Introduction
Colorectal cancer (CRC) is the second most common
cause of cancer-related death in the Western world. In the US, CRC
is the third most common cancer with nearly 150,000 new cases and
50,000 deaths estimated in 2009 (1). After surgery, the mainstay of
systemic therapy is a cytotoxic regimen based on 5-fluorouracil
combined with folinic acid, oxaliplatin and/ or irinotecan. To
this, targeted agents have been added such as the cetuximab or
bevacizumab monoclonal antibodies directed against epidermal growth
factor receptor (EGFR) or vascular endothelial growth factor
(VEGF), respectively. Despite improvements in cytotoxic
chemotherapy and the introduction of novel agents, up to 50% of
patients undergoing apparently curative surgery relapse and die of
metastatic disease. Moreover, a substantial proportion of CRC
patients (∼20%) present with metastatic disease for whom systemic
chemotherapy is palliative at best (2). Thus there is a sustained need for new
antitumor therapies that can be used either alone or in combination
with existing treatments.
The Aurora kinases are a family of three
serine/threonine kinases (Aurora A, B and C) that are important for
the accurate segregation of chromosomes into daughter cells during
mitosis (3). Aurora-A kinase (AAK)
localizes to the centrosomes and spindle poles where it is required
for correct spindle assembly. In contrast, Aurora-B kinase (ABK) is
a chromosome passenger protein required for phosphorylation of
histone H3 (phH3), correct alignment of chromosomes on the
metaphase plate and cytokinesis. Expression of the third family
member, Aurora-C kinase, is mostly restricted to the testis where
it may play a role in meiosis (4).
Expression of the Aurora kinases is frequently elevated in human
tumors including those of the colon and rectum (5). In particular, increased ABK mRNA
levels have been shown recently to be associated with poor overall
survival (OS) in patients with metastatic CRC (6).
In CRC, one of the earliest molecular events is loss
of function of the adenomatous polyposis coli (APC) gene product.
Somatic mutations in the APC gene occur in the majority of sporadic
CRC (7). Germline APC mutations
are found in the autosomal dominant condition, familial adenomatous
polyposis (FAP), where patients develop numerous colorectal
adenomas and early onset CRC (8).
APC plays a central role in the regulation of β-catenin, a key
intracellular mediator of the Wnt signaling pathway. In addition to
its critical role in the Wnt signaling pathway, the product of the
APC gene has been shown to be involved in multiple cellular
functions including chromosome segregation (9,10).
Recent studies, using CRC cell lines and patient samples, have
shown mutated APC upregulates expression of the anti-apoptotic
protein survivin (11) which in
turn activates ABK leading to catalysis of mitosis (12). This may explain the increase in
mitotic figures and cellular proliferation that is one of the
hallmarks of CRC pathology.
Targeting kinases, such as ABK, that are involved in
mitosis is an attractive therapeutic strategy, with the potential
to provide similar efficacy to conventional cytotoxic agents but
with fewer side effects, such as peripheral neuropathy. Barasertib
(formerly AZD1152) is a dihydrogen phosphate prodrug which is
rapidly converted to the hydroxyquinazoline pyrazol anilide
(barasertib-hQPA) in plasma. BarasertibhQPA is a highly potent and
selective inhibitor of ABK (Ki=0.36 nmol/l) compared to AAK
(Ki=1369 nmol/l). Consistent with what is known about the function
of ABK, treatment of tumor cells with barasertib hQPA either in
vitro or in vivo induces chromosome misalignment and
failure of cytokinesis resulting in polyploidy and cell death
(13).
Most previous studies on the effects of barasertib
on solid tumor growth have been carried out in heterotopic tumor
xenograft models where CRC cell lines have been grown
subcutaneously and not within the context of the GI tract
microenvironment. An alternate model that enables GI lesions that
develop in situ to be examined, is the
ApcMin/+ model of spontaneous intestinal
neoplasia. ApcMin/+ mice carry a germ line
mutation in the APC gene comparable to the mutation identified in
patients with inherited FAP (14).
Like patients with FAP, ApcMin/+ mice develop
multiple adenomas throughout the intestine which progress to
invasive tumors with a frequency that depends on genetic background
(15). Allowing for a close
association between the ApcMin/+ mouse model and
human CRC (14,16,17).
In the present report, we describe the antitumor
results of more extensive experiments evaluating barasertib
activity in a panel of human CRC cell lines grown in vitro
and as xenografts in immunocompromised nude mice; and in the
autochthonous ApcMin/+ mouse model. We confirm
our preliminary observations and show that barasertib-hQPA is a
potent inhibitor of CRC cell line growth both in vitro and
in vivo. As expected, growth inhibition is accompanied by
reduced phospho-histone H3 (phH3) levels, increased numbers of
polyploid cells and an increase in apoptotic cells. In the
ApcMin/+ model, treatment with barasertib
significantly reduced adenoma burden in the small bowel.
Materials and methods
Reagents
Chemicals were purchased from Sigma (Poole, UK),
unless otherwise stated. Barasertib-hQPA is an acetanilide
substituted pyrazole aminoquinazoline and barasertib is a
dihydrogen phosphate prodrug of barasertib-hQPA (18). Both were synthesized by AstraZeneca
Pharmaceuticals, Alderley Park, Macclesfield, Cheshire, UK.
Animals
All procedures, including breeding and pharmacology
studies using ApcMin/+ mice (originally obtained
from Amy R. Moser, University of Wisconsin, Madison, WI, USA) and
Swiss nude mice (nu/nu genotype; AstraZeneca, UK), were
approved by the Cancer Research UK and AstraZeneca animal ethics
committees, respectively and carried out in accordance with the UK
Home Office Animals (Scientific Procedures) Act, 1986.
Cell lines
The DMS114, HCT116, RKO, SW620, Colo205, WiDr, HCT8,
Colo320DM, and SW116 CRC cell lines were obtained from the European
Collection of Animal Cell Cultures (ECACC) or American Type Culture
Collection (ATCC). Cells were cultured in RPMI-1640 medium (Sigma)
supplemented with 10% fetal calf serum and 2 mmol/l L-glutamine and
maintained at 37°C in a humidified atmosphere of 5%
CO2/95% air as described previously (13).
Proliferation assay
Cells in log-phase growth were seeded in 96-well
tissue culture plates and allowed to adhere overnight. Cells were
then exposed to increasing concentrations of barasertib-hQPA
ranging from 0.003 to 3 μmol/l. After incubation for 96 h, cells
were fixed in 2% formaldehyde for 30 min, washed in PBS and stained
with Hoechst dye to visualize the nuclei. Replicate plates were
also stained at the start of dosing to provide a day zero reading.
Cells were then imaged on the ArrayScan II platform (Cellomics,
Pittsburgh, PA, USA) to determine cell number. Individual
GI50 values were calculated using the cell count
readings at time zero and at the end of the treatment period and
the data were summarized using the geometric mean (the average of
the logarithmic values converted back to a base 10 number,
n=2–3).
Apoptosis assay
The apoptotic effect was determined by measuring the
active form of BAK protein, an early marker of apoptosis. Cells in
96-well plates were exposed to increasing concentrations of
barasertib-hQPA for 24 h. After treatment, the compound was washed
out and the cells left to recover for 48, 96, 120 or 144 h. Cells
were then fixed and stained with Hoechst dye (1:10,000, Molecular
Probes, Eugene, CA, USA) to assess cell number and with anti-Bak
antibody (Y164)(1:1000, Abcam, Cambridge, UK) that recognizes the
active form of BAK to determine the number of apoptotic cells.
Images, from 3–4 replicate wells, were taken and endpoints
quantified using the Cellomics Arrayscan II platform (Cellomics) as
described previously (19).
Human tumor xenografts
Human tumor xenografts were established in 8- to
12-week old female nude mice by injecting 0.1 ml of a suspension of
3×105 cells (either SW620, Colo205 or HCT116) mixed
50:50 with Matrigel (BD Biosciences, UK) into the dorsal flank.
Once tumors were established (0.2–0.3 cm3) animals were
randomized into treatment groups (n=8–11 per group); barasertib
prepared in Tris buffer pH 9.0 or vehicle alone was administered
either as a continuous 48-h infusion via subcutaneous (s.c.)
implanted osmotic mini-pumps (Model 2001D, Durect Corp, USA: two 24
h pumps implanted sequentially) in accordance with the
manufacturer’s instructions or, in SW620, as an intra-peritoneal
(i.p.) injection daily for 4 consecutive days (Q1Dx4). Tumors were
measured up to three times per week with calipers, tumor volumes
were calculated and the data plotted using the geometric mean for
each group versus time. Tumor volume and percentage tumor growth
inhibition were calculated as described previously (20).
For pharmacodynamic (PD) studies, mice (3 per group)
were humanely culled at the end of barasertib administration and
tumor xenografts harvested and snap frozen. Cell suspensions were
prepared from the frozen tumors using a tissue disaggregation
system (Medimachine; BD Biosystems, Oxford, UK). Estimation of
cellular DNA content and phH3 expression and was undertaken using
previously described immunostain and flow cytometry protocols
(13,21). In brief, tumor cells were stained
using propidium iodide (Sigma), a rabbit polyclonal IgG anti-phH3
(Upstate Biotechnology, Millipore, Abingdon, UK) and a
fluorescein-conjugated goat anti-rabbit IgG (Jackson
ImmunoResearch, Stratech, Suffolk, UK). Samples were run on a
FACSCalibur flow cytometer and assessed using CellQuest software
(both Becton-Dickinson, Oxford, UK).
In vivo adenoma model
Eight-week old ApcMin/+ mice were
randomized into groups of 15 animals and treated with either
vehicle (see above) or barasertib (25 mg/kg) Q1Dx4 each week for 3
consecutive weeks. Samples were taken 2 h after the last dose of
barasertib or vehicle. To assess proliferation, bromodeoxyuridine
(BrdU, 50 mg/kg i.p.) was given 1 h before mice were humanely
culled.
Macroscopic assessment of tumor
burden
At necropsy, spleens were removed and weighed as a
marker of tumor load. The small bowel SB (duodenum, jejunum, and
ileum), and colon were removed and cleaned by rinsing with cold
PBS). The SB was divided into three equal sections: proximal (SB1),
middle (SB2) and distal (SB3). These sections and the colon were
dissected longitudinally using a recently described cutting guide
(22) and spread onto filter paper
before being fixed in Carnoy’s fluid for 1 h and stored in 70%
ethanol until analysis. Macroscopic adenoma quantification was
carried out using a dissecting microscope as described previously
(23) to assess the number, size
and overall tumor burden.
Microscopic adenoma assessment
After measurement of adenoma number and diameter,
fixed distal SB3 tissue from six mice from each group were rolled
and processed to paraffin blocks in a standard manner. Eight 4 μm
step sections, taken at 200 μm intervals, were stained with
haematoxylin and eosin (H&E) and scored morphologically.
Microadenomas were assigned to a category from 1–4: adenomas that
remained within the limits of a single villus were classified as
category 1; those within the space of >1-5 villi were classified
as category 2; >5-10 villi as category 3, and >10 villi as
category 4 (24).
Immunohistochemistry (IHC)
Sections of formalin-fixed paraffin-embedded tissue
specimens (distal small bowel) from vehicle and barasertib-treated
mice were stained for BrdU uptake using sequential incubation with
a mouse anti-BrdU monoclonal primary antibody (Abcam),
corresponding biotin-conjugated rabbit anti-mouse secondary
antibody and developed with streptavidin-peroxidase reagent and DAB
as previously described (24).
Proliferation activity was quantified using thirty well-oriented
crypts per animal (n=6 per group) also as described previously
(23,25).
Expression of phH3 was determined in sections cut
from specimens of the distal small intestine, using sequential
incubation with a mouse anti-pHH3-(Ser10) monoclonal primary
antibody (Sigma, St. Louis, MO, USA), a biotin-conjugated rabbit
anti-mouse polyclonal secondary antibody (DakoCytomation Ltd., Ely,
UK), and a streptavidin-peroxidase reagent that was developed in
DAB followed by counterstaining with haematoxylin. All cells within
25 hemi-crypts per animal in the distal intestine were examined at
high magnification (x40 objective) and DAB positive vs.
haematoxylin counterstained cells were counted (25). Six animals per group were
assessed.
Statistical analysis
All IHC measurements were scored with the slide
reader blinded to the treatment. Results are presented as group
mean ± standard error of the mean (SEM). One-way analysis of
variance (ANOVA) and Student’s t-tests were carried out to test for
statistical significance of any effects in barasertib-treated group
versus control (P-value of ≤0.05 was considered to be statistically
significant). All statistics were performed using Minitab
Statistical Software, Release 10.5 Xtra (Minitab Ltd., Coventry,
UK) and Graphpad Prism version 4.0b, 2004.
Results
Effect of barasertib-hQPA on cell growth
and survival in vitro
It has been shown previously that barasertib-hQPA is
a potent and selective inhibitor of ABK which inhibits histone H3
phosphorylation and induces polyploidy in CRC cell lines (HCT116
and SW620) (13,26). Here, we investigated the
anti-proliferative and cytotoxic effects of barasertib-hQPA in a
panel of CRC cell lines in vitro, exposed to increasing
concentrations of barasertib-hQPA for 96 h. BarasertibhQPA
treatment potently reduced the proliferation of tumor cells as
determined by cell number. In a panel of 9 tumor cell lines, the
concentrations of drug that reduced cell growth by 50% were
<0.015 μM in 5 of the lines and between 0.015–2.235 μM in the
remainder (Table I). To determine
whether barasertib-hQPA could also induce apoptosis, a BAK assay
was performed on four of the lines which had shown greatest
sensitivity in the proliferation assay. Exposure to barasertib-hQPA
for 24 h, followed by washout for up to 144 h resulted in
significant inhibition of cell growth, at drug concentrations ≥0.01
μM (Fig. 1A–D). This was
accompanied by increased BAK protein (Fig. 1E–H). Both the washout time required
for tumor cells to become apoptotic and the extent of apoptosis
varied across the cell lines tested. The induction of apoptosis was
particularly marked in the Colo-205 and RKO cell lines (Fig. 1E and H, respectively) where 40–60%
of cells stained positive for BAK at drug concentrations ≥0.1
μM.
 | Table IBarasertib-hQPA inhibits the
proliferation of a panel of colon tumor cell lines. |
Table I
Barasertib-hQPA inhibits the
proliferation of a panel of colon tumor cell lines.
| Cell line | Proliferation
GI50, μM |
|---|
| DMS114 | 0.003±0.001 |
| HCT116 | 0.010±0.002 |
| RKO | 0.012±0.008 |
| SW620 | 0.013±0.002 |
| Colo205 | 0.014±0.008 |
| WiDr | 0.109±0.006 |
| HCT8 | 0.503±0.076 |
| Colo320DM | 1.075±0.508 |
| SW116 | 2.235±0.658 |
Barasertib treatment leads to antitumor
and pharmacodynamic effects in a range of human CRC xenografts
Pharmacokinetic studies in rodents have previously
indicated that barasertib is rapidly converted in vivo to
the active moiety barasertib-hQPA (18). To further evaluate the antitumor
potential in gastrointestinal malignancies, barasertib (150
mg/kg/day; s.c. minipump infusion over 48 h) was tested in nude
mice subcutaneously implanted with CRC human tumor xenografts. In
SW620, HCT116 and Colo205 xenografts significant tumor growth
inhibitions of 79% (P<0.001, day 23), 60% (P<0.001, day 25)
and 81% (P<0.05, day 21) were observed, respectively (Fig. 2A–C). Colo205 xenografts appeared
the most sensitive to treatment with a mean tumor volume (± SEM) on
day 21 after cell implantation, of 0.42±0.19 cm3 for the
barasertib group compared to 2.24±0.75 cm3 (P<0.05)
for the vehicle control animals (Fig.
2C). A significant antitumor response was also observed in
SW620 xenografts when barasertib was administered using a
well-tolerated once-daily intraperitoneal (i.p) schedule (25 mg/kg;
i.p. Q1Dx4): the mean tumor volume (± SEM) on day 24 after cell
implantation for barasertib-treated mice was 0.74±0.07
cm3, compared to vehicle control treated mice, which had
a mean tumor volume of 1.26±0.13 cm3 (P<0.05)
(Fig. 2D).
To confirm that barasertib inhibited ABK activity
in vivo the CRC human tumor xenografts were analyzed 2 h
after termination for biomarker effects including phH3 expression,
a proximal downstream marker of ABK signaling (27) and DNA ploidy. The barasertib
infusion schedule (150 mg/kg/48 h) led to a significant reduction
in the proportion of phH3 positive tumor cells in all three CRC
xenografts (SW620, HCT116 and Colo205) when compared to respective
controls (Fig. 2E). Flow
cytometric analysis of the DNA content of SW620 and HCT116 tumors
also revealed an increased proportion of cells with a 4N and >4N
DNA content from animals that received barasertib versus vehicle
control treated mice (Fig. 2F and
G). This accumulation of cells with a 4N and >4N DNA content
is consistent with failed cytokinesis and continued cell cycle
progression and endoreduplication following inhibition of ABK
activity. Such a trend of increased DNA content was not as clear in
the Colo205 xenografts receiving barasertib, perhaps related to
their propensity to undergo apoptosis. A similar pharmacodynamic
trend (i.e. reduction in tumor PhH3 but increase in 4N and >4N
cells treated with barasertib versus control) was also observed in
the SW620 xenograft model following daily intraperitoneal
administration of barasertib (25 mg/kg/day) for 4 days (data not
shown).
Barasertib significantly reduces adenoma
burden in ApcMin/+ mice
To investigate the efficacy of inhibiting ABK
activity over time in the ApcMin/+ model we
adapted the i.p. barasertib schedule previously shown to be
efficacious in the SW620 tumor xenograft model.
ApcMin/+ mice were treated for three consecutive
weeks with either barasertib (25 mg/kg/day i.p. Q1Dx4 weekly, for 3
cycles) or vehicle alone, and at the end of the treatment phase,
intestinal tissue was examined for adenoma formation/development.
Barasertib, compared with vehicle-treated mice, reduced adenoma
number in each of the small bowel (SB) segments SB1, 2 and 3 (data
not shown). When the whole SB was considered (SB All) barasertib
reduced the mean number of adenomas by 39% from 52.31±6.18 to
31.87±5.64 (P<0.05); Table II.
There were much fewer adenomas in the colon than in the SB, due to
the lower incidence of adenomas occurring in the colon in this
model (22). Although a similar
reduction in the number of adenomas in the colon (38%) was seen in
barasertib versus vehicle-treated mice, this did not reach
statistical significance (Table
II).
| Table IIMacroadenoma analysis after a 3-week
treatment cycle of barasertib in ApcMin/+
mice. |
Table II
Macroadenoma analysis after a 3-week
treatment cycle of barasertib in ApcMin/+
mice.
| Adenoma survey
|
|---|
| Control | Barasertib (25
mg/kg/day) | Student’s t-test
P-value |
|---|
| No. of adenomas (SB
All) | 52.31±6.18 | 31.87±5.64 | 0.02 |
| No. of adenomas
(Colon) | 1.92±0.33 | 1.20±0.30 | NS |
| Diameter of
adenomas (mm) (SB All) | 0.94±0.02 | 0.78±0.08 | NS |
| Diameter of
adenomas (mm) (Colon) | 1.14±0.20 | 0.79±0.22 | NS |
| Tumor burden
(mm3) (SB All) | 22.09±3.14 | 12.67±2.13 | 0.02 |
| Tumor burden
(mm3) (Colon) | 7.68±3.58 | 3.49±1.72 | NS |
When compared with vehicle-treated controls, the
mean adenoma diameter in barasertib-treated mice appeared smaller
in both the SB and colon, although in neither case was this
statistically significant. Adenoma burden (a measure derived from
tumor number and diameter) in the small bowel was significantly
reduced by 43% from 22.09±3.14 to 12.67±2.13 mm3
(P<0.05) (Table II). Whilst
there was also a reduction in adenoma burden with barasertib in the
colon versus vehicle-treated ApcMin/+ mice of 55%
this was again not statistically significant.
In addition to analyzing the effects of barasertib
on macroscopic adenoma formation and growth, a survey of
microscopic adenomas was undertaken in which tumors were
categorized according to size: category 1 represented an adenoma
remaining within the limits of a single villus; category 2,
adenomas within the space of >1–5 villi; category 3, adenoma
size >5–10 villi; category 4, adenomas >10 villi in size. In
barasertib-treated animals there were significantly fewer larger
microadenomas (by 64, 68, 67% in categories 2, 3, 4, respectively)
suggesting that there are significant effects of barasertib on all,
but the very smallest adenomas (Table
III).
| Table IIIEffects of barasertib on microscopic
adenoma burden in ApcMin/+ mice. |
Table III
Effects of barasertib on microscopic
adenoma burden in ApcMin/+ mice.
| | Microadenoma no.
|
|---|
| Gut segment | Adenoma
category | Control | Barasertib (25
mg/kg/day) | Student’s t-test
P-value | Percentage
reduction (%) |
|---|
| Small bowel | 1 | 20.0±4.5 | 17.0±3.7 | NS | 15 |
| 2 | 35.2±6.4 | 12.8±3.5 | 0.01 | 64 |
| 3 | 19.5±4.2 | 6.3±1.7 | 0.01 | 68 |
| 4 | 7.5±2.1 | 2.5±0.7 | 0.04 | 67 |
Effect of barasertib on pharmacodynamic
endpoints and cell proliferation in vivo
Aurora-B phosphorylates histone H3 at Ser10,
exclusively during mitosis in mammalian cells (28). Samples for pharmacodynamic
endpoints were collected 2 h post the last dose of barasertib (i.p.
ApcMin/+). Absent IHC staining of phH3 (Ser10)
was taken as direct evidence of inhibition of ABK activity
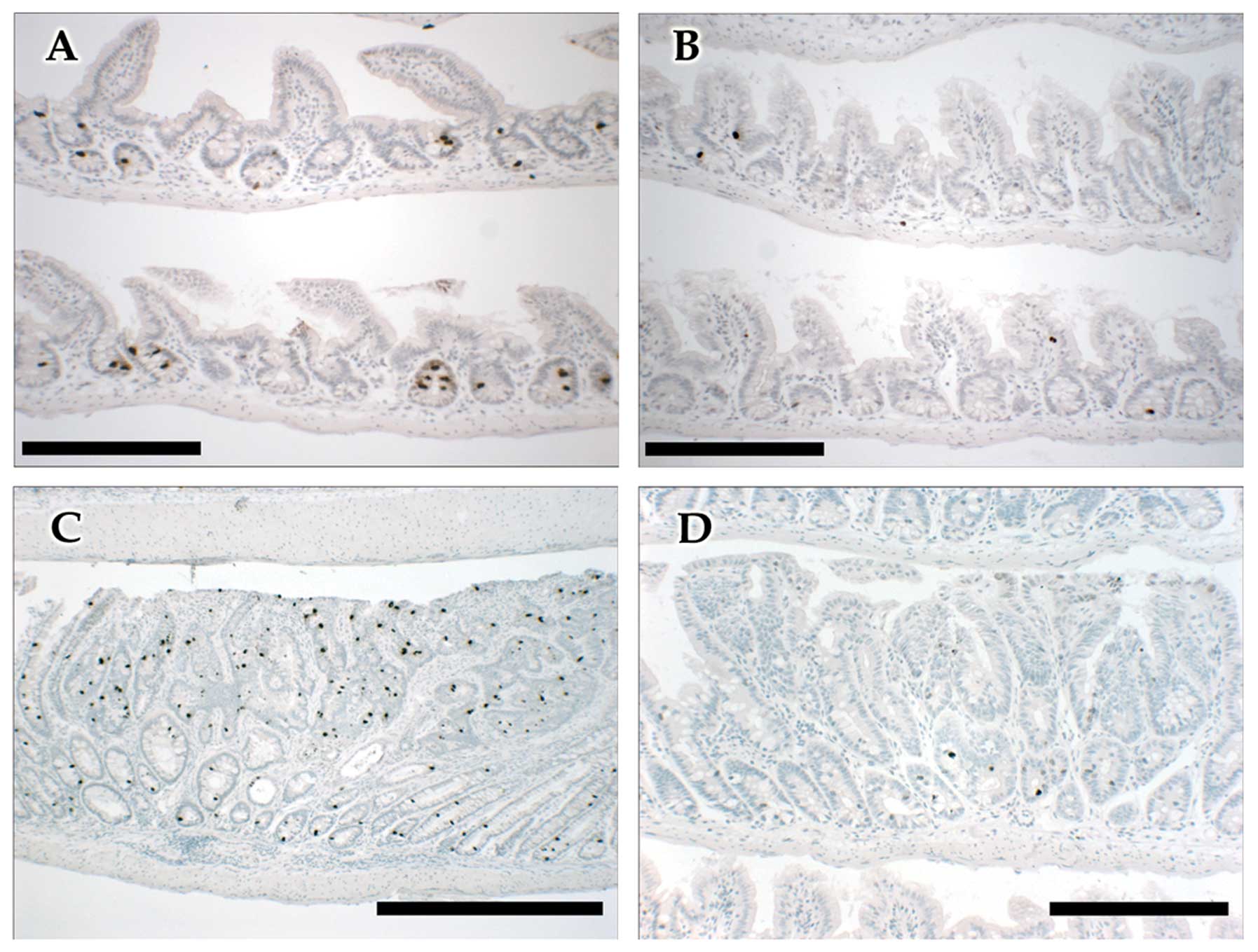
(Fig. 3).
In the adenomatous tissue, the number and proportion
(%) of phH3-positive cells in the barasertib versus the
vehicle-treated group were markedly reduced by 97% (P<0.001) and
94% (P<0.001), respectively; Table
IV, Fig. 3. Adenomas were 53%
smaller on average (P<0.001, Table
IV) in the barasertib versus vehicle-treated group and the
adenomas displayed a noticeable trend of a greater number of
apoptotic bodies when examined after H&E staining. Further, a
similar trend was observed following cleaved caspase-3 staining
(data not shown). BrdU uptake in the adenomas experienced a
decrease of 51% (P=0.01, Table
IV), indicating a possible lower division rate in the adenoma.
The comparison of BrdU labeling indices across treated and control
group did not reveal much difference in BrdU uptake, but this could
be explained by the smaller overall size of barasertib-treated
adenomas (Table IV). Normal
non-adenomatous tissue displayed similar effects but with no major
changes in tissue renewal (data not shown).
| Table IVThe effects of barasertib treatment
on proliferation markers in adenomas of the small bowel of
ApcMin/+. |
Table IV
The effects of barasertib treatment
on proliferation markers in adenomas of the small bowel of
ApcMin/+.
| Proliferation
assessment
|
|---|
| Control | Barasertib (25
mg/kg/day) | Student’s t-test
P-value |
|---|
| BrdU+
per adenoma | 241.23±36.05 | 117.01±31.66 | 0.015 |
| Mitoses per
adenoma | 7.24±1.10 | 5.98±1.60 | NS |
| phH3+
per adenoma | 37.76±5.19 | 1.00±0.34 | <0.001 |
| Cells per
adenoma | 1143.44±156.75 | 537.17±67.67 | 0.001 |
| BrdU labeling index
(%) | 21.10±1.18 | 21.78±1.34 | NS |
| Mitotic index
(%) | 0.63±0.10 | 1.11±0.27 | NS |
| phH3 labeling index
(%) | 3.30±0.34 | 0.19±0.09 | <0.001 |
Discussion
Previous research has shown that Aurora kinase
levels are elevated in several cancer types, including CRC and that
the Aurora kinase family may represent potential targets for
therapeutic intervention (29).
Studies in in vitro and in vivo tumor models support
this view, and have shown that inhibiting ABK activity produces
significant antitumor effects in both hematological (30) and solid tumors including
hepatocellular cancer (31).
Recent work in CRC has pointed towards a role for mutant APC in
activating ABK, via survivin; leading to increased mitotic rates
and a mechanism by which colon tumorigenesis is promoted (12). Here we investigate further the
potential of targeting GI tract cancers using the selective ABK
prodrug, barasertib. Barasertib-hQPA (the active moiety of
barasertib) inhibits tumor cell growth and induces apoptosis in a
concentration- and time-dependent manner across a panel of CRC cell
lines. Consistent with previous reports (32,33),
barasertib-hQPA caused a lower induction in apoptosis in HCT116
cells and required prolonged exposures (Fig. 1F). A similar effect was observed in
SW620 cells whereas in Colo205 and RKO cells the extent of
apoptosis was more pronounced, reaching 40–60% at concentrations of
0.1 μmol/l barasertib-hQPA. The fact that cell growth was inhibited
after a 24 h exposure to barasertib-hQPA followed by extended
washout periods suggests that an intermittent schedule may be an
effective treatment in CRC. The difference in response rates to
barasertib between the CRC cell lines may underlie differences in
the expression of proteins involved in cell cycle
checkpoint/apoptotic pathways as well as differences in growth
rate, % of polyploidy and /or DNA content of polyploidy cells.
Differences in intracellular drug uptake between cell lines could
also be responsible; previous studies on the effects of barasertib
in AML cells have indicated expression of the transporter proteins
P-glycoprotein and BCRP1 can have a negative impact on
barasertib-hQPA efficacy (34).
We also demonstrated that short-term treatment (48
h, osmotic minipump) with barasertib produced durable anti-tumor
effects in vivo in three established subcutaneous CRC
xenografts in nude mice. Similar preclinical activity has recently
been documented in both a CRC cell line-derived xenograft (HT29)
and 3 patient-derived explants with ENMDA-2076, a multi-targeted
anti-angiogenic and Aurora kinase inhibitor (35). Whilst Colo205 cells more readily
underwent apoptosis following barasertib treatment in vitro
and also appeared to be the most sensitive model in vivo,
barasertib induced significant antitumor activity in both SW620 and
HCT116 models. Differences between in vitro and in
vivo barasertib data may reflect the multi-cellular
contribution of the tumor microenvironment (including stroma) found
in tumor xenografts versus the mono-cellular conditions of cell
culture assays used in this study. Pharmacodynamic analysis in CRC
xenografts indicated the antitumor effects were associated with
reduced phH3 expression and accumulation of cells with 4N and
>4N DNA content. These findings are consistent with failed
chromosome separation and thus failed cytokinesis but continued
cell cycle progression following inhibition of ABK activity. These
data indicate that the mechanism of action of barasertib in
vivo recapitulates that observed with barasertib-hQPA in
vitro and with other ABK inhibitors (13,36).
Interestingly, pharmacodynamic analysis of barasertib-treated
Colo205 xenografts did not indicate such a strong trend for
enriched 4N and ploidy (>4N) tumor cells. Colo205 cells were
observed to be very pro-apoptotic in in vitro assays and
sensitive in in vivo growth inhibition studies. The weaker
trend in increased DNA content following barasertib treatment in
vivo may have reflected cells undergoing early apoptosis rather
than endo-reduplication. Consistent with this, flow cytometric
analysis of tumor cells from Colo205 xenografts showed there to be
a higher proportion with a sub-G1 DNA content, a marker of
apoptosis in this model (data not shown).
Inhibition of ABK activity in an autochthonous model
of CRC, such as the ApcMin/+ mouse which is
considered as well validated model of intestinal cancer, has not
yet been reported (37,38). The ApcMin/+ mouse
has several advantages over standard CRC in vivo models,
such as human tumor xenografts, in that the mouse strain is
immunocompetent and the adenomas grow in situ within the
context of the complex intestinal mucosal microenvironment. In the
ApcMin/+ mouse model we found a marked reduction
in the number of detectable adenomas throughout the SB following
the three-week treatment with barasertib, with the most pronounced
effect being seen in the distal segment where most of the lesions
occurred. In addition to the reduction in the number of adenomas,
the sizes of the lesions in the SB were also reduced leading to a
significant reduction in tumor burden. Tumor burden in the colon
was also reduced, but this did not reach statistical significance
and is most likely due to the relatively small number of adenomas
that are found in the colon. The number of colonic adenomas was low
in the current study, but it is known that the number of colonic
adenomas in the ApcMin/+ mouse is variable, and
may be in part be a reflection the inflammatory environment in the
intestines which can be affected by gut micro-flora which in turn
can be influenced by specific pathogen-free conditions of a
vivarium (39,40).
Microscopic analysis refined the quantification of
adenomas and confirmed that the ApcMin/+ model
developed adenomas of various sizes, and demonstrated that the
incidence of all but the smallest adenomas (those occupying space
within a single villus) were substantially reduced by treatment
with barasertib. It is important to note that under treatment
larger adenomas were less numerous (category 3 and 4) potentially
due to the pro-apoptotic and anti-proliferative effects of
barasertib. It is also possible that barasertib treatment could
de-bulk larger adenomas to smaller sizes (e.g. from category 3 or 4
to category 1), and therefore account for the reason why the
difference in category 1 adenomas between barasertib and vehicle
control ApcMin/+ mice was not statistically
significant in this study. From these data we cannot fully
ascertain whether the effects of barasertib are to prevent adenoma
growth, to shrink adenomas, or both. Further work (including
short-term exposures) would be required to investigate this
further.
Scoring the histological sections of SB from
barasertib treated ApcMin/+ mice revealed a
substantial suppression of ABK activity in both histologically
normal crypts and adenomas, as measured by phH3 immunostaining.
Mitotic index was also increased in both normal crypts and
adenomas. Taken together, the in vivo data indicate that the
biological response of intestinal tissue in
ApcMin/+ mice parallels the response of SW620 and
HCT116 xenografts following inhibition of ABK activity by
barasertib. Although not directly comparable, it is noteworthy that
the extent of reduced phH3 staining following barasertib in the
SW620 xenograft by flow cytometry appeared substantially less than
that seen in the adenomas/crypts from ApcMin/+
mice. However, this may have reflected differences in the
methodologies used and the duration of barasertib treatment which
was much shorter in the CRC xenografts. Furthermore, the tumor
burden was much larger in the CRC xenograft studies compared with
the ApcMin/+ mouse study, which could in part
contribute to a smaller effect on phH3 levels. Recent work by Zhang
et al address the relationship of APC regulating ABK
activity and its effect of phH3 distribution in colonic crypt via
survivin, an increase in ABK activity leads to higher phH3 position
within APC mutated crypts and adenoma (12). Interestingly, the IHC phH3 staining
pattern in the vehicle control sections of the
ApcMin/+ mice (Fig.
3) was reminiscent of the clinical counterparts described the
Zhang et al study (12).
Namely, phH3 staining confined to lower intestinal crypts in the
normal tissue and a more stochastic distribution throughout the
adenomas.
The lack of an apparent effect of barasertib on BrdU
labeling index is likely to have resulted from smaller adenomas and
the rapid apoptosis of cells undergoing aberrant mitosis. Since
apoptotic cells are likely to be promptly eliminated from the
adenoma tissue (and therefore not contribute significantly to the
assessment of labeling index), the overall proportion of cells in
S-phase could appear relatively unaffected, in particular if the
tumor was shrinking in response to drug treatment. Although we did
not directly assess tumor shrinkage in the present study, the
number of cells per adenoma was significantly lower in
barasertib-treated animals compared with controls. Assessment of
cleaved caspase-3 showed a trend in apoptosis accumulation
following treatment although this effect was not significant (data
not shown). The later may be explained due to the nature of the
intestinal gut, being a site of high cell turnover, which would
have allowed for the clearing of apoptotic cells prior to analysis.
Future work taking earlier samples for analysis may give additional
insights into these results.
Our data in CRC cell lines and xenografts and in
ApcMin+ mice provide supportive evidence that
barasertib treatment inhibits ABK and reduces phH3 levels in
dividing tumor cells. This results in aberrant mitosis, aneuploidy,
polyploidy and apoptosis thereby suppressing the growth of tumor
xenografts or intestinal adenomas. The results of the present study
confirm that ABK activity plays a substantial role in the growth of
established CRC tumor models, but more interestingly it also plays
a role in the growth of very early pre-carcinogenic lesions in the
intestinal tract of ApcMin+ mice. Selective ABK
inhibitors are currently being evaluated in various clinical trials
and the present study provides a rationale for extending the scope
of clinical evaluation to include earlier stages of CRC. Barasertib
has been tested in both solid and hematological cancer patients. In
the case of solid tumors a 2-h infusion schedule, every 7 or 14
days, was investigated (41).
Neutropenia was the most frequently reported adverse event/
dose-limiting toxicity and the best observed responses in this
trial were prolonged disease stabilizations in some patients. As
with other therapies targeted against cycling cells, care and
attention will be needed to identify the optimum dosing regime in
order to achieve a good therapeutic margin.
Abbreviations:
|
CRC
|
colorectal cancer
|
|
APC
|
adenomatous polyposis coli
|
|
Min
|
Multiple intestinal neoplasia
ApcMin/+
|
|
ABK
|
Aurora-B kinase
|
|
phH3
|
phospho-histone H3
|
|
Barasertib-hQPA
|
barasertibhydroxy quinazoline pyrazol
anilide
|
Acknowledgements
We thank Dr Kate Byth at AstraZeneca
for helpful feedback and discussions during the drafting of this
manuscript. We acknowledge the assistance of both the CDMG
(AstraZeneca) and Clare Hall (CRUK) for in vivo efficacy
studies. Further, we thank George Elia for immunohistochemistry
support. Financial support for this study was provided by
AstraZeneca. D.A. is funded by a Biology and Biotechnology Science
Research Council (BBSRC) CASE research studentship in conjunction
with AstraZeneca.
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar
|
|
2
|
Chee CE and Sinicrope FA: Targeted
therapeutic agents for colorectal cancer. Gastroenterol Clin North
Am. 39:601–613. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Keen N and Taylor S: Aurora-kinase
inhibitors as anticancer agents. Nat Rev Cancer. 4:927–936. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang KT, Li SK, Chang CC, et al: Aurora-C
kinase deficiency causes cytokinesis failure in meiosis I and
production of large polyploid oocytes in mice. Mol Biol Cell.
21:2371–2383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bischoff JR, Anderson L, Zhu Y, et al: A
homologue of Drosophila Aurora kinase is oncogenic and amplified in
human colorectal cancers. EMBO J. 17:3052–3065. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pohl A, Azuma M, Zhang W, et al:
Pharmacogenetic profiling of Aurora kinase B is associated with
overall survival in metastatic colorectal cancer. Pharmacogenomics
J. 11:93–99. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ilyas M, Straub J, Tomlinson IP and Bodmer
WF: Genetic pathways in colorectal and other cancers. Eur J Cancer.
35:1986–2002. 1999. View Article : Google Scholar
|
|
9
|
Senda T, Iizuka-Kogo A, Onouchi T and
Shimomura A: Adenomatous polyposis coli (APC) plays multiple roles
in the intestinal and colorectal epithelia. Med Mol Morphol.
40:68–81. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hanson CA and Miller JR: Non-traditional
roles for the Adenomatous Polyposis Coli (APC) tumor suppressor
protein. Gene. 361:1–12. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Boman BM, Walters R, Fields JZ, et al:
Colonic crypt changes during adenoma development in familial
adenomatous polyposis: immunohistochemical evidence for expansion
of the crypt base cell population. Am J Pathol. 165:1489–1498.
2004. View Article : Google Scholar
|
|
12
|
Zhang T, Fields JZ, Opdenaker L, et al:
Survivin-induced Aurora-B kinase activation: A mechanism by which
APC mutations contribute to increased mitoses during colon cancer
development. Am J Pathol. 177:2816–2826. 2010. View Article : Google Scholar
|
|
13
|
Wilkinson RW, Odedra R, Heaton SP, et al:
AZD1152, a selective inhibitor of Aurora B kinase, inhibits human
tumor xenograft growth by inducing apoptosis. Clin Cancer Res.
13:3682–3688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Preston S, Leedham S, Oukrif D, et al: The
development of duodenal microadenomas in FAP patients: the human
correlate of the Min mouse. J Pathol. 214:294–301. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Minde DP, Anvarian Z, Rüdiger SG and
Maurice MM: Messing up disorder: how do missense mutations in the
tumor suppressor protein APC lead to cancer? Mol Cancer.
10:1012011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen LC, Hao CY, Chiu YS, et al:
Alteration of gene expression in normal-appearing colon mucosa of
APC(min) mice and human cancer patients. Cancer Res. 64:3694–3700.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Corpet DE and Pierre F: How good are
rodent models of carcinogenesis in predicting efficacy in humans? A
systematic review and meta-analysis of colon chemoprevention in
rats, mice and men. Eur J Cancer. 41:1911–1922. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mortlock AA, Foote KM, Heron NM, et al:
Discovery, synthesis, and in vivo activity of a new class of
pyrazoloquinazolines as selective inhibitors of aurora B kinase. J
Med Chem. 50:2213–2224. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cummings J, Hodgkinson C, Odedra R, et al:
Preclinical evaluation of M30 and M65 ELISAs as biomarkers of drug
induced tumor cell death and antitumor activity. Mol Cancer Ther.
7:455–463. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wedge SR, Ogilvie DJ, Dukes M, et al:
ZD4190: an orally active inhibitor of vascular endothelial growth
factor signaling with broad-spectrum antitumor efficacy. Cancer
Res. 60:970–975. 2000.
|
|
21
|
Widrow RJ, Rabinovitch PS, Cho K and Laird
CD: Separation of cells at different times within G2 and mitosis by
cyclin B1 flow cytometry. Cytometry. 27:250–254. 1997. View Article : Google Scholar
|
|
22
|
Alferez D, Ryan A, Goodlad R, Wright N and
Wilkinson R: Effects of vandetanib on adenoma formation in a
dextran sodium sulphate enhanced ApcMIN/+ mouse model.
Int J Oncol. 37:767–772. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alferez D, Wilkinson RW, Watkins J, et al:
Dual inhibition of VEGFR and EGFR signaling reduces the incidence
and size of intestinal adenomas in Apc(Min/+) mice. Mol Cancer
Ther. 7:590–598. 2008.PubMed/NCBI
|
|
24
|
Goodlad RA, Ryan AJ, Wedge SR, et al:
Inhibiting vascular endothelial growth factor receptor-2 signaling
reduces tumor burden in the ApcMin/+ mouse model of early
intestinal cancer. Carcinogenesis. 27:2133–2139. 2006.PubMed/NCBI
|
|
25
|
Alferez D and Goodlad R: To best measure
cell proliferation in samples from the intestine. Cell Prolif.
40:231–240. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nair JS, Ho AL, Tse AN, et al: Aurora B
kinase regulates the postmitotic endoreduplication checkpoint via
phosphorylation of the retinoblastoma protein at serine 780. Mol
Biol Cell. 20:2218–2228. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ditchfield C, Johnson VL, Tighe A, et al:
Aurora B couples chromosome alignment with anaphase by targeting
BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 161:267–280.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ota T, Suto S, Katayama H, et al:
Increased mitotic phosphorylation of histone H3 attributable to
AIM-1/Aurora-B overexpression contributes to chromosome number
instability. Cancer Res. 62:5168–5177. 2002.PubMed/NCBI
|
|
29
|
Lens SM, Voest EE and Medema RH: Shared
and separate functions of polo-like kinases and aurora kinases in
cancer. Nat Rev Cancer. 10:825–841. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oke A, Pearce D, Wilkinson RW, et al:
AZD1152 rapidly and negatively affects the growth and survival of
human acute myeloid leukemia cells in vitro and in vivo. Cancer
Res. 69:4150–4158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aihara A, Tanaka S, Yasen M, et al: The
selective Aurora B kinase inhibitor AZD1152 as a novel treatment
for hepatocellular carcinoma. J Hepatol. 52:63–71. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nair JS, de Stanchina E and Schwartz GK:
The topoisomerase I poison CPT-11 enhances the effect of the aurora
B kinase inhibitor AZD1152 both in vitro and in vivo. Clin Cancer
Res. 15:2022–2030. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Azzariti A, Bocci G, Porcelli L, et al:
Aurora B kinase inhibitor AZD1152: determinants of action and
ability to enhance chemotherapeutics effectiveness in pancreatic
and colon cancer. Br J Cancer. 104:769–780. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Grundy M, Seedhouse C, Russell NH and
Pallis M: P-glycoprotein and breast cancer resistance protein in
acute myeloid leukaemia cells treated with the aurora-B kinase
inhibitor barasertib-hQPA. BMC Cancer. 11:2542011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tentler JJ, Bradshaw-Pierce EL, Serkova
NJ, et al: Assessment of the in vivo antitumor effects of
ENMD-2076, a novel multi-targeted kinase inhibitor, against primary
and cell line-derived human colorectal cancer xenograft models.
Clin Cancer Res. 16:2989–2998. 2010. View Article : Google Scholar
|
|
36
|
Girdler F, Gascoigne KE, Eyers PA, et al:
Validating Aurora B as an anti-cancer drug target. J Cell Sci.
119:3664–3675. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Boivin GP, Washington K, Yang K, et al:
Pathology of mouse models of intestinal cancer: consensus report
and recommendations. Gastroenterology. 124:762–777. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Paulsen JE: Modulation by dietary factors
in murine FAP models. Toxicol Lett. 112–113:403–409.
2000.PubMed/NCBI
|
|
39
|
Rhodes JM and Campbell BJ: Inflammation
and colorectal cancer: IBD-associated and sporadic cancer compared.
Trends Mol Med. 8:10–16. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Alison M: The Cancer Handbook. John Wiley
& Sons, Chichester; West Sussex, Hoboken, NJ: 2007
|
|
41
|
Boss DS, Witteveen PO, van der Sar J, et
al: Clinical evaluation of AZD1152, an i.v. inhibitor of Aurora B
kinase, in patients with solid malignant tumors. Ann Oncol.
22:431–437. 2011. View Article : Google Scholar : PubMed/NCBI
|