Introduction
Multicellular organisms stringently control the
activities of their cells and tissues by a complex network of
signaling pathways. This network guarantees that cells grow,
proliferate and divide in a systematic manner within the organism.
Conditions leading to the deregulation of these signaling pathways
often result in cancer. The Ras-Raf-MAPK signaling cascade is
crucial in maintaining cell homeostasis, as it plays a diverse role
in various biological processes (1). The overexpression or constitutive
activation of any one of a large number of receptor tyrosine
kinases (RTK) leads to Ras activation. The Ras proteins are
products of one of the earliest identified oncogenes, and ∼30% of
human epithelial tumors frequently possess activating mutations of
a ras gene (H-, N-or K-ras) (2,3).
Activation of Ras results in the stimulation of downstream
signaling cascades of which the Raf-MEK-ERK cascade is the best
characterized candidate. Ras has proven to be problematic for
intervention by pharmacological agents (4). Consequently, Raf and MEK are regarded
as key protein kinase candidates for anticancer drug design. Ras
interacts with and activates the Raf family of kinases, which in
turn phosphorylate and activate the mitogen-activated protein
kinase kinase 1/2 (MEK1/2). MEK ultimately phosphorylates and
activates intracellular serine/threonine kinases termed
mitogen-activated protein kinases or extracellular signal-regulated
kinases (MAPK/ERK). The MAPK cascade is an important regulator of
cell proliferation, differentiation and apoptosis (5). Thus, blocking the MAPK cascade may
provide the means to increase cancer cell death.
Raf kinase is directly downstream of Ras in the MAPK
cascade. The Raf kinase family comprises a-Raf, b-Raf and Raf-1,
which are thought to have both overlapping and unique regulatory
functions (6). Active mutants of
Raf-1 and b-Raf possess transforming activity in vitro and
have also been identified in a number of human tumors (7,8).
Activating mutations in Ras or RTKs also result in activation of
the Raf-1 kinase, thus conferring a growth advantage on the cancer
cell. Evidence from mouse knockouts has demonstrated a previously
unknown pro-survival role for Raf-1 and b-Raf (9–12).
These kinases are essential in the prevention of apoptosis during
mouse development. Thus, inhibition of Raf-1 or b-Raf kinases may
sensitize cancer cells to apoptotic stimuli. The pharmacological
inhibition of Raf kinases has been extensively explored with
limited success, and results of anti-c-Raf RNA aptamers and
peptides that block Ras-Raf interaction have shown some
promise.
To evaluate Raf kinases as targets to block growth
or enhance cancer cell death, the expression of Raf-1 and b-Raf was
silenced by RNA interference (RNAi) and the effects of this
depletion on growth and apoptosis were determined. The effects of
MEK inhibition were evaluated in assays used in this study. The
results showed that silencing Raf-1 or b-Raf or MEK alone causes a
modest increase in cancer cell death. However, a combination
treatment that depletes both Raf-1 and MEK provides a significant
increase in colon cancer cell death. These data suggest that tumors
with activated ras or a hyperactive MAPK cascade (perhaps
via constitutively active Ras or b-Raf mutants) may be sensitive to
a multi-pronged attack on the MAPK cascade with Raf and MEK
inhibitors.
Materials and methods
Cell culture
Cos-7 cells and cancer cell lines HCT116, HT29,
Colo205, ME180 and MCF-7 were purchased from ATCC and propagated in
Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented
with 10% fetal bovine serum (FBS) (JRH Biosciences) and 1x
penicillin/streptomycin (Invitrogen) at 37°C in 10%
CO2.
RNA interference
Cells (3x105/well) were plated in 6-well
plates (Nalgene). Oligonucleotides encoding the RNA of choice (IDT,
Coralville, IA, USA) were subcloned into pSUPER or pSUPER-retro
(Oligoengine, Seattle, WA, USA) using the Oligoengine protocol.
Cancer cells were grown overnight at 37°C in 10% CO2 and
then transfected with 1 μg of pSUPER-Raf-1 RNAi
(GATCCCCGTGATGCTGTCCACTCGGATTCAAGAGAT CCGAGTGGACAGCATCACTTTTTGGAAA)
or pSUPER vector alone or pSUPER -b-Raf wt RNAi (GATCCCCGCTA
CAGTGAAATCTCGATTTCAAGAGAATCGAGATTTCA CTGTAGCTTTTTGGAAA) or
pSUPER-b-Raf VE RNAi (GATCCCCGCTACAGAGAAATCTCGATTTCAAGAGAA
TCGAGATTTCTCTGTAGCTTTTTGGAAA), using Lipofectamine™ 2000
(Invitrogen), according to the manufacturer’s instructions. After
72 h, cells were lysed in 1% NP40 lysis buffer and cytosolic
lysates were separated by 10% SDS-PAGE and immunoblotted to detect
Raf-1, b-Raf or actin.
Western blotting
Cytosolic lysates were prepared in 1% NP40 lysis
buffer (150 mM NaCl, 10 mM HEPES pH 7.45, 1% NP40, 5 mM each of
Na4P2O7 and NaF, 2 mM
Na3VO4, 10 μg/ml each of Aprotinin and
Leupeptin, 1 mM PMSF) and protein concentration was determined
using the Bio-Rad DC™ protein assay. Protein (25
μg) was loaded in each well, resolved by 10% SDS-PAGE and
transferred to nitrocellulose membranes (Schleicher & Schuell).
Western blotting was carried out using the following antibodies:
Raf-1 and b-Raf (sc-133, sc-5284; Santa Cruz Biotechnology, Inc.),
actin (AC-40; Sigma), phospho-MEK and phospho-ERK (9121 and 9101;
Cell Signaling), cytochrome c (65981A; BD-Pharmingen), GST
(05-311, Upstate Biotechnology). The blots were developed using
enhanced chemiluminescence (ECL, Amersham Biosciences, Inc.) and
quantified using Bio-Rad Fluor-Max 700.
Cell cycle and apoptosis
Cancer cells transfected with Raf-1/b-Raf/luciferase
shRNA were treated for 24 h with 5 μM MEK inhibitor (U0126)
or vehicle (DMSO). Medium containing floating cells was collected
and pooled with trypsinized cells, and total cells were obtained by
centrifugation at 1000 rpm at 4°C. Cell pellets were washed with 1X
PBS, resuspended and fixed overnight in 70% ethanol at 4°C.
Subsequently, the cells were stained with propidium iodide and
analyzed by flow cytometry for cell cycle and apoptosis. Similar
experiments were conducted in combination with chemotherapeutic
drugs (10 nM paclitaxel, 10 μM cisplatin).
Cell growth assay
Cells stably transfected with Raf-1/b-Raf/luciferase
shRNA/empty vector were selected and seeded at 5x102
cells/well in a 96-well plate. After 24 h, the MTS reagent
(CellTiter, Promega, WI, USA) was added to each well (333
μg/ml MTS) and cells were incubated at 37°C, 10%
CO2, 4 h. Bioreduction of formazan by endogenous
esterases in viable cells was colorimetrically quantified using a
spectrophotometer at 490 nm. The amount of converted formazan was
directly proportional to the number of viable cells in culture.
Soft agar assay
Cells were transfected with 1 μg pSUPER or
pSUPER-retro plasmid (Raf-1 shRNA, b-Raf shRNA or luciferase shRNA)
and 0.1 μg of pBabe-puromycin plasmid, and selected with 2
μg/ml puromycin. Clones were selected and expanded for 1
month and analyzed for Raf-1 or b-Raf protein by western blotting.
Cells (2x104) in DMEM/10% FBS were suspended in 1.5 ml
of 0.3% agarose and seeded in triplicate in 35-mm plates coated
with 1.5 ml of 0.6% agarose/DMEM/10% FBS. Cells were grown at 37°C
in 10% CO2 and the number of colonies formed was scored
after 2 weeks.
Results
Raf-1 and b-Raf silencing by RNAi
To determine whether therapeutic intervention in the
MAPK cascade at Raf is feasible and potentially effective in
killing cancer cells, we initially silenced Raf-1 expression using
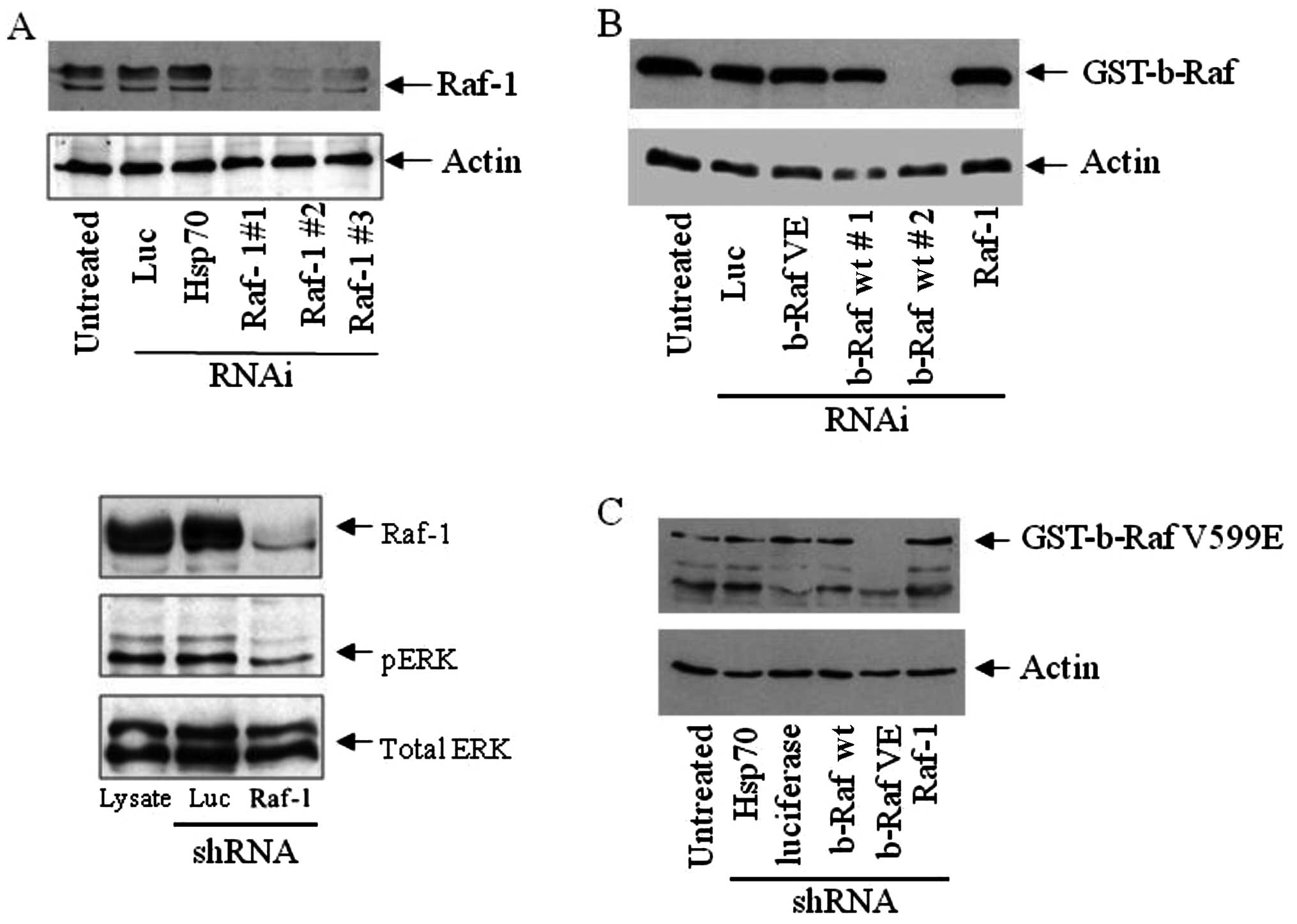
RNAi. As shown in Fig. 1A, Raf-1
was effectively silenced in epithelial cancer cells using three
different target sites for shRNAs directed against c-Raf. A control
shRNA vector targeting Hsp70 or luciferase did not deplete Raf-1,
revealing the specificity of Raf-1 silencing. The expression of
shRNA targeting b-Raf wt did not alter Raf-1 protein levels, and
vice versa (Fig. 2B and Fig. 1B). Subsequent experiments were
carried out using the shRNA vector, which targets site 1 (Raf-1 #1
in Fig. 1A). Raf-1 silencing
resulted in a modest decrease in ERK1/2 phosphorylation but did not
change the total ERK expression (Fig.
1A). Additionally, shRNA constructs targeting b-Raf wt
(Fig. 1B) and b-Raf VE (Fig. 1C) selectively inhibited the
specific protein expression.
Raf-1 depletion in HCT116 does not alter
proliferation but increases apoptosis
Raf-1 is a key mediator of the MAPK cascade in cells
and its activation ultimately facilitates cell growth,
differentiation and proliferation. Moroever, it is reported to
protect cells from apoptosis. Based on these data, we expected that
silencing of Raf-1 would decrease cancer cell growth and/or
increase their apoptotic index. To test this model, we utilized
HCT116 colon cancer cells that either stably expressed pSUPER-retro
targeting luciferase or Raf-1 or b-Raf wt under puromycin
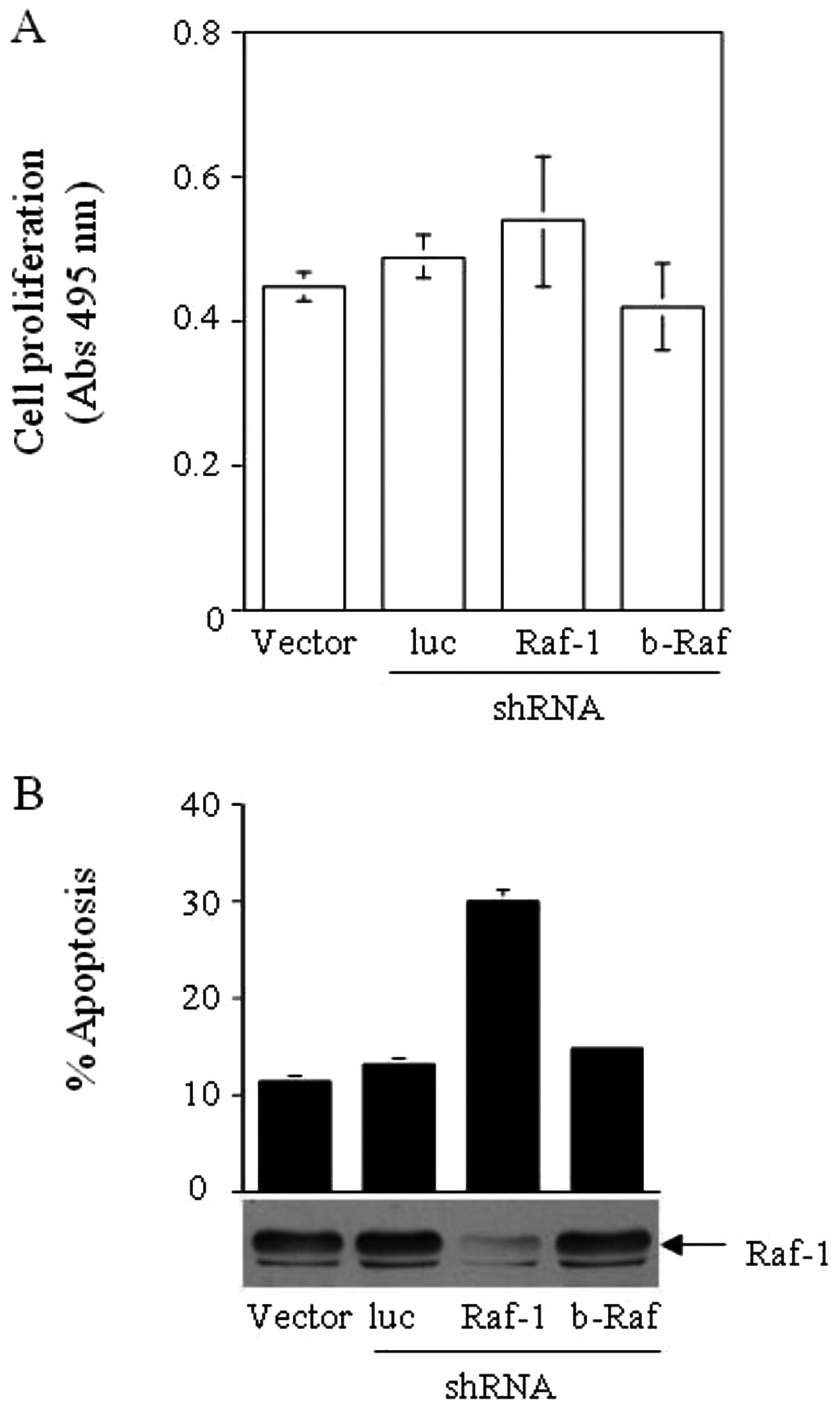
selection, and, using an MTS assay, examined whether these cell
lines exhibited any differences in growth rate and viability. The
growth rate of HCT116 (K-Ras G13D) did not decrease significantly
upon the expression of shRNA directed against Raf-1, b-Raf or
luciferase (Fig. 2A). Cell cycle
analysis revealed that the Raf-1-deprived HCT116 cells had higher
levels of apoptosis, observed as an increased sub-G0 population
(Fig. 2B). Cells with decreased
b-Raf and control cells expressing an shRNA to luciferase,
exhibited similar levels of apoptosis as the untreated cells.
Increase in cell apoptosis upon b-Raf and
Raf-1 silencing
Using mouse knockout models, two Raf genes
(b- and c-Raf) were previously shown to play a key
role in preventing cell apoptosis during development (9–11).
However, these mouse knockouts were embryonic lethal. To determine
whether these Raf kinases protect differentiated cells from
apoptosis, we silenced either Raf-1 or b-Raf expression using RNAi
in HCT116, HT29 and Colo205 cells and determined cell apoptosis by
DNA content-based flow cytometry. These cells express either mutant
K-Ras G13D (HCT116) or mutant b-Raf V599E (HT29 and Colo205). DNA
content-based flow cytometry analysis revealed that HCT116 (K-Ras
G13D/Raf-1 wt/b-Raf wt) had increased apoptosis when Raf-1 was
silenced, but b-Raf wt or b-Raf VE silencing did not increase
apoptosis above control levels (Fig.
3A). Cisplatin treatment was used as a positive control for
cell apoptosis, while treatment with pSUPER-luciferase was the
negative control. HT29 cells (Ras wt/Raf-1 wt/b-Raf wt: VE), which
are heterozygous for the activating b-Raf mutation V599E, showed
increased apoptosis when b-Raf VE was silenced as compared to when
Raf-1 or b-Raf wt were silenced (Fig.
3B). However, Colo205 (Ras wt/Raf-1 wt/b-Raf VE: VE), which are
homozygous for b-Raf V599E, were unresponsive to the depletion of
Raf-1 or b-Raf wt. Notably, silencing of b-Raf V599E in Colo205
cells caused a marked increase in apoptosis as compared to the
control (Fig. 3C). Our results
demonstrate that inhibition of the specific Raf kinases in cancer
cells dependent on oncogenes such as ras or b-Raf,
are likely to increase the apoptotic index and contribute to cancer
cell death.
Raf-1 and b-Raf enhance
anchorage-independent growth in soft agar
To test the potential of Raf kinase inhibition in
cancer therapy, we carried out soft agar colony formation assays.
Raf-1 or b-Raf was silenced by RNAi in HCT116 cells and stable cell
lines were generated as described earlier. These stable cell lines
were grown in soft agar to detect their ability to form colonies in
an anchorage-independent manner. Cell lines with
luciferase-targeted shRNA formed many small colonies in soft agar
within two weeks (Fig. 4, upper
panel). However, silencing of Raf-1 in HCT116 cells caused a
notable decrease in soft agar colony formation, confirming the
importance of Raf-1 kinase activity for anchorage-independent cell
growth (Fig. 4, middle panel).
However, b-Raf silencing in HCT116 cells was not as effective at
inhibiting soft agar colony formation, although it modestly
decreased soft-agar growth (Fig.
4, lower panel). Previous experiments proved that silencing
Raf-1 or b-Raf did not significantly alter the growth rate of these
cells (Fig. 2A). Conversely,
analysis of cell apoptosis by flow cytometry demonstrated that
Raf-1 silencing resulted in increased apoptosis of the cells used
in the soft agar assay (Fig. 2B).
These results may explain the decreased rate of colony formation in
soft agar upon Raf-1 silencing.
Combination of Raf-1 depletion and MEK
inhibition increases cancer cell death
Previous studies have shown that Raf-1 activation
enhances cellular resistance to chemotherapeutic drug treatments
(13,14). We tested whether Raf-1 was
important for colon cancer cell sensitivity to the chemotherapeutic
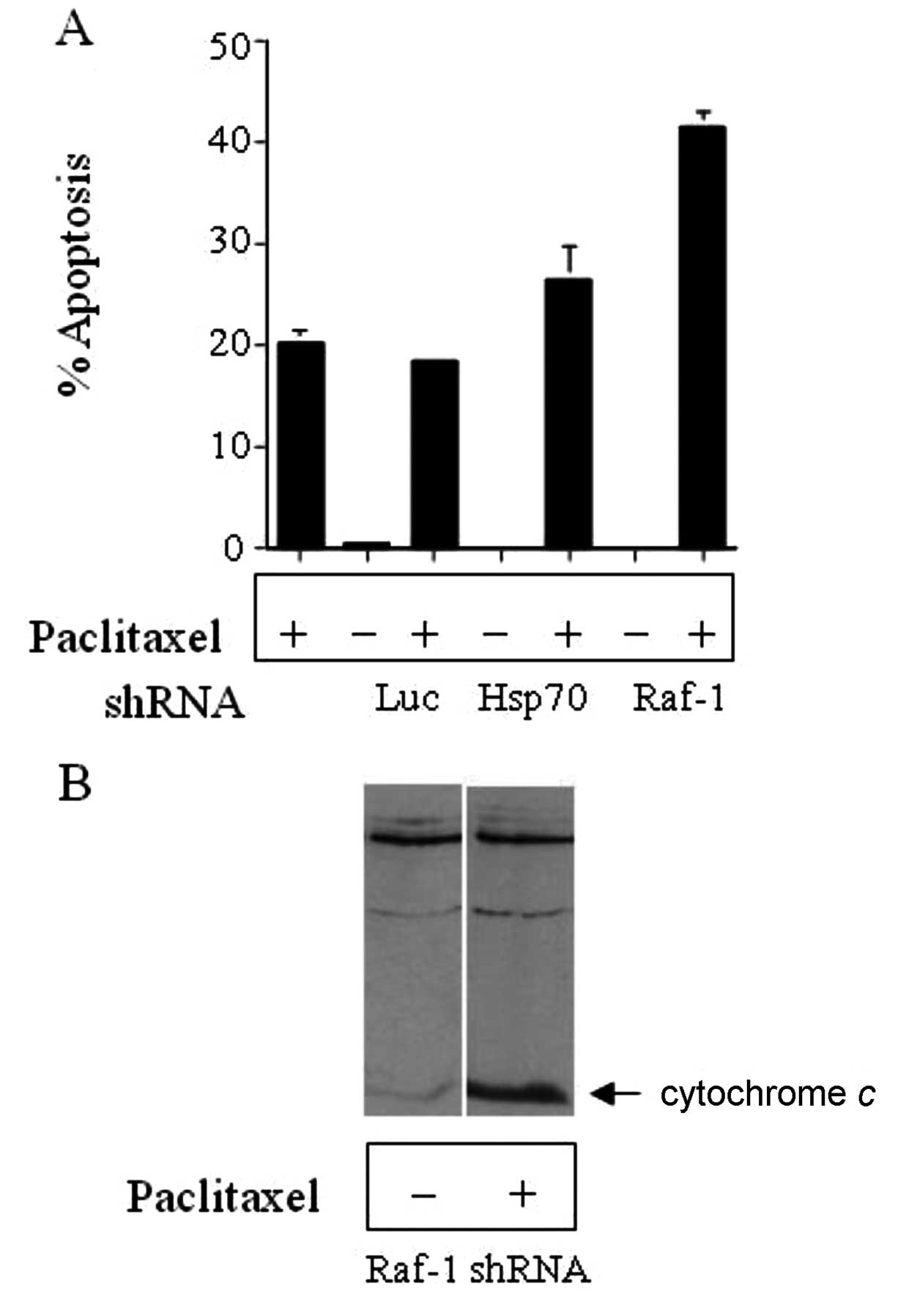
agent, paclitaxel. HCT116 colon cancer cells, with a stable
expression of either pSUPER-luciferase (Luc) or pSUPER-Hsp70 shRNA
as negative control or pSUPERRaf-1 shRNA, were treated with 10 nM
paclitaxel or DMSO for 16 h. After 16 h of paclitaxel treatment,
the cells were fixed, DNA was stained with propidium iodide and
cells were analyzed for apoptosis by flow cytometry (Fig. 5A). Silencing of Raf-1 resulted in
increased sensitivity to paclitaxel as evidenced by the increased
apoptosis in these cells compared to cells treated with paclitaxel
only, and this corresponded to increased cytochrome c
release (Fig. 5B).
In previous studies, Raf-1 was found to have a
MEK-independent role in signaling. Conversely, MEK/MAPK signaling
potentially occurs independent of Raf-1 (15,16).
Therefore, we assessed whether a multi-pronged inhibition of the
Ras-Raf-MAPK cascade enhances cancer cell death when compared to
Raf or MEK-1 inhibition alone. HCT116 (colon cancer) and ME-180
(cervical cancer) cells were employed to test this hypothesis by
silencing Raf-1 with shRNA and treating the cells with a MEK
inhibitor (5 μM U0126). Cells treated with Raf-1 shRNA and
vehicle (DMSO) had a moderate 2-fold increase in apoptosis compared
to the vehicle and luciferase shRNA or Hsp70 shRNA-treated cells
(Fig. 6A and B). MEK-1 inhibition
results in greater apoptosis than Raf-1 silencing alone in both
HCT116 and ME-180 cells. Notably, the co-treatment of cancer cells
with Raf-1 shRNA and MEK inhibitor resulted in a marked increase in
cell apoptosis in HCT116 (Fig. 6A)
and ME-180 (Fig. 6B) cells. Thus,
cancer cells may possess a MEK-independent signaling function for
Raf-1. Such a phenomenon has the potential to be exploited for
therapeutic purposes by efficiently blocking the entire MAPK
cascade simultaneously with inhibitors for Raf kinase as well as
MEK kinase.
Discussion
Cancer is known to be a multistage process (17) caused by the progressive
accumulation of activating mutations in dominant growth-enhancing
genes/oncogenes and inactivating mutations in
growth-suppressing/tumor suppressor genes (18). To maintain cell homeostasis, cancer
cells may be addicted to the activity of specific oncogenes as
opposed to normal cells. This oncogene addiction is beneficial to
the cancer cell by way of conferring certain growth advantages upon
it. However, inhibition of the oncogene function may lead to
collapse of cell homeostasis and cancer cell death (19–22).
Approximately 30% of solid tumors carry activating Ras mutations,
and epithelial tumors have been shown to possess a hyperactive
Ras-Raf-MAPK pathway (23).
Recently, activating mutations in b-Raf (particularly V599E) have
been found in over 60% of melanomas and in approximately 15% of
colon cancers (8). These data led
to the hypothesis that inhibition at any point of the Ras-Raf-MAPK
pathway in cancer cells is likely to diminish growth-promoting
signals, and thus tilt the cellular balance in favor of
death-promoting pathways. To test this hypothesis, we targeted the
Ras-Raf-MAPK pathway in cancer cells at two separate points by
silencing the Raf protein expression using RNAi together with the
pharmacological inhibition of MEK.
In this study, we have demonstrated that Raf kinases
are effectively depleted or silenced in cancer cells by RNAi. The
targeting of Raf-1 and b-Raf by RNA interference was highly
specific as evidenced by the lack of Raf kinase depletion by RNAi
of Hsp70 or luciferase (Fig. 1).
In addition, RNAi of Raf-1 reduces but does not eliminate
downstream effector signaling such as ERK phosphorylation (Fig. 1A), indicating that Raf-1 may not be
the only molecule signaling via ERK. ERK is important in modulating
cell growth and proliferation as well as resistance to apoptosis
(24,25), and ERK is hyperactive in colon
cancers possessing activated K-Ras (26). Raf-1 signaling via ERK is also
associated with increased mdr1 or drug efflux pump expression,
which is able to increase cellular resistance to chemotherapeutic
drug treatment by higher drug efflux from the cell (27). Of note, ERK activation has been
shown to result in a positive feedback to Raf-1 thereby amplifying
the Raf-MAPK cascade and promoting cell survival and growth
(28). Thus, even a modest
reduction in ERK activation is likely to result in decreased cell
growth, survival and increased susceptibility of the cancer cells
to apoptotic stimuli.
In our experiments, silencing of Raf-1 reduced ERK
phosphorylation, but did not decrease cancer cell growth. MTS
assays demonstrated that cancer cells such as HCT116 or MCF-7, with
or without a lower expression of Raf-1/b-Raf, had similar culture
growth rates. These data suggest that Raf kinases are not crucial
for cell growth. However, it is possible that the low level of ERK
activation still present is sufficient for cell growth. Cyclin
B-Cdc2, a master mitosis regulator, has been reported to activate
MEK1 during mitosis without subsequent ERK activation (29). Thus, it is possible that Raf
kinases have a reduced role during cell division and growth.
Recent mouse knockout models for Raf kinases have
shown that Raf-1 and b-Raf play critical roles during development
(9,10), primarily in preventing apoptosis.
ERK activation is also involved in the phosphorylation and
proteasomal degradation of the pro-apoptotic BH3-only protein Bim
(30), thus preventing cell death.
Our data support these findings since the depletion of Raf-1 or
b-Raf by RNAi increased cellular apoptosis. Notably, we found that
cancer cells with certain potential oncogene addictions, such as
activated K-Ras, were more susceptible to apoptosis via the
inhibition of Raf-1 kinase as compared to b-Raf. Similarly, cancer
cells with the activating b-Raf mutant (V599E) were better targeted
by b-Raf RNAi compared to the RNAi of Raf-1. HCT116 cells that
possess activated K-Ras (G13D) exhibited greater apoptosis when
Raf-1 was depleted, suggesting that Raf-1 is the primary MAPKKK for
Ras-mediated signaling in these cells. However, HCT116 cells were
refractory to b-Raf silencing, confirming previous studies that
cancers with an activating mutation in ras seldom possess an
accompanying mutation in b-Raf (12), since they probably preferentially
signal via alternate molecules such as Raf-1. Colon cancer cells
which were either heterozygous (HT29) or homozygous (Colo205) for
the activating b-Raf mutation (V599E) exhibited greater sensitivity
to abrogation of signaling from the b-Raf mutant. Similarly, b-Raf
V599E-dependent melanoma cells (A375) were sensitive to the
RNAi-mediated silencing of b-Raf V599E (data not shown). These data
suggest that the primary role of Raf kinases is to protect cells
from apoptosis. Results obtained from the soft agar assays
supported this hypothesis. This assay is a stringent test of the
ability of cancer cells to grow in an anchorage-independent manner.
A strong correlation exists between the ability of cells to grow in
soft agar and their ability to form tumors in vivo, although
the correlation is not absolute. Comparison of soft agar growth in
the presence and absence of Raf-1 or b-Raf demonstrated noteworthy
differences. Specifically, HCT116 and MCF-7 cells depleted of Raf-1
showed greater apoptosis by flow cytometry and much fewer colonies
in soft agar compared to control or b-Raf depletion, suggesting
that these cells are primarily dependent on Raf-1 for MAPK
signaling and protection from apoptosis. Additionally, the
molecular targeting of cancers based on the activated oncogene is a
potentially viable therapeutic strategy that could contribute to a
greater understanding of personalized medicine.
Since Raf participates in preventing cell death, it
follows that depletion/silencing of Raf may sensitize cancer cells
to concomitant chemotherapeutic drug treatment. Results of this
study have shown that Raf-1 silencing in HCT116 or ME-180 cells
confers markedly greater susceptibility to paclitaxel treatment.
Previous studies have shown that cervical cancer cells with
inherently low levels of Raf-1 exhibit paclitaxel sensitivity
(13). In this study, cancer cell
lines with high Raf-1 expression levels were used and modest levels
of apoptosis with paclitaxel treatment were observed. However,
silencing of Raf-1 expression by RNAi increased paclitaxel-mediated
cytotoxicity. A possible explanation is that Raf depletion may
decrease mdr1 expression and efflux of paclitaxel. This reiterates
the need for effective Raf kinase inhibitors, so that combination
treatment regimens may be employed in the treatment of such
cancers.
The dual specificity kinase MEK plays a key role in
integrating extracellular signals into the MAPK cascade. Activating
ras mutations in cancers result in hyperactive MEK, and MEK
activity could be targeted to prevent growth of these cancer cells.
Kinase inhibitors targeting MEK have been shown to suppress colon
tumor growth in vivo (31),
however results of clinical trials have been disappointing. MEK
inhibitors have been shown to be primarily cytostatic and not
cytotoxic to cancer cells. Recent evidence has, however, raised
questions pertaining to the exclusivity of the Raf/MEK/ERK pathway.
Mouse knockouts of Raf-1 are embryonic lethal, however, the knockin
of an allele of c-Raf, mutant at Y340/341F, rendered the
knockouts viable. These mice lacked detectable MEK activity but
exhibited normal ERK activity and cell proliferation (15). Raf has also been involved in direct
translocation to the mitochondria and subsequent phosphorylation of
Bad, thus enhancing 14-3-3 binding and blocking of Bad-mediated
apoptotic activity (32–35). ASK1, a stress-induced pro-apoptotic
protein, is another noteworthy binding partner of Raf-1. Notably
ASK1 is inhibited by either wild-type or kinase dead Raf-1
(36). Raf-1 has also been
reported to induce NF-κB via MEKK1 bypassing MEK (37). Activating mutations of MEK were
shown to be capable of transforming NIH3T3 cells in the absence of
ERK activation (38). Therefore,
mounting evidence indicates a possible MEK-independent role for
Raf-1, which suggests that blockade of the MAPK pathway at either
Raf-1 or MEK may not be sufficient to block cell survival and
induce apoptosis. Thus, we tested whether combination therapy
targeting both Raf-1 and MEK was able to increase apoptosis in
cancer cells. Our findings have shown that simultaneous Raf-1 RNAi
and pharmacological MEK inhibition results in greater cell death in
HCT116 colon cancer cells and ME-180 cervical cancer cells than
either treatment alone. These are some of the first results
demonstrating that dual targeting of a single signaling pathway at
two points or nodes may be more efficacious than any single
inhibitor alone. Thus, Raf-1 and b-Raf kinases potentially serve as
focal points for the inhibition of cancer cell survival.
In summary, our study suggests that multi-pronged
targeting of the MAPK pathway in cancer cells with activating
mutations in Ras or b-Raf is likely to increase the apoptosis of
these cancer cells, and it stresses the need for better
pharmacological inhibitors of Raf kinases.
Acknowledgements
We thank Dr Jean Zhao, Dr Kai Xia and
Dr Radha Narsimhan and members of the Roberts Lab for helpful and
enlightening discussions.
References
|
1.
|
Downward J: Ras signalling and apoptosis.
Curr Opin Genet Dev. 8:49–54. 1998. View Article : Google Scholar
|
|
2.
|
Barbacid M: Ras genes. Annu Rev Biochem.
56:779–827. 1987. View Article : Google Scholar
|
|
3.
|
Kiaris H and Spandidos D: Mutations of
ras genes in human tumours (Review). Int J Oncol. 7:413–421.
1995.
|
|
4.
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Fang JY and Richardson BC: The MAPK
signalling pathways and colorectal cancer. Lancet Oncol. 6:322–327.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Chong H, Vikis HG and Guan K-L: Mechanisms
of regulating the Raf kinase family. Cell Signal. 15:463–469. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Storm SM, Brennscheidt U, Sithanandam G
and Rapp UR: Raf oncogenes in carcinogenesis. Crit Rev Oncog.
2:1–8. 1990.
|
|
8.
|
Davies H, Bignell GR, Cox C, et al:
Mutations of the BRAF gene in human cancer. Nature. 417:949–954.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Mikula M, Schreiber M, Husak Z, et al:
Embryonic lethality and fetal liver apoptosis in mice lacking the
c-raf-1 gene. EMBO J. 20:1952–1962. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Wojnowski L, Zimmer AM, Beck TW, et al:
Endothelial apoptosis in Braf-deficient mice. Nat Genet.
16:293–297. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Wojnowski L, Stancato LF, Larner AC, Rapp
UR and Zimmer A: Overlapping and specific functions of Braf and
Craf-1 proto-oncogenes during mouse embryogenesis. Mech Dev.
91:97–104. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Rajagopalan H, Bardelli A, Lengauer C,
Kinzler KW, Vogelstein B and Velculescu VE: Tumorigenesis: RAF/RAS
oncogenes and mismatch-repair status. Nature. 418:9342002.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Britten RA, Perdue S, Opoku J and
Craighead P: paclitaxel is preferentially cytotoxic to human
cervical tumor cells with low Raf-1 kinase activity: implications
for paclitaxel-based chemoradiation regimens. Radiother Oncol.
48:329–334. 1998. View Article : Google Scholar
|
|
14.
|
Rasouli-Nia A, Liu D, Perdue S and Britten
RA: High Raf-1 kinase activity protects human tumor cells against
paclitaxel-induced cytotoxicity. Clin Cancer Res. 4:1111–1116.
1998.PubMed/NCBI
|
|
15.
|
Huser M, Luckett J, Chiloeches A, et al:
MEK kinase activity is not necessary for Raf-1 function. EMBO J.
20:1940–1951. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Murakami MS and Morrison DK: Raf-1 without
MEK? Sci STKE. 2001:e302001.
|
|
17.
|
Nowell PC: The clonal evolution of tumor
cell populations. Science. 194:23–28. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Kinzler KW and Vogelstein B: Landscaping
the cancer terrain. Science. 280:1036–1037. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Jain M, Arvanitis C, Chu K, et al:
Sustained loss of a neoplastic phenotype by brief inactivation of
MYC. Science. 297:102–104. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Felsher DW and Bishop JM: Reversible
tumorigenesis by MYC in hematopoietic lineages. Mol Cell.
4:199–207. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Pelengaris S, Littlewood T, Khan M, Elia G
and Evan G: Reversible activation of c-Myc in skin: induction of a
complex neoplastic phenotype by a single oncogenic lesion. Mol
Cell. 3:565–577. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Chin L, Tam A, Pomerantz J, Wong M, et al:
Essential role for oncogenic Ras in tumour maintenance. Nature.
400:468–472. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Barbacid M: Ras oncogenes: their role in
neoplasia. Eur J Clin Invest. 20:225–235. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Anderson P: Kinase cascades regulating
entry into apoptosis. Microbiol Mol Biol Rev. 61:33–46.
1997.PubMed/NCBI
|
|
25.
|
Rice PL, Goldberg RJ, Ray EC, Driggers LJ
and Ahnen DJ: Inhibition of extracellular signal-regulated kinase
1/2 phosphorylation and induction of apoptosis by sulindac
metabolites. Cancer Res. 61:1541–1547. 2001.PubMed/NCBI
|
|
26.
|
Bos JL, Fearon ER, Hamilton SR, et al:
Prevalence of ras gene mutations in human colorectal cancers.
Nature. 327:293–297. 1987. View
Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Zhou G and Kuo MT: NF-κB-mediated
induction of mdr1b expression by insulin in rat hepatoma cells. J
Biol Chem. 272:15174–15183. 1997.
|
|
28.
|
Zimmermann S, Rommel C, Ziogas A, Lovric
J, Moelling K and Radziwill G: MEK1 mediates a positive feedback on
Raf-1 activity independently of Ras and Src. Oncogene.
15:1503–1511. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Harding A, Giles N, Burgess A, Hancock JF
and Gabrielli BG: Mechanism of mitosis-specific activation of MEK1.
J Biol Chem. 278:16747–16754. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Ley R, Balmanno K, Hadfield K, Weston C
and Cook SJ: Activation of the ERK1/2 signaling pathway promotes
phosphorylation and proteasome-dependent degradation of the
BH3-only protein. Bim. J Biol Chem. 278:18811–18816. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Sebolt-Leopold JS, Dudley DT, Herrera R,
et al: Blockade of the MAP kinase pathway suppresses growth of
colon tumors in vivo. Nat Med. 5:810–816. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Majewski M, Nieborowska-Skorska M,
Salomoni P, et al: Activation of mitochondrial Raf-1 is involved in
the antiapoptotic effects of Akt. Cancer Res. 59:2815–2819.
1999.PubMed/NCBI
|
|
33.
|
Fang X, Yu S, Eder A, et al: Regulation of
BAD phosphorylation at serine 112 by the Rasmitogen-activated
protein kinase pathway. Oncogene. 18:6635–6640. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Zhong J, Troppmair J and Rapp UR:
Independent control of cell survival by Raf-1 and Bcl-2 at the
mitochondria. Oncogene. 20:4807–4816. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Subramanian RR, Masters SC, Zhang H and Fu
H: Functional conservation of 14-3-3 isoforms in inhibiting
bad-induced apoptosis. Exp Cell Res. 271:142–151. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Chen J, Fujii K, Zhang L, Roberts T and Fu
H: Raf-1 promotes cell survival by antagonizing apoptosis
signal-regulating kinase 1 through a MEK-ERK independent mechanism.
Proc Natl Acad Sci USA. 98:7783–7788. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Baumann B, Weber CK, Troppmair J, et al:
Raf induces NF-kappaB by membrane shuttle kinase MEKK1, a signaling
pathway critical for transformation. Proc Natl Acad Sci USA.
97:4615–4620. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Alessandrini A, Greulich H, Huang W and
Erikson RL: Mek1 phosphorylation site mutants activate Raf-1 in NIH
3T3 cells. J Biol Chem. 271:31612–31618. 1996. View Article : Google Scholar : PubMed/NCBI
|