Introduction
Prostate cancer (PCa) is a leading cause of cancer
mortality among men in the United States and is the second leading
cause of cancer mortality in men worldwide (1). Current treatments include hormonal
therapy, targeted radiation, and radical prostatectomy. The only
alternative is watchful waiting. The limitations and side-effects
associated with these therapies, however, warrant the development
of new therapies (2).
Testosterone (T) regulates normal cell growth in the
prostate gland and is secreted by testicular Leydig cells. The
action of T is mediated by the androgen receptor (AR), a nuclear
receptor expressed in the stromal and luminal prostate gland
epithelial cells. Androgen-dependent and independent neoplastic PCa
cells depend on T, its more potent 5 α-reductase metabolite, 5
α-dihydrotestosterone (DHT), and AR (3). Huggins (4) demonstrated PCa dependency on T and,
based on his early findings that showed reduced prostate growth
following removal of androgen, local and metastatic cancers are
currently treated with endocrine therapies aimed at reducing
androgen production and/or blocking AR with anti-androgens. These
therapies limit tumor growth and reduce tumor mass but the cancer
may recur as androgen-insensitive (AI) disease (5).
Multiple direct and indirect mechanisms are thought
to cause the AI state through changes in AR activity (6–8).
These changes include AR signaling modulation by a kinase receptor
such as human epidermal growth factor receptor 2 (HER-2/neu)
tyrosine kinase (9);
overexpression of, or mutations in, the AR gene (10); increased AR sensitivity (11); or activation of the AR by growth
factors such as insulin-like growth factor 1, epidermal growth
factor and kerationcyte growth factor (6,12).
Additional indirect and equally important mechanisms by which PCa
cell growth circumvent androgen-dependency involve overexpression
of GPCRs (13,14). An effective cure for AI is
currently unavailable despite the intense research in this area
(15).
Since mutations in the AR are responsible for a
small number of prostate cancers, indirect mechanisms are likely to
be involved in the majority of androgen-refractory cases. One such
mechanism is activation and overexpression of GPCRs (8). Several studies showed that GPCRs are
overexpressed in various cancer types and contribute to tumor
growth when activated by circulating or locally produced peptide
ligands (13). These ligands
induce cell proliferation and prevent apoptosis of
hormone-independent PCa cells, which suggest a critical role in PCa
progression (14,16–20).
Normal GPCR functions are altered in malignant cells leading to
autonomous cell proliferation (13). Hence, GPCR-targeted therapy for the
treatment of PCa is actively pursued (21).
GPCR actions in PCa involve cell growth enhancement
via ERK6, a mitogen-activated protein kinase-related
serine/threonine kinase (MAPK)(22). ERK can phosphorylate AR at several
sites thereby upregulating AR target genes (13). GPCRs that signal through
stimulatory G proteins α (Gsα) can also transactivate AR through
cyclic adenosine monophosphate (cAMP) production (23,24)
and protein kinase A (PKA)(25).
In addition, the activated Gsα subunit can directly activate AR and
act synergistically with low androgen concentrations to increase AR
activity in PCa cells (24).
Several studies suggest a role for at least some of
the MCRs in cancer development. An association between MC1R and PCa
risk was reported in human (26)
and central MCR blockade with SHU9119-reversed PCa anorexia in
Wistar rats (27). Additionally,
MC5R is widely expressed in normal rodent exocrine glands including
the prostate (28).
MC2R, activated by ACTH, is involved in androgen and
steriodogenic enzyme synthesis. It is normally expressed in the
adrenal gland cortex and epidermis as well as in basal cell
carcinoma tissue and cultured melanocytes. As with other GPCRs,
MC2R binding ACTH activates adenylyl cyclase with subsequent
production of cAMP. Cyclic AMP can then directly activate or
interact with various proteins such as ion-channels, transcription
factors, guanine nucleotide exchange factors, and protein kinases
such as PKA. PKA and cAMP play critical roles in the progression
and differentiation of prostate carcinogenesis (25). Activation of MC2R can also result
in phosphorylation of MAPK and the MC2R accessory protein isoform β
(MRAP-β) in cells stably expressing MC2R (29). Similarly, ectopic production of
ACTH, the putative ligand for MC2R, has been reported in cases of
prostate carcinoma associated with Cushing’s syndrome (30).
Currently, the role of MCRs in PCa is unclear. The
objective of this study was to investigate the MCRs expression in
human PCa cell lines and their effect on cell proliferation, with
particular emphasis on MC2R.
Materials and methods
Reagents
ACTH and DHT were obtained from Sigma (St. Louis,
MO). Melanocortin peptides, NDP-MSH (a potent α-MCR analogue for
all MCRs except MC2R) and THIQ (highly selective MC4R agonist),
D-Trp8-γ-MSH (highly selective MC3R agonist), and
SHU9119 (a universal MCR blocker) were obtained from Tocris
Bioscience, Ellisville, MO. A selective MC4R inverse agonist (Ipsen
5i) was synthesized by Enzo Life Sciences International Inc.,
Plymouth Meeting, PA. RPMI-1640 media with and without phenol red,
Dulbecco’s phosphate buffered saline (DPBS), without calcium and
magnesium, Forskolin (cAMP inducer), and penicillin streptomycin
antibiotics were purchased from Lonza (Portsmouth, NH).
Heat-inactivated and charcoal-dextran-treated fetal calf serum
(FCS) was obtained from Atlanta Biological (Atlanta, GA). Cell
culture flasks and other disposable cell culture supplies were
purchased from VWR International, LCC (Atlanta, GA). MTT cell
viability assay kits were purchased from the American Type Culture
Collection (ATCC). Primary antibodies and control peptides for MC2R
(SA-639), and AR (Sc-816) were obtained from Enzo Life Science
(Farmingdale, NY) and Santa Cruz Biotechnology (Santa Cruz, CA),
respectively. Dylite 488 conjugated goat anti-rabbit IgG (Jackson
Immunoresearch Laboratories, PA) was used for the detection of
primary binding in immunocytochemistry assays.
Cell culture
The human prostate normal epithelial cells, RWPE-1,
and human prostate adenocarcinoma cell lines, LNCaP, PC3, and
DU-145, were obtained from the American Type Culture Collection
(Manassas, VA). The RWPE-1 cells were propagated in base medium
containing bovine pituitary extract and human recombinant epidermal
growth factor (Invitrogen, Carlsbad, CA). PCa cell lines were
maintained in RPMI-1640 media supplemented with 1% (v/v)
streptomycin-penicillin antibiotics (Invitrogen), and 10% (v/v)
heat-inactivated FCS. Cells were grown in T-75 vented filter cap
tissue culture flasks until they reached approximately 75–90%
confluency at 37°C in a humidified incubator under a 5%
CO2 atmosphere. Detachment of the cells from the T-75
flasks was accomplished with trypsin after the cells were washed
with DPBS to remove residual serum. Cells were then counted using
an automated coulter cell counter and cultured at the desired
concentration. For culture cell proliferation/viability
experiments, ligands were diluted in PBS-BSA and added in 20 μl
volumes containing various concentrations. Twenty-four hours before
treatments, cells were depleted of androgen by replacing growth
media with phenol red-free RPMI-1640 supplemented with 5%
charcoal-dextran-treated FCS. Treatment peptides were replenished
every 24 h for 72 h. Cells were seeded at a density of
2x105 cells per well when grown in 6-well culture plates
(for protein and RNA isolation), and at 2x103 cells per
well when grown in flat-bottom 96-well plates for MTT cell
viability assay. Passage number was recorded with each split and
cells were used between passages 5 and 38.
MTT cell viability assay
Cells in log-phase growth were harvested via
trypsinization and counted using a coulter counter. The effect of
test drugs on cell viability/proliferation was determined using the
3-(4,5-dimethylthiazol-2-yl)-2-5-diphenyltetrazolium bromide (MTT)
cell viability assay ATCC kit according to the manufacturer’s
instructions. This colometric assay measures the reduction of a
tetrazolium component (MTT) into an insoluble formazan product by
the mitochondria of metabolically active cells. Forty-eight to 72 h
after test drugs were added (eight replicates per treatment), 10 μl
MTT reagent was added per well for 2 h at 37°C. Next, 100 μl
detergent solution was added to lyse the cells and solubilize
colored crystals. Plates were then incubated in the dark for 6 h at
room temperature (RT). Optical density (OD) was obtained using
Spectramax Plus microtiter plate reader at 570 nm wavelength. The
amount of purple color produced in this test is directly
proportional to the number of viable and metabolically active
cells. The effect on cell proliferation/viability was calculated
based on three independent experiments.
Conventional and real-time PCR
Total RNA was isolated using TRIzol reagent
(Invitrogen-Life Technologies Inc., Carlsbad, CA, USA), according
to the manufacturer’s protocol and as described in our previous
study (31). RNA concentrations
were determined at 260 nm wavelength and the ratio of 260/280 was
obtained using UV spectrophotometry (DU640, Beckman Coulter,
Fullerton, CA, USA). Samples with 260/280 ratio of ≥1.8 were used.
First strand synthesis was performed with SABiosciences first
strand kit C-03 (SABiosciences, Frederic, MD, USA). Conventional
PCR was used initially to determine the presence of MCR (1–5)
genes in LNCaP, PC-3, and DU-145 using validated human primer sets
obtained from SABiosciences (Table
I). RT-PCR was performed using ReactionReady™ HotStart ‘Sweet’
PCR master mix (SABiosciences). PCR products were analyzed in
parallel with GAPDH housekeeping gene. Final end PCR products were
fractionated and visualized on 2% ethedium bromide-stained agarose
gel. Next, real-time PCR was used to determine the expression level
of the MCRs (1–5) in LNCaP, PC-3 and DU-145 cancer cells
relative to RWPE 1 normal prostate epithelial cells. The assay was
carried out as we previously described (31). Briefly, reactions were performed in
25 μl reaction mixture containing 12.5 μl RT2 real-time
SYBR/Fluorescein Green PCR master mix with final concentrations of
10 mM Tris-HCl, 50 mM KCl, 2.0 mM MgCl2, 0.2 mM dNTPs,
and 2.5 units of HotStart Taq DNA polymerase (SABiosciences), 1 μl
first strand cDNA, 1 μl RT2 validated primer sets, and 10.5 μl
PCR-grade water. Reactions were run in 96-well PCR plates using
Bio-Rad PCR cycler (Bio-Rad, MyiQ, Hercules, CA, USA). Reactions
were run in duplicates and the results were normalized to GAPDH.
The amplification protocol was set at 95°C for 15 min, and 40
cycles each at 95°C for 30 sec, 55°C for 30 sec, and 72°C for 30
sec.
 | Table IExpected PCR product size and
accession nos. of human MCR genes. |
Table I
Expected PCR product size and
accession nos. of human MCR genes.
| Gene symbol | Expected PCR
product size | RefSeq accession
no. | Reference
positions |
|---|
| MC1R | 179 | NM_002386.3 | 2933–2951 |
| MC2R | 159 | NM_000529.2 | 180–202 |
| MC3R | 146 | NM_019888.3 | 639–658 |
| MC4R | 186 | NM_005912.2 | 717–741 |
| MC5R | 90 | NM_005913.1 | 103–121 |
Western blotting (WB)
WB was performed as previously described by us
(31). Briefly, cell lysates were
prepared by homogenization in RIPA buffer (150 mM sodium chloride,
50 mM Tris-HCl, pH 7.4, 1 mM ethylenediaminetetraacetic acid, 1 mM
phenylmethylsulfonyl flouride, 1% Triton X-100, 1% sodium
deoxycholic acid, 0.1% sodium dodecylsulfate) containing 5 μg/ml of
aprotinin, and 5 μg/ml of leupeptin. Cell debris was removed by
centrifugation. Protein concentration was determined with the
Bio-Rad protein assay. Cell lysates were boiled for 5 min in 1X SDS
sample buffer (50 mM Tris-HCl pH 6.8, 12.5% glycerol, 1% sodium
dodecylsulfate, 0.01% bromophenol blue) containing 5%
β-mercaptoethanol. Adrenal cell lysate (positive control for MC2R)
was obtained from VWR.
Immunofluorescence microscopy
Cells were grown on Lab-Tek II chamber slides for 24
h. Appropriate treatments were added as described in the figure
legends. Cells were then rinsed 3x in PBS and fixed in buffered 4%
paraformaldehyde (30 min). Following fixation, cells were rinsed 2x
in dH2O and transferred to absolute methanol for 10 min
followed by equilibration in PBS (3x for 3 min each). Before adding
the primary antibody, cells were incubated in appropriate blocker
(5% normal goat serum, 2% BSA, in PBS, pH 7.3). Rabbit polyclonal
anti-MC2R (extracellular) and anti-AR (nuclear) were incubated
(1:100) in blocking solution overnight at RT. Secondary,
fluorescence-labeled goat anti-rabbit, IgG antibody was incubated
at RT for 1 h. Slides were mounted with Vectashield mounting medium
and the preparations were sealed with clear nail polish.
Epifluorescence and DIC images were acquired with a Nikon E600
microscope equipped with a Retiga EX CCD digital camera (Q Imaging,
Burnaby, Canada). Images were processed and saved with NIS-Elements
AR 3.2 software. Exposure times were identical among the treatment
groups. Confocal images were captured with a Nikon TE2000E confocal
microscope (Nikon, Mississauga, Canada) operated with EZ-C1 3.91
software.
Cyclic AMP
ACTH-induced cAMP was determined in three PCa cell
lines. Briefly, PC3, DU-145, and LNCaP cells were plated in 6-well
plates and allowed to attach for 16 h. The following day, the cells
were washed twice with warm Waymouth/BSA. Then fresh Waymouth/BSA
containing 0.5 mM isobutylmethylxanthine (IBMX) (a cAMP promoter
and a non-specific inhibitor of cAMP and cGMP phosphodiesterases,
Sigma) was added to each well. After 15 min of incubation at 37°C,
different concentrations of ACTH or [Nle4,
D-Phe7]-α-melanocyte stimulating hormone (NDP-MSH) were
added and the cells were incubated for another hour. Final
concentrations of ligands are indicated in Fig. 7. Intracellular cAMP was extracted
using 0.5 N perchloric acid containing 180 μg/ml theophylline
(Sigma), and measured using radioimmunoassay (32). All determinations were performed in
triplicate. The cAMP levels were calculated as fold increase over
basal levels of each cell line. Maximal response (Rmax) and
EC50 values were calculated with GraphPad software.
Statistical analysis
Data from cell culture MTT cell viability assay were
expressed as the mean ± SD. The data were analyzed by Student’s
t-test, one-way ANOVA followed by Dunnett’s post hoc test (Graph
Pad Prism 5.0, San Diego, CA). P<0.05 was considered to indicate
statistically significant differences. Statistical analysis for the
real-time PCR data was performed using a modification of the ΔΔCt
method (ΔΔCt) as described in our previous study (33) .
Results
MCR (1–5)
transcripts are expressed in PCa cell lines
We used conventional and real-time RT-PCR analysis
to determine that MCR (1–5) transcripts are expressed in LNCaP and
PC3 cells. DU-145 cells expressed the MCR 1–3, and 5 transcripts,
but did not express MC4R mRNA (Fig.
1A). Comparative real-time PCR analysis of MCR (1–5)
showed higher expression in cancer cells compared with RWPE-1
normal prostate epithelial cells (Fig.
1B). Similar to conventional RT-PCR data, the MC4R level in
DU-145 cancer cells was undetectable by the real-time PCR
technique. Table I shows the
expected PCR product size and the accession numbers of MCR genes in
Genbank.
ACTH (MC2R specific ligand) increases
cell proliferation in human PCa cells and SHU9119 (a universal MCR
blocker) partially blunts this effect
Because increased GPCRs expression in PCa cells was
thought to provide cell survival signal in PCa (34). We used several MCR synthetic
peptide agonists and antagonists in cell viability/proliferation
assays to test the function of the MCRs in PCa cells. These
compounds were: NDP-MSH, the potent α-MSH analogue for all MCRs
except MC2R; THIQ, the highly selective agonist of MC4R;
D-Trp8-γ-MSH, the highly selective agonist of MC3R;
SHU9119, a universal MCR antagonist; and Ipsen 5i, an inverse MC4R
agonist. In LNCaP, only ACTH induced proliferation and enhanced
cell viability (Fig. 2). Similar
findings were obtained in PC3 and DU-145 cells (data not shown).
The stimulatory effects of ACTH were concentration-dependent in
both androgen-dependent LNCaP and androgen-independent PC3 and
DU-145 cells (Fig. 3A and B). The
maximal cell proliferation increase was between 55 and 170% above
the control. Cell proliferation increased incrementally with
continuous ACTH treatment for up to 8 days (Fig. 3C). Treatment with the universal MCR
blocker SHU9119 inhibited cell proliferation in ACTH treated LNCaP
cells by approximately 50% (Fig.
3D).
Indirect immunocytochemistry showed a punctate label
pattern on PC3 cell membranes (Fig.
4A). The labeling pattern was similar to that described in
transfected HEK293-related cells (293/FRT cell line) (29). Immunoblots incubated with MC2R
antibody that was pre-absorbed with commercially available MC2R
polypeptide showed no band (Fig.
4B). Routine western blot analysis showed higher MC2R protein
expression in LNCaP, PC3, DU-145 cells compared with normal
non-tumorigenic RWPE2 prostate cells (Fig. 4C).
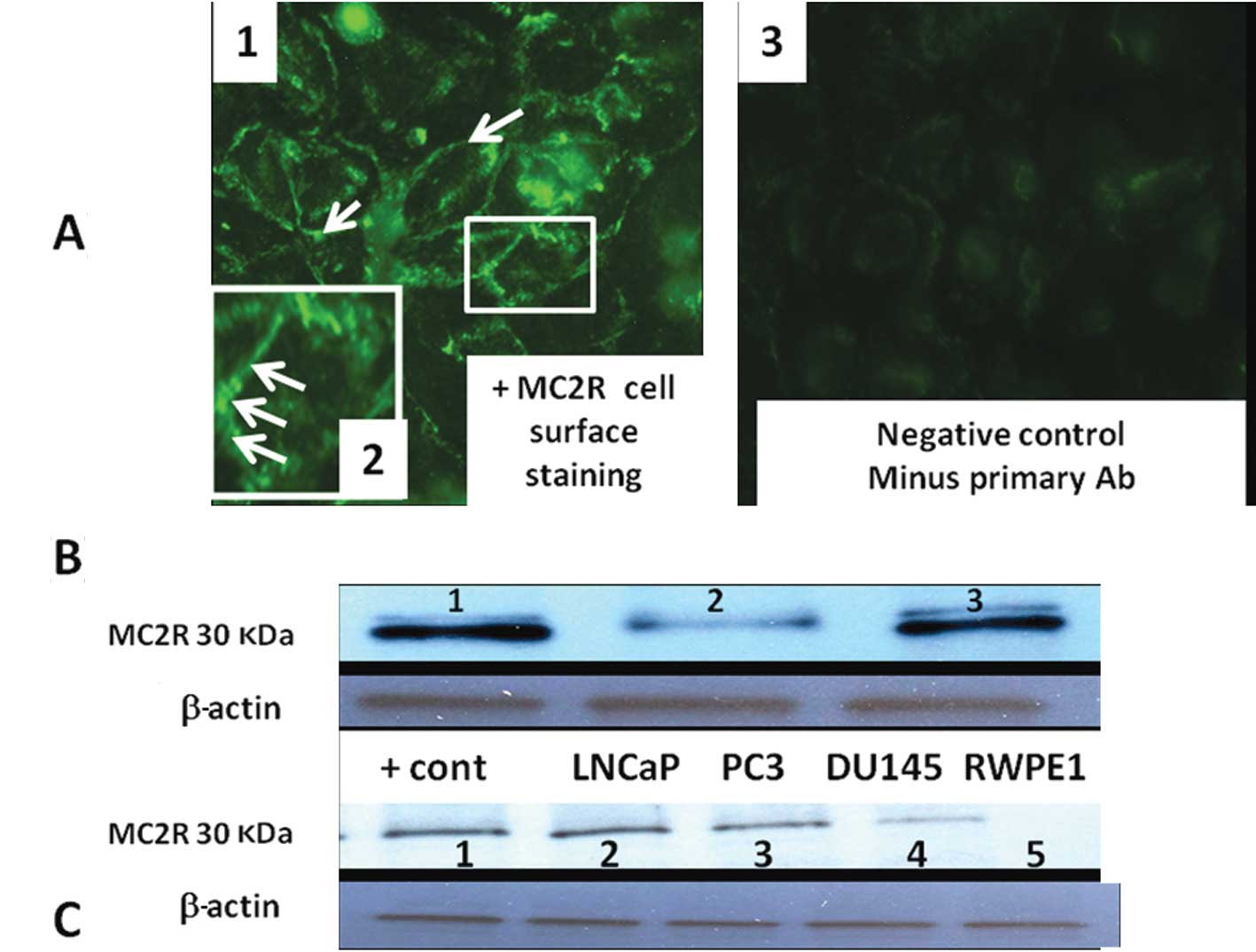 | Figure 4(A) Immunohistochemical localization
of MC2R in PC3 using rabbit anti-melanocortin receptor 2
(extracellular) antibody captured with fluorescence-labeled goat
anti-rabbit secondary antibody. Note specific cell surface
expression of MC2R in panel (1).
Insert (panel 2) is the magnification of a cell membrane to show
punctuated and discrete plasma membrane sub-domains. Little or no
staining is present in negative control (minus primary Ab, panel
3). (B) MC2R protein is present in LNCaP lysate as shown by western
blotting and was masked by MC2R specific peptide blocker. Lane 1,
MC2R protein; lane 2, MC2R band was partially abolished when MC2R
antibody was pre-incubated with a control MC2R peptide; lane 3,
positive MC2R control (35 μg adrenal cell lysate). (C) MC2R protein
in LNCaP, PC3, DU-145 PCa cell lines and RWPE1 normal prostate
cells. Lane 1, adrenal lysate (positive control); lane 2, LNCaP;
lane 3, PC3; lane 4, DU-145; lane 5, RWPE1. |
ACTH proliferative effect is additive
with DHT stimulation
Owing to the fact that adrenal steroids,
dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate
(DHEAS), and androstenedione, are induced by ACTH and are linked to
PCa recurrence, especially in patients undergoing long-term
androgen ablation (6,35), we investigated a possible
interaction with ACTH and androgen. ACTH stimulation of LNCaP cells
in combination with DHT, a potent AR agonist, induced a 150–200%
increase in cell proliferation compared with ACTH or DHT alone
(Fig. 5).
ACTH treatment enhances AR protein
nuclear translocation and AR protein expression in LNCaP
Production of cAMP/PKA via GPCR activation can
transactivate AR in prostate carcinoma (25). Therefore, we considered whether
ACTH stimulation of LNCaP cells affected the amount of nuclear AR
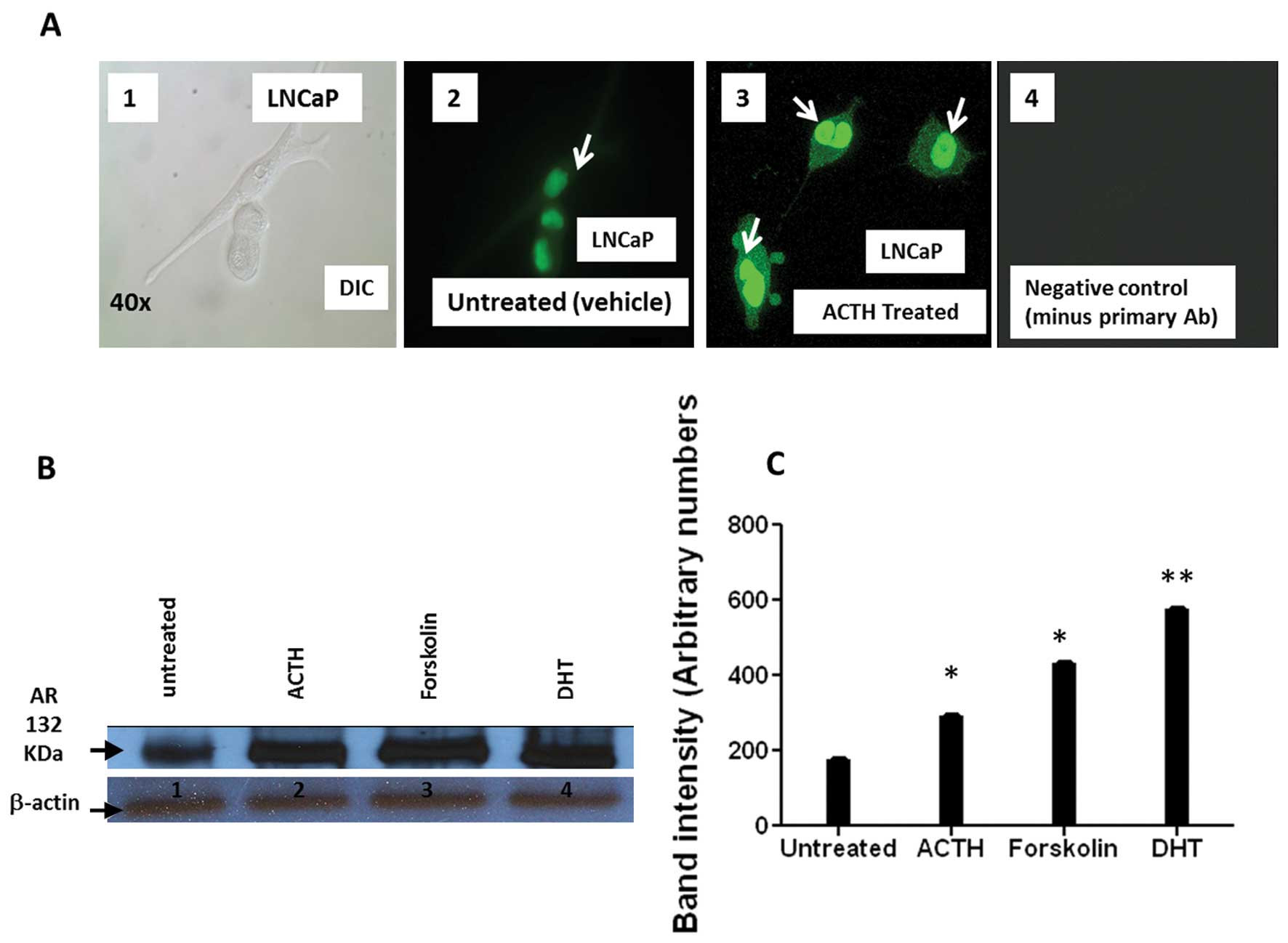
protein expression. Immunocytochemistry showed increased AR nuclear
signal intensity following ACTH treatment compared to that seen in
vehicle treated cells (Fig. 6A).
Similarly, western blot analysis of ACTH treated LNCaP cells
confirmed an increase in total AR expression (Fig. 6B and C). Forskolin, a cAMP inducer
and AR activator that mimics ACTH action, and DHT, a potent AR
agonist, were included as positive controls. The results of these
experiments indicate that ACTH treatment enhances AR nuclear
translocation and increases AR protein in PCa cells (Fig. 6A–C).
ACTH treatment increases cAMP
concentration
Cyclic AMP is a critical mediator of cell
proliferation and differentiation; it induces cAMP-dependent PKA
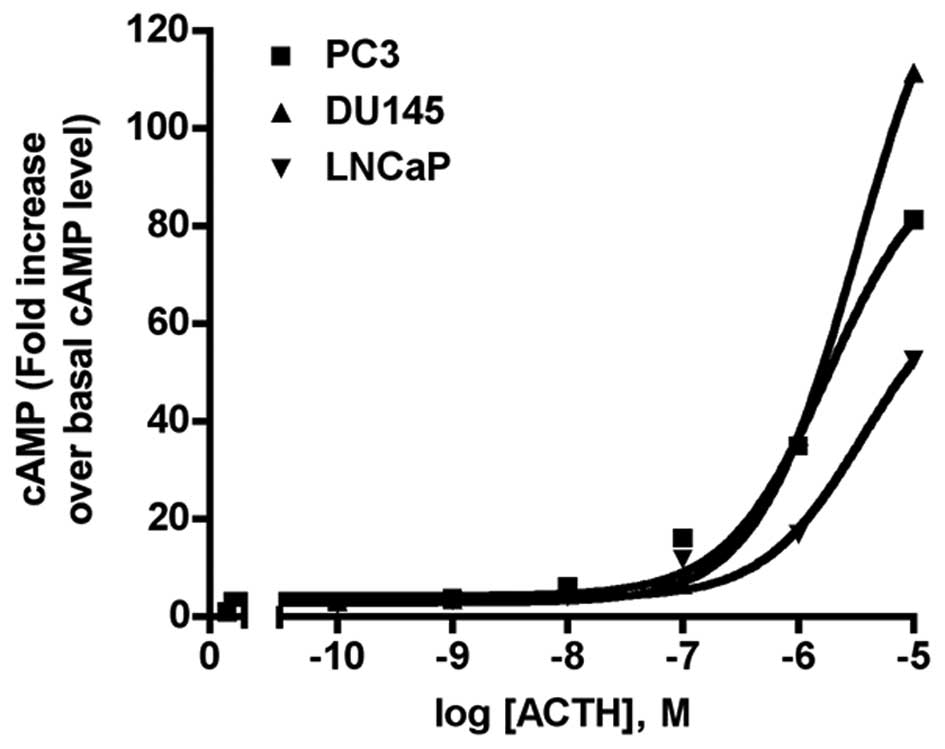
production in various cell types. As shown in Fig. 7, all three PCa cell lines responded
to ACTH stimulation with concentration-dependent cAMP production
increases. The mean EC50, from three independent
experiments was, 1.79 μM for PC-3; 3.20 μM for DU-145; and 3.67 μM
for LNCaP. The highest ACTH concentration (10 μM) increased the
cAMP level 81-fold in PC3 cells, 111-fold in DU-145 cells, and
52-fold in LNCaP cells. There was no increase in cAMP production
following NDP-MSH treatment (n=3, data not shown).
MRAP-α and -β are expressed in PCa
cells
As coexpression of MC2R accessory protein isoforms α
and β (MRAP-α and -β) is required for cAMP signaling via MC2R in
some cells such as human embryonic kidney 293 cells (36), we examined whether MRAP-α and
MRAP-β mRNA transcripts are expressed in LNCaP, PC3, and DU-145 PCa
cells. RT-PCR analysis showed amplification of both transcripts in
all three cell lines (Fig. 8).
Positive control cDNA from the adrenal gland was amplified in
parallel for validation of the PCR products.
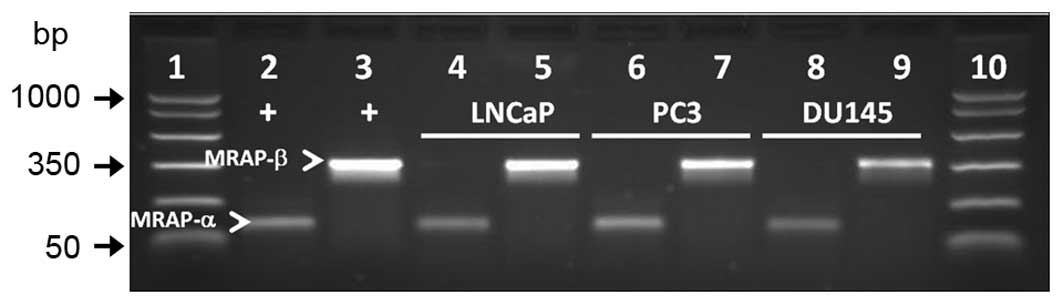 | Figure 8MRAP-α and MRAP-β (arrowheads) are
expressed in LNCaP, PC3, and DU-145 human PCa cell lines. PCR
product sizes for MRAP-α and MRAP-β are 100 and 350 bp,
respectively. Lane 1, molecular weight DNA markers; lanes 2 and 3,
positive controls for MRAP-α and MRAP-β, respectively (adrenal
DNA). Lanes 4 and 5, LNCaP; lanes 6 and 7, PC3; lanes 8 and 9,
DU-145. Lane 10, molecular weight DNA markers. |
Discussion
The findings reported here strongly link the
ACTH-MC2R signaling pathway to PCa progression. This conclusion is
based on several observations. Transcripts for MCRs, as well as
MC2R protein, are expressed in LNCaP, PC3 and DU-145 human PCa cell
lines. ACTH, the primary ligand for MC2R, increased cell
proliferation/viability in the androgen dependent LNCaP and the
androgen-independent PC3 and DU-145 cells by upregulating cAMP.
ACTH-MC2R-induced cell proliferation was partly abrogated by
SHU9119, a universal MCR blocker. Finally, AR was transactivated
and its nuclear translocation increased in LNCaP cells following
treatment with exogenous ACTH.
Physiological and sub-nanomolar ACTH concentrations
stimulated PCa cell proliferation under serum-free conditions and
in the absence of an exogenous cAMP inducer. The mitogenic effect
was above 100% that of the control. NDP-MSH, a potent α MSH
analogue and agonist of all but MC2R, as well as
D-Trp8-γ-MSH and THIQ, highly selective agonists for
MC3R- and MC4R respectively, failed to stimulate cell
proliferation. These results were predictable since it is known
that all five MCRs respond to ACTH (37) but, among the MCRs, MC2R binds only
ACTH and lacks affinity for other melanocortins. The other MCRs
are, however, activated by both MSH and ACTH. The additional
observation that the universal MCR blocker SHU9119 reduces but does
not cancel the mitogenic effect of ACTH suggests that proliferation
is at least partially mediated by MC2R.
Such trophic effects of ACTH were reported in human
adrenal gland cells and skin melanocytes (38). Several studies documented what was
referred to as ‘ectopic ACTH syndrome’ whereby several cancers from
endocrine sites such as the pituitary gland (39) and diverse non-endocrine sites
including the prostate gland, lung, liver, pancreas, and neuronal
tissue, secrete ACTH that drives cell proliferation in these
tissues (40–42).
ACTH-driven cell proliferation was likely mediated
via cAMP induction since ACTH is known to bind MC2R, a G
protein-coupled receptor, which subsequently activates adenylyl
cyclase and results in cAMP production (43). The significant cAMP increases at
high ACTH concentrations (above 1 μM) reported here indicated that
an endogenous ACTH receptor is present in all three PCa cell lines.
Since NDP-MSH is a superpotent agonist for other MCRs, except for
MC2R, we reasoned that if the PCa cells cannot respond to NDP-MSH,
the observed ACTH-dependent cAMP production in our three PCa cell
lines should be caused by direct MC2R activation by ACTH. Indeed,
there was no cAMP production by NDP-MSH stimulation (data not
shown), suggesting that MC2R is expressed in all three cell
lines.
The enhanced transactivation of AR via this pathway
suggests cross-talk between MC2R and AR and supports the view of a
critical downstream AR role in driving PCa cell growth in
androgen-dependent and androgen-independent PCa cell growth
(5). The increased AR protein
expression following ACTH treatment suggests that the ACTH-MC2R
signaling transduction pathway contributes to enhanced AR-DNA
binding. That increased binding could promote PCa cell growth, an
effect that would be particularly important under low androgen
conditions. Such a conclusion is consonant with studies showing
that several PCa cell GPCRs are indirectly involved in AR
transactivation (13). In PCa,
several GPCRs that signal through Gαs, such as prostaglandin E2
receptors (EP2 and EP4), and α-adrenergic receptor, can
transactivate AR and induce cAMP and PKA activation which, in turn,
can enhance AR sensitivity to low androgen levels. Our data
indicate that MC2R is another GPCR that contributes to PCa cell
proliferation and survival.
The central feature of the study reported here
relates to PCa cell proliferation by ACTH-MC2R interaction that was
partially abrogated by the universal MCR blocker SHU9119. This
strongly suggests a paracrine role for ACTH and its MC2R in
regulating PCa cell growth. The fact that there are two
approximately equivalent androgen sources acting in the prostate
gland, namely T from the testis and androgen derived from the
adrenal gland, makes a strong case for including MC2R blockade as
part of a combination therapy that involves inhibitors of androgen
and other hormones.
Abbreviations:
|
MCRs
|
melanocortin receptors
|
|
ACTH
|
adreno - corticotropic hormone
|
|
AR
|
androgen receptor
|
|
MC2R
|
melanocortin receptor 2
|
|
GPCRs
|
G protein coupled-receptors
|
|
PCa
|
prostate cancer
|
Acknowledgements
This study was partially supported by
funds from the interdepartmental Research Grants Program,
Scott-Richey Research Center and the Boshell Diabetes and Metabolic
Diseases Program, College of Veterinary Medicine, Auburn
University, Auburn, AL, USA.
References
|
1
|
Woolf SH: Screening for prostate cancer
with prostate-specific antigen. An examination of the evidence. N
Engl J Med. 333:1401–1405. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Oh WK and Kantoff PW: Management of
hormone refractory prostate cancer: Current standards and future
prospects. J Urol. 160:1220–1229. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li TH, Zhao H, Peng Y, Beliakoff J, Brooks
JD and Sun Z: A promoting role of androgen receptor in
androgen-sensitive and -insensitive prostate cancer cells. Nucleic
Acids Res. 35:2767–2776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huggins C: Endocrine-induced regression of
cancers. Cancer Res. 27:1925–1930. 1967.PubMed/NCBI
|
|
5
|
Debes JD and Tindall DJ: Mechanisms of
androgen-refractory prostate cancer. N Engl J Med. 351:1488–1490.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Feldman BJ and Feldman D: The development
of androgen-independent prostate cancer. Nat Rev Cancer. 1:34–45.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nguyen MM and Wang Z: Manipulation of
androgens and alterations in the androgen receptor axis in prostate
cancer. Minerva Urol Nefrol. 60:15–29. 2008.PubMed/NCBI
|
|
8
|
Grossmann ME, Huang H and Tindall DJ:
Androgen receptor signaling in androgen-refractory prostate cancer.
J Natl Cancer Inst. 93:1687–1697. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Craft N, Shostak Y, Carey M and Sawyers
CL: A mechanism for hormone-independent prostate cancer through
modulation of androgen receptor signaling by the HER-2/neu tyrosine
kinase. Nat Med. 5:280–285. 1999. View
Article : Google Scholar
|
|
10
|
Linja MJ and Visakorpi T: Alterations of
androgen receptor in prostate cancer. J Steroid Biochem Mol Biol.
92:255–264. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vis AN and Schröder FH: Key targets of
hormonal treatment of prostate cancer. Part 1: the androgen
receptor and steroidogenic pathways. BJU Int. 104:438–448. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Culig Z, Hobisch A, Cronauer MV, et al:
Androgen receptor activation in prostatic tumor cell lines by
insulin-like growth factor-I, keratinocyte growth factor and
epidermal growth factor. Eur Urol. 27(Suppl 2): 45–47. 1995.
|
|
13
|
Dorsam RT and Gutkind JS:
G-protein-coupled receptors and cancer. Nat Rev Cancer. 7:79–94.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Daaka Y: G proteins in cancer: The
prostate cancer paradigm. Sci STKE: re2. 2004.PubMed/NCBI
|
|
15
|
Mendiratta P, Armstrong AJ and George DJ:
Current standard and investigational approaches to the management
of hormone-refractory prostate cancer. Rev Urol. 9:S9–S19.
2007.PubMed/NCBI
|
|
16
|
Raj GV, Barki-Harrington L, Kue PF and
Daaka Y: Guanosine phosphate binding protein coupled receptors in
prostate cancer: A review. J Urol. 167:1458–1463. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xia C, Ma W, Wang F, Hua SB and Liu M:
Identification of a prostate-specific G-protein coupled receptor in
prostate cancer. Oncogene. 13:5903–5907. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang J, Weng J, Cai Y, Penland R, Liu M
and Ittmann M: The prostate-specific G-protein coupled receptors
PSGR and PSGR2 are prostate cancer biomarkers that are
complementary to alpha-methylacyl-CoA racemase. Prostate.
1:847–857. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weng J, Wang J, Cai Y, et al: Increased
expression of prostate-specific G-protein-coupled receptor in human
prostate intraepithelial neoplasia and prostate cancers. Int J
Cancer. 113:811–818. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yowell CW and Daaka Y: G protein-coupled
receptors provide survival signals in prostate cancer. Clin
Prostate Cancer. 1:177–181. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun L, Luo J, Mackey LV, et al:
Investigation of cancer cell lines for peptide receptor-targeted
drug development. J Drug Target. 19:719–730. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gutkind JS: Regulation of
mitogen-activated protein kinase signaling networks by G
protein-coupled receptors. Sci STKE: re1. 2000.PubMed/NCBI
|
|
23
|
McDonnell TJ, Troncoso P, Brisbay SM, et
al: Expression of the Protooncogene bcl-2 in the Prostate and Its
Association with Emergence of Androgen-independent Prostate Cancer.
Cancer Res. 52:6940–6944. 1992.PubMed/NCBI
|
|
24
|
Kasbohm EA, Guo R, Yowell CW, et al:
Androgen receptor activation by G (s) signaling in prostate cancer
cells. J Biol Chem. 280:11583–11589. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Merkle D and Hoffmann R: Roles of cAMP and
cAMP-dependent protein kinase in the progression of prostate
cancer: Cross-talk with the androgen receptor. Cell Signal.
23:507–515. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Luscombe CJ, French ME, Liu S, et al:
Prostate cancer risk: Associations with ultraviolet radiation,
tyrosinase and melanocortin-1 receptor genotypes. Br J Cancer.
85:1504–1509. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wisse BE, Frayo RS, Schwartz MW and
Cummings DE: Reversal of cancer anorexia by blockade of central
melanocortin receptors in rats. Endocrinology. 142:3292–3301. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen W, Kelly MA, Opitz-Araya X, Thomas
RE, Low MJ and Cone RD: Exocrine gland dysfunction in
MC5-R-deficient mice: Evidence for coordinated regulation of
exocrine gland function by melanocortin peptides. Cell. 91:789–798.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Roy S, Pinard S, Chouinard L and
Gallo-Payet N: Adreno-corticotropin hormone (ACTH) effects on MAPK
phosphorylation in human fasciculata cells and in embryonic kidney
293 cells expressing human melanocortin 2 receptor (MC2R) and MC2R
accessory protein (MRAP)β. Mol Cell Endocrinol. 336:31–40.
2011.
|
|
30
|
Alwani RA, Neggers SJ, van der Klift M, et
al: Cushing’s syndrome due to ectopic ACTH production by
(neuroendocrine) prostate carcinoma. Pituitary. 12:280–283.
2009.
|
|
31
|
Mansour M, Schwartz D, Judd R, et al:
Thiazolidinediones/PPARγ agonists and fatty acid synthase
inhibitors as an experimental combination therapy for prostate
cancer. Int J Oncol. 38:537–546. 2011.
|
|
32
|
Tao YX, Huang H, Wang ZQ, Yang F, Williams
JN and Nikiforovich GV: Constitutive activity of neural
melanocortin receptors. Methods Enzymol. 484:267–279. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mansour M, White D, Wernette C, et al:
Pancreatic neuronal melanocortin-4 receptor modulates serum insulin
levels independent of leptin receptor. Endocrine. 37:220–230. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Marinissen MJ and Gutkind JS:
G-protein-coupled receptors and signaling networks: emerging
paradigms. Trends Pharmacol Sci. 22:368–376. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Labrie F, Belanger A, Dupont A, Luu-The V,
Simard J and Labrie C: Science behind total androgen blockade: from
gene to combination therapy. Clin Invest Med. 16:475–492.
1993.PubMed/NCBI
|
|
36
|
Roy S, Rached M and Gallo-Payet N:
Differential regulation of the human adrenocorticotropin receptor
[melanocortin-2 receptor (MC2R)] by human MC2R accessory protein
isoforms alpha and beta in isogenic human embryonic kidney 293
cells. Mol Endocrinol. 21:1656–1669. 2007.
|
|
37
|
Cone RD: Studies on the physiological
functions of the melanocortin system. Endocr Rev. 27:736–749. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Abdel-Malek Z, Swope VB, Suzuki I, et al:
Mitogenic and melanogenic stimulation of normal human melanocytes
by melanotropic peptides. Proc Natl Acad Sci USA. 92:1789–1793.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Melmed S: Pathogenesis of pituitary
tumors. Nat Rev Endocrinol. 7:257–266. 2011. View Article : Google Scholar
|
|
40
|
Liddle GW, Island DP, Ney RL, Nicholson WE
and Shimizu N: Nonpituitary neoplasms and cushing’s syndrome:
Ectopic ‘adrenocorticotropin’ produced by nonpituitary neoplasms as
a cause of Cushing’s syndrome. Arch Intern Med. 111:471–475.
1963.
|
|
41
|
Gewirtz G and Yalow RS: Ectopic ACTH
production in carcinoma of the lung. J Clin Invest. 53:1022–1032.
1974. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Singer W, Kovacs K, Ryan N and Horvath E:
Ectopic ACTH syndrome: clinicopathological correlations. J Clin
Pathol. 31:591–598. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Halkerston ID: Cyclic AMP and
adrenocortical function. Adv Cyclic Nucleotide Res. 6:99–136.
1975.PubMed/NCBI
|