencing (3).
HDAC inhibitors (HDACis) are a new group of
anticancer agents and are currently evaluated in several phase I
and phase II clinical trials in patients with hematologic and solid
malignancies (4,5). Five chemical classes of HDACis have
been characterized including benzamides (MS-275), hydroxamic acids
(SAHA and trichostatin A), short-chain fatty acids (sodium butyrate
and phenylbutyrate), cyclic tetrapeptide containing a
2-amino-8-oxo-9,10-epoxy-decanoyl moiety (trapoxin A) and cyclic
peptides without the 2-amino-8-oxo-9,10-epoxy-decanoyl moiety
(FK228). Recent studies have shown that HDAC inhibitors induce cell
cycle arrest, differentiation, and apoptosis in vitro and
in vivo (6). HDACis also
inhibit endothelial cell proliferation and angiogenesis by
down-regulating angiogenesis-related gene expression (7). Two HDACis, SAHA and romidepsin
(depsipeptide or FK228), have been approved by the US Food and Drug
Administration for the treatment of cutaneous T cell lymphoma
(8,9). Since breast cancer is the highest
cause of tumor death in women, the antitumoral potency of HDACis on
human breast tumor cells has been widely investigated.
In breast cancer cell lines, SAHA has been
demonstrated to suppress the growth of tumor cells in vitro
at low micromolar concentrations (10). Growth inhibition was associated
with cell cycle arrest, morphological and functional
differentiation and apoptosis. In vivo, in tumor xenograft
models, SAHA significantly inhibits tumor growth of human breast
cancer cells without apparent toxicity in animals (11).
Several studies have demonstrated that HDACis can
sensitize cancer cells to apoptosis induced by Apo2L/TRAIL,
Gleevec, chemotherapeutic agents and ionizing radiations (12,13),
and also sensitization of breast cancer cells to topoisomerase II
inhibitors by SAHA treatment has been reported, indicating that
HDACis may be capable of enhancing the effectiveness of
apoptosis-inducing agents used clinically (14). Although many studies have
demonstrated the effect of histone acetylation on the re-expression
of ERα receptors (15), the
activation/repression by HDACis of apoptotic genes which could
counteract the aggressive behaviour of ER(−) breast cancer cells is
still matter of investigation (16,17).
This study was designed to analyze genes involved in
SAHA-induced cell death pathways. In fact we observed a different
antitumoral potency of SAHA in vivo and in vitro
cytotoxicity experiments on the human breast cancer cell line
MDA-MB-231, which is ER(−) and expresses mutant p53, when compared
to the ER(+)/wild-type p53 MCF7 cell line. Here, we found that SAHA
has different effects on the two breast cancer cell lines, as
regard to H4 histone acetylation, cell cycle, apoptosis kinetics,
but above all on the expression of apoptotic genes. In particular
TNF superfamily molecules (TRAIL, CD137, CD70/CD27) are strongly
upregulated in MDA-MB-231 cells and we demonstrated that the
combination of SAHA with the soluble CD137 receptor exerts a
synergistic effect on MDA-MB-231 cell death. On the contrary, in
MCF7 cells, apoptosis seems to be TNFR-independent and CD137
upregulation is absent. Thus, these results enlarge the field of
application of the therapeutical approach which combines HDACis and
TNF receptor family molecules, suggesting that the combination of
HDACis plus soluble CD137 receptor could be useful in killing
CD137L positive human breast cancer cells.
Materials and methods
Reagents and chemicals
Suberoylanilide hydroxamic acid (SAHA, Vorinostat),
was synthesized in the Department of Chemistry of Menarini Ricerche
SpA (Pomezia, Italy). DMSO was purchased from Sigma (St. Louis, MO,
USA). Anti-CD137-APC and anti-CD137L-PE antibodies and APC-and
PE-stained isotype controls were purchased from BD Pharmingen (San
Diego, CA, USA). FcR blocking reagent and 10% BSA solution were
purchased from Miltenyi Biotech (Bergish Gladback, Germany).
Cell culture
The human mammary adenocarcinoma cell line ER(−),
MDA-MB-231, was purchased from ATCC (American Type Culture
Collection, VA, USA). The human mammary adenocarcinoma cell line
ER(+), MCF7, was kindly provided by Dr F. Zunino (Istituto
Nazionale Tumori, Milan, Italy). MDA- MB-231 cells were propagated
in Leibovitz’s L-15 medium supplemented with 10% foetal bovine
serum (FBS) and 2 mM L-glutamine (Gibco). MCF7 cells were
propagated in Eagle’s minimum essential medium supplemented with
0.01 mg/ml bovine insulin, 10% FBS and 2 mM L-glutamine. Cells were
cultured at 37°C in a 5% CO2/95% air humidified
atmosphere.
Cell treatments
In apoptosis experiments, sub-confluent cells were
cultured for 24- or 48-h in the presence of equitoxic
concentrations of SAHA, corresponding to the IC90
previously determined for each cell line (50 and 10 μM for
MDA-MB-231 and MCF7 cells, respectively). Experiments to detect
acetylated lysine were performed in the presence of SAHA
IC50 (5 and 1 μM for MDA-MB-231 and MCF7 cells,
respectively), incubating cells for 24 h in the continous presence
of the drug.
In vitro cytotoxicity assay
Drug-induced cytotoxic effects were evaluated by
using Alamar Blue assay (18). In
brief, MCF7 and MDA-MB-231 cells were seeded into 96-well
microtitration plates at 4×103 cells/well to ensure a
logarithmic growth throughout the experiments. After 24-h of cell
attachment, fresh medium containing SAHA at different
concentrations was added to each well for further 72 h. At the end
of the incubation period, 20 μl of Alamar Blue (Biosource
International, Camarillo, CA, USA) was added to each well and the
plates were further incubated for 4 h. Fluorescence emission was
monitored in a multilabel counter Victor 1420 (Wallak, Finland) at
530/590 nm excitation/emission wavelength. CD137 receptor and CD137
ligand cytotoxic effects were evaluated as follows: MDA-MB-231
cells were plated in 96-well plates at 4×103 cells/ well
and incubated at 37°C in a humidified atmosphere with 5%
CO2. After 24 h, cells were treated in duplicate with
fresh medium containing 5 μM SAHA alone or in separate mixtures
with the following recombinant proteins and antibodies: 1 μg/ well
CD137-hIg, 1 μg/well CD137L-muCD8, 2 μg/well mouse anti-CD137 mAb
(clone 4B4-1) from Ancell (Bayport, MN, USA), 1 μg/well mouse
anti-CD137L mAb (clone C65-485) and 1–2 μg/well mouse IgG1 control
(clone MOPC-21) from BD Bioscience (Franklin Lakes, NJ, USA). Cells
were incubated for 0, 6, 24, 48, 72 h at 37°C, 5% CO2.
At the end of the incubation period, 20 μl of Alamar Blue was added
to each well and the plates were further incubated for 4 h.
Fluorescence emission was monitored in a multilabel counter Victor
1420 (Wallak, Finland) at 530/590 nm excitation/emission
wavelength.
Tumor xenograft
Female athymic nude mice (6–8 weeks old) were
purchased from Charles River (Calco, Lecco, Italy), maintained in
microisolator cages and supplied with sterile materials under
standard conditions, according to United Kingdom Co-ordinating
Committee on Cancer Research guidelines (19). For the generation of MCF7 tumors,
female nude mice were first implanted s.c. in the left flank with
17β-estradiol-sustained release pellets (Innovative Research,
Sarasota, FL) and after one week implanted s.c. in the control
lateral flank with MCF7 tumor cells (20×106
cells/flank/0.2 ml of 0.9% NaCl sterile solution). Human tumor
MDA-MB-231 originated from s.c. in vivo injection of tumor
cells (20×106 cells/flank/0.2 ml) in the right flank of
adult athymic female nude mice. Tumor volumes were monitored weekly
by caliper measurement of length and width twice weekly. Tumor
volume (TV) was calculated by using the formula: volume
(mm3) = width2 × length/2 (20). SAHA was dissolved in DMSO and
stored at −80°C. This stock solution was diluted just before dosing
with D5W (dextrose 5%) to a final DMSO concentration not superior
to 1%. SAHA was admini stered i.v. at a dose of 50 mg/kg in a
volume corresponding to 10 ml/kg body weight. Tumor-bearing mice
were treated with SAHA or vehicle once daily, 5 days/week, for a
total of 10 doses, starting when tumors were approximately 50
mm3 in volume. No adverse or toxic effect were observed
at this dose level. All animal experiments were reviewed and
approved by the Ethics commission at Menarini Ricerche according to
the guidelines of the European Directive for the protection of
vertebrate animals used for experimental and other scientific
purposes (2010/63/UE).
Apotox assay
MDA-MB-231 cells were plated in 96-well plates at
4×103 cells/well and incubated at 37°C in a humidified
atmosphere with 5% CO2. After 24 h, cells were incubated
in duplicate with fresh medium containing 1 μM SAHA (synthesized at
the Chemistry Department of Menarini Ricerche Pomezia, Rome,
Italy), 1 μg/well CD137-hIg (Ancell, Bayport, MN, USA), 10 μM
staurosporine (Sigma) or the mix of 1 μM SAHA with 1 μg/well
CD137-Ig, for 0, 6, 24, 48, 72 and 96-h time points. Cell survival
was determined through the ApoTox-Glo Triplex Assay (Promega,
Madison, WI, USA), according to the manufacturer’s
instructions.
Flow cytometric detection of apoptotic
cells
Apoptosis was measured by Annexin V-PE/7-ADD
staining method (21). Briefly,
adherent and floating cells were collected, washed twice with cold
PBS and then resuspended in PBS + 0.5% BSA (staining buffer) at
1×106 cells/ml, 3×105 cells were transferred
in 5 ml culture tube, stained with 15 μl of Annexin V-PE and 15 μl
of 7-ADD, vortexed and incubated for 15 min at room temperature in
the dark. At the end of the incubation time, 200 μl of staining
buffer was added to each tube and cells analyzed by flow cytometry
collecting 10,000 events on a FACSCanto flow cytometer (BD
Biosciences). Data analysis was performed using FacsDiva 6.1.2
version software. Percentage of apoptotic cells was determined
setting quadrant gates on the unstained control.
Quantitative analysis of acetylated
lysine by flow cytometry
After treatment with the HDACi, cells were
trypsinized and fixed in 4% paraformaldehyde for 10 min at room
temperature. Paraformaldehyde was used for fixation of cells since
it has been found to preserve the structure of acetylated chromatin
organization. After fixation, cells were washed three times and
permeabilized with PBS containing 0.4% Triton X-100 and 0.1% BSA.
5x105 cells/tube resuspended in PBS + 0.5% BSA were
labelled with a polyclonal rabbit antibody which recognizes
hyperacetylated histone H4 (Upstate Biotechnology) at a dilution of
1:50 for 1 h at 4°C. Subsequently, cells were washed twice,
incubated with a FITC-conjugated polyclonal anti-rabbit antibody
(Santa Cruz Biotechnology) for at least 30 min at 4°C and then
resuspended in 1 ml PBS + 0.5% BSA. As background controls we used
both a specific rabbit IgG and a sample stained only with the
secondary antibody. Fluorescence of acetylated lysine was
determined on a FacsCanto flow cytometer (BD Biosciences) and
expressed in logarithmic arbitrary units as mean fluorescence
intensity (MFI). Data analysis was performed using FacsDiva 6.1.2
version software.
Flow cytometric determination of CD137
and CD137L expression
MDA-MB-231 and MCF7 cells were seeded at
5×105 cells in T25 flasks, allowed to adhere overnight
and next day incubated with 5 or 1 μM SAHA for 24 h. Cells were
washed with PBS and detached using Trypsin/EDTA solution, counted
and resuspended in PBS + 0.5% BSA (staining buffer) at
107 cells/ml. Cells were incubated with FcR blocking
buffer for 15 min at room temperature and then 5×105
cells transferred in flow cytometric tubes and incubated with 20 μl
of anti-CD137-APC or anti-CD137L-PE antibody or with the isotype
matched antibodies and incubated for 30 min at 4°C. Cells were
washed twice with PBS + 0.5% BSA (staining buffer) and resuspended
in 0.5 ml of the same buffer. Flow cytometric acquisition was
performed analyzing 10,000 events on a FacsCanto flow cytometer (BD
Biosciences). Background staining for both treated and untreated
cells was assessed by incubation of cells with mouse
fluorochrome-isotype controls. Background and unstained controls
were used to correctly set analysis gates so that CD137- and
CD137L-positive quadrants in isotype samples do not contain more
than 2% of cells. Data analysis was performed using FacsDiva 6.1.2
version software.
HDAC activity in cellular extracts
HDAC activity in nuclear extracts was measured
according to the manufacturer’s instructions (HDAC Fluorescent
assay, Biomol International LP, PA). Nuclear extracts were prepared
using the kit NE-PER (Pierce Biotechnology) and incubated (5
μg/well) at 25°C with 40 μM of Fluor de Lys™ substrate. Reactions
were stopped after 30 min with Fluor de Lys™ developer and
fluorescence measured using a Victor1420 microplate reader (Wallac,
Finland) at ex/em 355/460 nm. Results are the mean of three
independent experiments ± SD.
RNA extraction and cDNA synthesis
Total RNA was extracted from cells using the SV
Total RNA Isolation System (Cat no. Z3100, Promega, WI, USA).
Briefly, cells from 1.5×106 to a maximum of
2.5×106 per treatment were lysed in 175 μl lysis buffer.
Lysates were subjected to purification with silica columns
according to the instructions of the manufacturer. The yield of
total RNA was determined spectrophotometrically at 260 nm, while
its integrity was determined by denaturing agarose gel
electrophoresis. For each sample, 5 μg total RNA were copied into
cDNA with the RT2 First Strand Kit (Cat no. C-03, SuperArray, MD,
USA). Finished reactions had a volume of 20 μl and were stored at
−20°C until they were used in real-time PCR analysis.
Gene array analysis by real-time PCR
The cDNA samples were pre-diluted to 100 μl and
mixed with RT2 qPCR Master Mix (Cat no. PA-012, SuperArray) to a
final volume of 2550 μl. The master mix contains all the reagents
and buffers required for the real-time PCR in our 7300 real-time
PCR system (Applied Biosystems, CA, USA), including the SYBR Green
dye and the ROX reference dye. The gene expression analysis was
performed by adding 25 μl of the experimental cocktail to each well
of a 96-well RT2 Profiler PCR Array for human apoptosis (Cat no.
PHAS-012A, SuperArray). In this array, each well contains a primer
pair specific for one of 84 key genes involved in human apoptosis,
besides housekeeping genes and control wells (Fig. 1). After an initial denaturation at
95°C for 10 min, the cycling program was 95°C, 15 sec and 60°C, 1
min for 40 cycles. Raw data were analyzed with the ΔΔCt method
using an Excel-based PCR Array Data Analysis Template (SuperArray).
Data from control cells were set as calibrator for the relative
quantification.
CD137 ligand mRNA by real-time PCR
To determine the mRNA expression levels of target
genes, duplicates of the cDNA samples, corresponding to 150 ng of
total RNA, were amplified in the presence of 12.5 μl of 2X TaqMan
Universal PCR Master Mix and 1.25 μl of 20X inventoried TaqMan Gene
Expression Assay (Applied Biosystems) to a final volume of 25 μl.
The TaqMan Gene Expression Assays were: Hs00155512_m1 for TNFRSF9
(CD137), Hs00169409_m1 for TNFSF9 (CD137L) and Hs99999905_m1 for
GAPDH. The samples of cDNA were also amplified in the presence of
Hs99999903_m1 for human β-actin to normalize mRNA quantities. After
10 min of incubation at 95°C for denaturation, samples were
subjected to 45 cycles of PCR. Each cycle was 95°C for 15 sec and
60°C for 1 min. The amplification took place in a 7300 real-time
PCR system (Applied Biosystems). The collected data of gene
expression were the number of cycles needed to reach a fixed
threshold fluorescence (Ct). The gene targets expression in
MDA-MB-231 cells treated with 5 μM SAHA was then compared to that
of untreated cells, selected as calibrator, by processing the
obtained Ct values with the 2−ΔΔCt method (22).
Statistical analysis
All results were expressed as the mean ± SD of data
obtained from 2 to 3 separate experiments. The data were entered
into Instat 2.03 GraphPad software (GraphPad Software Inc., CA,
USA) to perform the Student’s t-test or the Tukey-Kramer multiple
comparison test. Means were considered significantly different at a
confidence level of p<0.05. Synergy calculations and
extrapolation of CI were performed using the Calcusyn 2.0 version
software.
Results
SAHA-induced cell death in in vitro and
in vivo models
In our in vitro Alamar Blue assay, SAHA
showed a cytotoxic activity with an IC50 of 5 and 1 μM
in MDA-MB-231 and MCF7 cells, respectively (Fig. 1A). When xenografted nude mice were
treated with SAHA 50 mg/kg per os, both tumor models, characterised
by a different estrogen receptor and p53 status, appeared sensitive
to the inhibitory effect of SAHA (Fig.
1B and C). A more pronounced and long-lasting inhibition of
tumor growth of MDA-MB-231 cells with respect to MCF7 tumor cell
line was observed.
Since in several studies it has been described that
SAHA exerts its antitumor effect through apoptosis induction, we
investigated the presence of apoptotic cell death using the
IC90 concentration of this HDACi. Table I shows that, in MCF7 cells, 10 μM
SAHA causes a slower increase of apoptosis, which at 24 h measures
17%, compared to the 43% of 50 μM SAHA-treated MDA-MB-231 cells. At
48 h MCF7 cells showed a drastic increase of apoptosis (45%), while
the ERα negative cells remained quite stable (40%). Thus, the
kinetics of SAHA-induced apoptosis in these cell lines seems to be
quite different, probably because of the distinct pathways involved
(see below). Flow cytometric analysis of cell cycle phases in
MDA-MB-231 and MCF7 cells at 50 and 10 μM of SAHA, respectively,
for 24 h, revealed only a moderate block in G2/M phase and a
reduction of S-phase in MDA-MB-231 cells and a more potent growth
arrest and G2/M block in MCF7 cells (data not shown). These results
are in agreement with the cytotoxicity assay, confirming a lower
in vitro sensitivity of MDA-MB-231 cells also to
SAHA-induced apoptotic cell death.
 | Table IApoptosis detection by flow cytometry
using Annexin/ 7-ADD method (Materials and methods).a |
Table I
Apoptosis detection by flow cytometry
using Annexin/ 7-ADD method (Materials and methods).a
| % Apoptosis
|
|---|
| Cells | Untreated | +SAHA 24 h | +SAHA 48 h |
|---|
| MDA-MB-231 | 15 | 43 | 40 |
| MCF7 | 16 | 17 | 45 |
H4 histone acetylation and inhibition of
HDAC activity
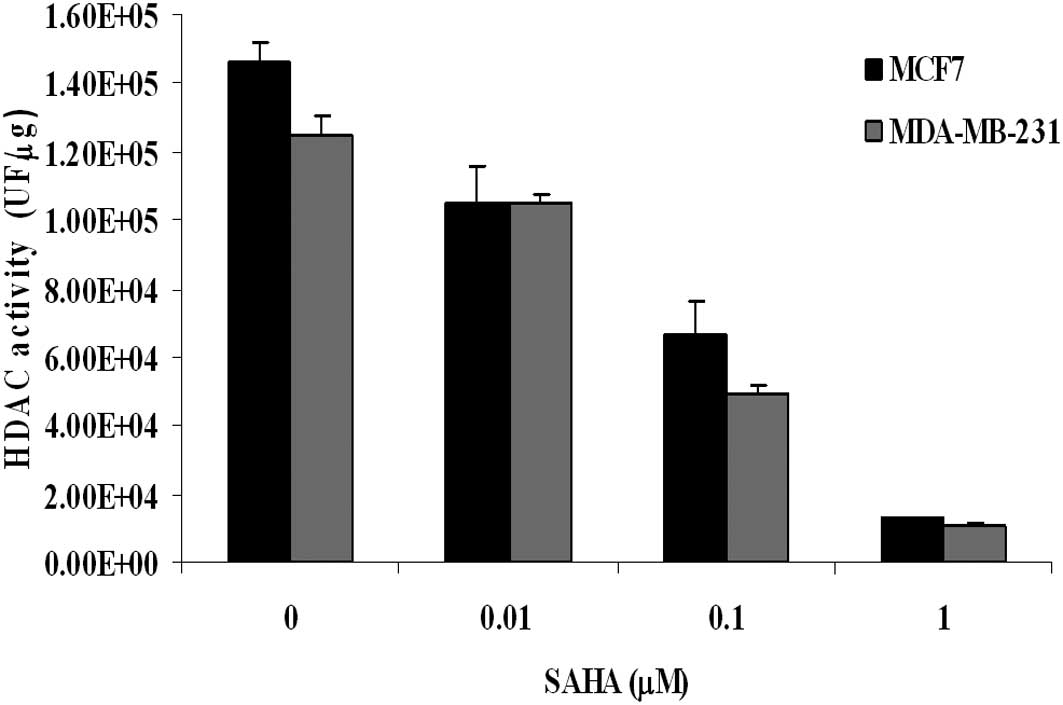
As shown in Fig. 2,
when HDAC activity was measured in nuclear extracts, we found that
SAHA exerted an inhibitory effect that was quite similar between
MDA-MB-231 and MCF7 cells. The flow cytometric analysis of H4
histone acetylation revealed that, in untreated MDA-MB-231 cells,
the basal level of acetylation was higher than that measured in
MCF7 cells. When cells were incubated for 24 h with equitoxic
concentration of SAHA (IC50), an increase of histone
acetylation was observed, and again it was more pronounced in
MDA-MB-231 than in MCF7 cells (percentage increase of 75% and 21%,
respectively) (Table II). Since
histone acetylation favours chromatin relaxation and gene
expression, this observation seems to correlate well with the
earlier induction of apoptosis seen at 24 h in MDA-MB-231
cells.
| Table IIFlow cytometric determination of H4
histone acetylation.a |
Table II
Flow cytometric determination of H4
histone acetylation.a
| Treatment | MDA-MB-231
(MFI) | MCF7 (MFI) |
|---|
| Blank | 26 | 14 |
| No drug | 306 | 75 |
| SAHA | 536 (+75%) | 91 (+21%) |
Effect of SAHA on apoptotic gene
expression
When we investigated the pattern of apoptotic genes
induced or repressed by SAHA (Fig.
3A), we found that, in the two human breast tumor cell lines,
mRNAs related to different apoptotic pathways were expressed.
Variations with more than 3-fold increase/decrease have been
considered significant. In MDA-MB-231 cells an upregulation of
genes which are associated to the extrinsic apoptotic death
receptor pathway was observed, such as TNFSF10 (TRAIL), TNFSF8
(CD30L), CD27, CD70, TNFRSF25 (DR3), caspase 8. In particular a
strong upregulation (more than 10-fold) of TNFRSF9 (CD137 receptor)
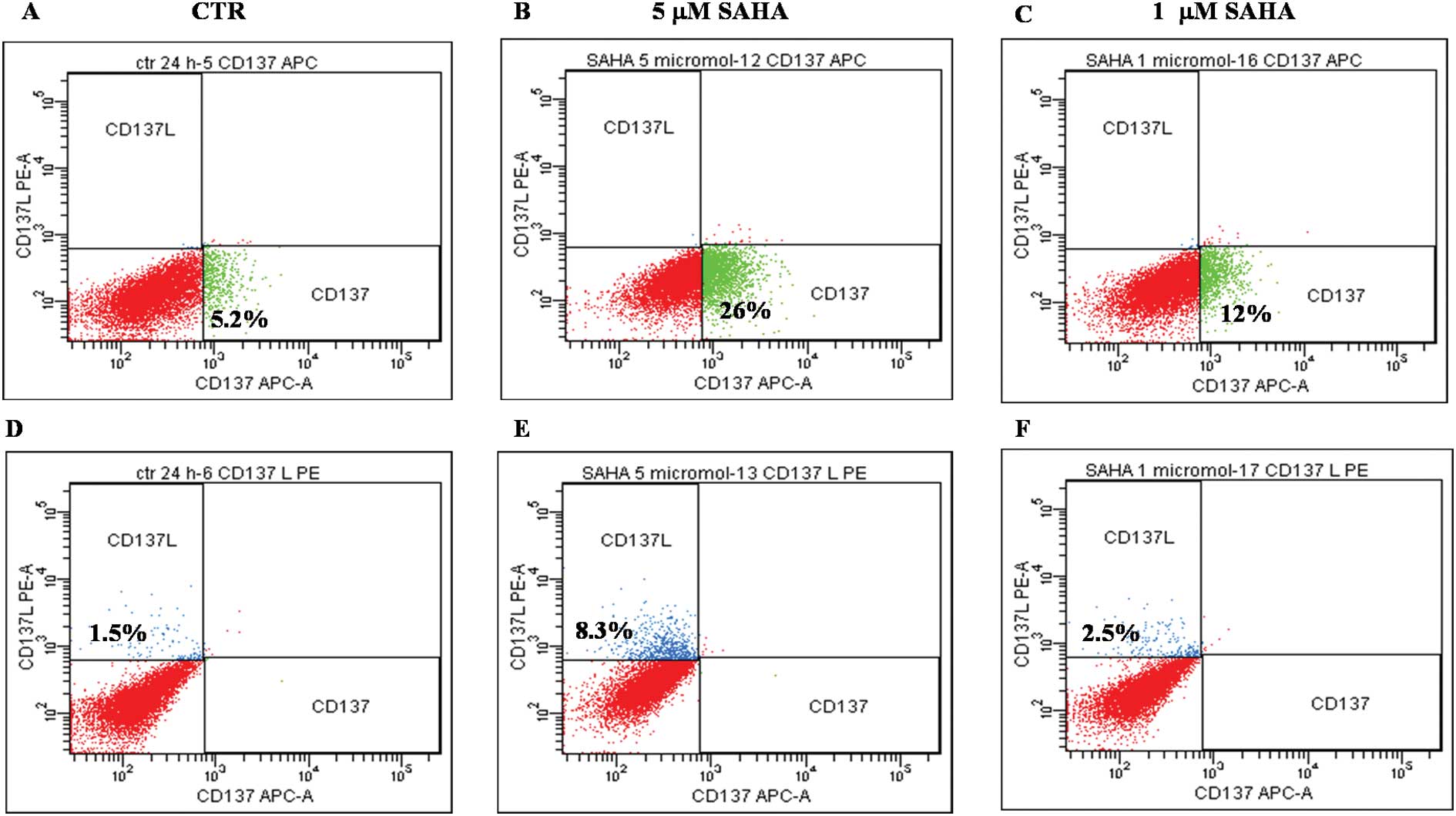
and of CD137 ligand transcription has been measured (Fig. 3) and protein expression confirmed
by flow cytometry (Fig. 4).
Factors involved into the mitochondrial pathway were not
particularly affected in this cell line. On the contrary, in MCF7
cells, the intrinsic apoptotic pathway seems to be involved: in
fact the TNF-superfamily genes were not induced by SAHA treatment,
but genes encoding for proapoptotic factors such as p53 binding
protein 2 (TP53BP2) and BNIP3 were upregulated. Furthermore, in
MDA-MB-231 cells, caspases 4, 5 and 7 rather then caspase 3 seem to
be activated while in MCF7 cells, which are caspase 3-deprived, we
observed a strong increase of caspase 14 mRNA, suggesting that in
this cell line this caspase could become the final effector of the
apoptotic signalling.
Effect of the combination of SAHA and
soluble CD137 on cytotoxicity/apoptosis in MDA-MB-231 cells
To assess the existence of a functional significance
of CD137 receptor/ligand upregulation, we combined treatment of
SAHA with soluble CD137 receptor and related molecules. Fig. 5A shows that the simultaneous
administration of 5 μM SAHA with the soluble form of CD137 receptor
induced the most significant enhancement of cell death, when
compared to single-drug conditions or to the combination with other
agonists (Fig. 5B–D). When 1 μM
SAHA was assayed in the Apotox assay in order to reduce the
cytotoxicity of single-drug treatment, a more pronounced effect was
observed, with a synergism of action (CI 0.24) at the 72-h time
point (Fig. 6).
Discussion
In the present study we showed for the first time
that an HDACi promotes CD137/CD137L upregulation in a solid tumor
in particular in a breast cancer cell line. Moreover, we found that
the simultaneous administration of SAHA and soluble CD137 receptor
exerts a synergistic effect on cancer cell destruction.
Many investigators have demonstrated that HDACis
exert their anticancer activity by inducing apoptosis and
modulating the in vivo environment of tumor cells (7,23,24).
Furthermore, it has been also described that HDACis sensitize
breast cancer cells to TRAIL induced apoptosis (25,26),
while a recent paper of Vire et al reported CD137/CD137L
induction by HDACis in haematological cancer cells (27), however, many aspects of their in
vivo cytotoxic action remain to be elucidated. Our findings
demonstrated that, in MCF7 cells, SAHA activates the intrinsic
apoptotic pathway through induction of genes related to p53
signalling, while, in MDA-MB-231 cells, SAHA treatment increases
the transcription of genes involved in the cell death pathway, such
as TNFSF10 (TRAIL), caspase 8, and even more the genes encoding for
the CD137 and CD70 receptor/ligand system. The CD137 receptor
(TNFRSF9, 4-1BB, ILA) is a member of the tumor necrosis factor
receptor family, and a potent T cell co-stimulatory molecule. Its
natural ligand, CD137 ligand (TNFSF9, CD137L-4-1BBL), has been
detected on professional APCs including B cells, macrophages,
dendritic cells and can transduce signals, a process known as
‘reverse signalling’ (28,29). The bidirectional signalling of
CD137L usually induces activation, proliferation and migration of
monocytes and hematopoietic progenitor cells. However, a recent
study reported that CD137L engagement can also inhibit cell
proliferation and induce apoptosis in multiple myeloma cell lines
(30). Other receptors, such as
CD40, have shown a similar double behaviour, suggesting that the
final effect of these receptor/ ligand systems is probably
dependent on tumor cell type and on the induction of other
bifunctional molecules (31). Our
data showed also that the stimulation of CD137 receptor was not
particularly effective in increasing cell death, despite its higher
expression on MDA-MB-231 cells, and no synergism or additive effect
was measured following combination of SAHA with CD137L or with an
anti-CD137 antibody. Thus these results seem to confirm the
observations of Gullo et al on the prevalent role of CD137L
in activating a cell death signal in human tumor cells (30). Furthermore, the presence of a
mutated p53 in this cell line, could account for the activation of
the extrinsic apoptotic pathway and for its in vitro reduced
sensitivity to SAHA. Recent studies reported that re-expression of
CD70 in tumor cells may induce an anti-tumor response by
T-lymphocytes and that the epigenetic down-regulation of CD70
receptor by DNA hypermethylation is a strategy of breast cancer
cells to escape immune response (32). Our observation that SAHA-induced
histone hyperacetylation determines also an overexpression of CD70
and CD27 in MDA-MB-231 cells, supports the idea that SAHA treatment
could restore an immune response against cancer cells. So our
initial results showing that MDA-MB-231 cells are more sensitive to
SAHA treatment in the in vivo tumor xenograft model than in
in vitro assays, when compared to the MCF7 cell line, could
be explained by the concurrent overexpression of multiple death
receptors/ligands. Since in solid tumors the presence of soluble
forms of CD137, TRAIL and of autocrine and paracrine factors which
affect cancer development has been documented (33,34),
it is possible that the stimulation of overexpressed
receptors/ligands contributes to the enhancement of apoptosis and
to the regression of tumor mass.
In conclusion, our study demonstrated that SAHA can
activate different apoptotic genes and targets depending on the
type of breast cancer cell line studied, and that promotes, in a
p53-mutated breast cancer cell line, the expression of cell death
molecules such as CD137/CD137L. These findings suggest that the
induction of death receptors could contribute to enhancing in
vivo SAHA activity on breast tumors and that the combination of
SAHA with soluble CD137 receptor could be a new further approach in
breast cancer therapy.
Abbreviations:
|
7-ADD
|
7-aminoactinomycin D
|
|
APC
|
allophycocyanin
|
|
Casp
|
caspase
|
|
CI
|
combination index
|
|
ER
|
estrogen receptor
|
|
IC
|
inhibitory concentration
|
|
PE
|
R-phycoerythrin
|
|
sCD137
|
soluble CD137 receptor
|
|
TNF
|
tumor necrosis factor
|
|
TNFRSF
|
TNF receptor superfamily
|
|
TRAIL
|
TNF-related apoptosis inducing
ligand
|
Acknowledgements
We would like to thank the Department
of Chemistry of Menarini Ricerche, Pomezia, for the synthesis of
SAHA.
References
|
1
|
Grunstein M: Histone acetylation in
chromatin structure and transcription. Nature. 389:349–352. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Glass CK and Rosenfeld MG: The coregulator
exchange in transcriptional functions of nuclear receptors. Genes
Dev. 14:121–141. 2000.PubMed/NCBI
|
|
3
|
Grignani F, De Matteis S, Nervi C,
Tomassoni L, Gelmetti V, Cioce M, Fanelli M, Ruthardt M, Ferrara
FF, Zamir I, Seiser C, Grignani F, Lazar MA, Minucci S and Pelicci
PG: Fusion proteins of the retinoic acid receptor-α recruit histone
deacetylase in promyelocytic leukaemia. Nature. 391:815–818.
1998.
|
|
4
|
Garcia-Manero G, Yang H, Bueso-Ramos C, et
al: Phase 1 study of the histone deacetylase inhibitor vorinostat
(suberoylanilide hydroxamic acid [SAHA]) in patients with advanced
leukemias and myelodysplastic syndromes. Blood. 111:1060–1066.
2008.
|
|
5
|
Blumenschein GR Jr, Kies MS,
Papadimitrakopoulou VA, Lu C, Kumar AJ, Ricker JL, Chiao JH, Chen C
and Frankel SR: Phase II trial of the histone deacetylase inhibitor
(Zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with
recurrent and/or metastatic head and neck cancer. Invest New Drugs.
26:81–87. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu WS, Parmigiani RB and Marks PA: Histone
deacetylase inhibitors: molecular mechanisms of action. Oncogene.
26:5541–5552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qian DZ, Kato Y, Shabbeer S, Wei Y,
Verheul HM, Salumbides B, Sanni T, Atadja P and Pili R: Targeting
tumor angiogenesis with histone deacetylase inhibitors: the
hydroxamic acid derivative LBH589. Clin Cancer Res. 12:634–642.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bertino E and Otterson GA: Romidepsin: a
novel histone deacetylase inhibitor for cancer. Expert Opin
Investig Drugs. 20:1151–1158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ramalingam SS, Kummar S, Sarantopoulos J,
Shibata S, Lo Russo P, Yerk M, Holleran J, Lin Y, Beumer JH, Harvey
RD, Ivy SP, Belani CP and Egorin MJ: Phase I study of vorinostat in
patients with advanced solid tumors and hepatic dysfunction: a
National Cancer Institute Organ Dysfunction Working Group study. J
Clin Oncol. 28:4507–4512. 2010. View Article : Google Scholar
|
|
10
|
Butler LM, Zhou X, Xu WS, Scher HI,
Rifkind RA, Marks PA and Richon VM: The histone deacetylase
inhibitor SAHA arrests cancer cell growth, up-regulates
thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc
Natl Acad Sci USA. 99:11700–11705. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cohen LA, Marks PA, Rifkind RA, Amin S,
Desai D, Pittman B and Richon VM: Suberoylanilide hydroxamic acid
(SAHA), a histone deacetylase inhibitor, suppresses the growth of
carcinogen-induced mammary tumors. Anticancer Res. 22:1497–1504.
2002.PubMed/NCBI
|
|
12
|
Nakata S, Yoshida T, Horinaka M, Shiraishi
T, Wakada M and Sakai T: Histone deacetylase inhibitors upregulate
death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/
APO2-L in human malignant tumor cells. Oncogene. 23:6261–6271.
2004. View Article : Google Scholar
|
|
13
|
Guo F, Sigua C, Tao J, Bali P, George P,
Li Y, Wittmann S, Moscinski L, Atadja P and Bhalla K: Cotreatment
with histone deacetylase inhibitor LAQ824 enhances Apo-2L/tumor
necrosis factor-related apoptosis inducing ligand-induced death
inducing signaling complex activity and apoptosis of human acute
leukemia cells. Cancer Res. 64:2580–2589. 2004. View Article : Google Scholar
|
|
14
|
Kim MS, Blake M, Baek JH, Kohlhagen G,
Pommier Y and Carrier F: Inhibition of histone deacetylase
increases cytotoxicity to anticancer drugs targeting DNA. Cancer
Res. 63:7291–7300. 2003.PubMed/NCBI
|
|
15
|
Jang ER, Lim SJ, Lee ES, Jeong G, Kim TY,
Bang YJ and Lee JS: The histone deacetylase inhibitor trichostatin
A sensitizes estrogen receptor α-negative breast cancer cells to
tamoxifen. Oncogene. 23:1724–1736. 2004.PubMed/NCBI
|
|
16
|
Licznar A, Caporali S, Lucas A, Weisz A,
Vignon F and Lazennec G: Identification of genes involved in growth
inhibition of breast cancer cells transduced with estrogen
receptor. FEBS Lett. 553:445–450. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Srivastava RK, Kurzrock R and Shankar S:
MS-275 sensitizes TRAIL-resistant breast cancer cells, inhibits
angiogenesis and metastasis and reverses epithelial-mesenchymal
transition in vivo. Mol Cancer Ther. 9:3254–3266. 2010. View Article : Google Scholar
|
|
18
|
Page B, Page M and Noel C: A new
fluorimetric assay for cytotoxicity measurements in vitro. Int J
Oncol. 3:473–476. 1993.PubMed/NCBI
|
|
19
|
Workman P, Aboagye EO, Balkwill F, et al:
Guidelines for the welfare and use of animals in cancer research.
Br J Cancer. 102:1555–1577. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Plowman J, Dykes DJ, Melinda H,
Simpson-Hellen L and Alley MC: Human tumor xenograft models in NCI
drug development. Anticancer Drug Development Guide. Teicher BA:
Humana Press; Totowa, NJ: pp. 101–126. 1997, View Article : Google Scholar
|
|
21
|
Koopman G, Reutelingsperger CP, Kuijten
GA, Keehnen RM, Pals ST and van Oers MH: Annexin V for flow
cytometric detection of phosphatidylserine expression on B cells
undergoing apoptosis. Blood. 84:1415–1420. 1994.PubMed/NCBI
|
|
22
|
Livak KJ: ABI Prism 7700 Sequence
Detection System. User Bulletin No. 2. PE Applied Biosystems AB
website, Bulletin Reference 4303859B 77802-002,. 1997.
|
|
23
|
Archer SY, Johnson J, Kim HJ, Ma Q, Mou H,
Daesety V, Meng S and Hodin RA: The histone deacetylase inhibitor
butyrate downregulates cyclin B1 gene expression via a p21/
WAF-1-dependent mechanism in human colon cancer cells. Am J Physiol
Gastrointest Liver Physiol. 289:G696–G703. 2005.
|
|
24
|
Hellebrekers DM, Melotte V, Viré E,
Langenkamp E, Molema G, Fuks F, Herman JG, van Criekinge W,
Griffioen AW and van Engeland M: Identification of epigenetically
silenced genes in tumor endothelial cells. Cancer Res.
67:4138–4148. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim HR, Kim E-J, Yang S-H, Jeong E-T, Park
C, Lee J-H, Youn M-J, So H-S and Park R: Trichostatin A induces
apoptosis in lung cancer cells via simultaneous activation of the
death receptor-mediated and mitochondrial pathway. Exp Mol Med.
38:616–624. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shankar S, Davis R, Singh KP, Kurzrock R,
Ross DD and Srivastava RK: Suberoylanilide hydroxamic acid
(Zolinza/vorinostat) sensitizes TRAIL-resistant breast cancer cells
orthotopically implanted in BALB/c nude mice. Mol Cancer Ther.
8:1596–1605. 2009. View Article : Google Scholar
|
|
27
|
Vire B, De Walque S, Restouin A, Olive D,
van Lint C and Collette Y: Anti-leukemia activity of MS-275 histone
deacetylase inhibitor implicates 4-1BBL/4-1BB immunomodulatory
functions. PLoS One. 4:e70852009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun Y, Chen JH and Fu Y: Immunotherapy
with agonistic anti-CD137: two sides of a coin. Cell Mol Immunol.
1:31–36. 2004.PubMed/NCBI
|
|
29
|
Shao Z and Schwarz H: CD137 ligand, a
member of the tumor necrosis factor family, regulates immune
responses via reverse signal transduction. J Leukoc Biol. 89:21–29.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gullo C, Koh LK, Pang WL, Ho KT, Tan SH
and Schwarz H: Inhibition of proliferation and induction of
apoptosis in multiple myeloma cell lines by CD137 ligand signaling.
PLoS One. 5:e108452010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tai YT, Catley LP, Mitsiades CS, Burger R,
Podar K, Shringpaure R, Hideshima T, Chauhan D, Hamasaki M,
Ishitsuka K, Richardson P, Treon SP, Munshi NC and Anderson KC:
Mechanisms by which SGN-40, a humanized anti-CD40 antibody, induces
cytotoxicity in human multiple myeloma cells: clinical
implications. Cancer Res. 64:2846–2852. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu SE, Park SH and Jang YK: Epigenetic
silencing of TNFSF7 (CD70) by DNA methylation during progression to
breast cancer. Mol Cells. 29:217–221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dimberg J, Hugander A and Wagsater D:
Expression of CD137 and CD137L in colorectal cancer patients. Oncol
Rep. 15:1197–1200. 2006.PubMed/NCBI
|
|
34
|
Nicolin V and Narducci P: Soluble TRAIL
could enhance bone destruction acting on Rank-ligand in
estrogen-independent human breast cancer cell line MDA-MB-231. Acta
Histochem. 112:1189–1192. 2010. View Article : Google Scholar : PubMed/NCBI
|