Introduction
Prostate cancer remains the second leading cause of
cancer death for Northern American men. According to the National
Cancer Institute, it is estimated that there will be 241,740 new
cases and 28,170 deaths from prostate cancer in the United States
in 2012 (http://www.cancer.gov/cancertopics/types/prostate).
Initially, prostate cancer cells depend on androgen stimulation for
growth and proliferation, and sensitive to androgen deprivation
therapy. Unfortunately most recurrent tumors return within two
years with an androgen-independent state and an aggressive,
metastatic phenotype. As of yet there is no effective treatment for
hormone-refractory prostate cancer (1,2).
Androgens and the androgen receptor (AR) are
critical components that govern the development, differentiation,
and functionality of the prostate gland and accessory reproductive
tissues, and are also involved in prostate tumorigenesis (3). These pathological functions are
dependent on the androgen-activated androgen receptor, which can
act as a transcription factor to regulate the expression of
multiple genes. Recently, several possible mechanisms (4,5) have
been proposed for the progression of androgen-independent prostate
cancer during androgen-deprivation therapy. Of them, mutation,
amplification (5,6), and expression alternative-splice
variants (7) of the androgen
receptor are related to the adaptation of recurrent prostate cancer
to low levels of androgen during androgen-deprivation therapy.
Androgen deprivation also induces neuroendocrine differentiation
(8) of prostate cancer cells
leading to autocrine/paracrine signaling pathways for survival. In
addition, androgen-independent prostate cancer cells have been
characterized to exhibit stem cell-like properties (9,10).
Prostate-specific membrane antigen (PSMA), a type II
membrane protein, has been found to be upregulated and strongly
expressed on prostate tumor cells and as a consequence, it has
attracted significant attention not only as a tumor marker for
disease progression but also as both an imaging and therapeutic
target for prostate cancer (11).
Although previous studies revealed that PSMA expression was
negatively regulated by androgen stimulation in AR-positive cells
(12–14) and downregulation of AR expression
was mediated by androgen-deprivation in an established
androgen-independent subline derived from the androgen-sensitive
LNCaP cell line (4), there are no
reports which investigate the expression of both AR and PSMA at the
protein level after long-term androgen deprivation. Therefore, this
study was designed to identify the effect of long-term
androgen-deprived growth on these two prostate cancer markers with
respect to possible implications on diagnostic and therapeutic
strategies for the androgen-independent disease-state. First,
androgen-sensitive LNCaP cells were cultured in androgen depleted
media up to 20 passages. Second, the altered expression of PSMA and
AR at the protein levels induced by androgen deprivation was
determined by western blotting, fluorescence imaging analysis, and
enzymatic activity. Our data suggest that long-term androgen
deprivation may induce the concomitant time-dependent
downregulation of both AR and PSMA to progress toward a more
aggressive, androgen-independent prostate cancer disease state.
Materials and methods
Cell lines and reagents
The human prostate cancer cell lines LNCaP and PC-3
were obtained from the American Type Culture Collection (Manassas,
VA, USA). The rabbit polyclonal anti-AR antibody (N-20) was
obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
The goat anti-rabbit secondary antibody-FITC and the mouse
monoclonal anti-GAPDH antibody (clone GAPDH-71.1) were obtained
from Sigma-Aldrich (St. Louis, MO, USA). The mouse monoclonal
anti-PSMA antibody 7E11 was graciously provided by Cytogen Corp.
(Princeton, NJ, USA). Protein blocking solution was obtained from
BioGenex (San Ramon, CA, USA). Hoechst 33342 was obtained from
Invitrogen-Molecular Probes (Carlsbad, CA, USA). Cy5.5-CTT-54.2 was
prepared by our lab as described previously (15). Halt Protease Inhibitor Cocktail
(100X) was purchased from Thermo Fisher Scientific (Rockford, IL,
USA). All other chemicals and cell-culture reagents were purchased
from Fisher Scientific (Sommerville, NJ, USA) or Sigma-Aldrich.
Cell culture
LNCaP and PC-3 cells were grown in T-75 flasks with
normal growth media [RPMI-1640 containing 10% heat-inactivated
fetal calf serum (FBS), 100 U of penicillin and 100 μg/ml
streptomycin] in a humidified incubator at 37°C with 5%
CO2. Otherwise, for androgen-deprivation growth, cells
were cultured with conditioned media [RPMI-1640 containing 10%
charcoal-stripped fetal bovine serum, 100 U of penicillin and 100
μg/ml streptomycin]. Confluent cells were detached with a
0.25% trypsin 0.53 mM EDTA solution, harvested, and plated in
two-well slide chambers at a density of 4×104
cells/well. Cells were grown for three days before conducting the
following experiments.
Immunofluorescence detection of AR
The LNCaP cells, grown under androgen deprivation
condition for over time (5, 10 and 20 passages), were cultured for
3 days on the slides in the conditioned media. For 2-day
androgen-deprivation treatment, LNCaP cells were seeded on slides
with normal growth media for 1-day growth, and replaced with
conditioned media for another 2-day growth. Normal LNCaP cells and
PC-3 cells were used for the AR-positive and AR-negative control
respectively. These cells were seeded on slides with normal growth
media for 3 days. Slides with 3-day growth cells in normal growth
media or conditioned media were washed twice in PBS buffer
(phosphate buffered saline), fixed in 4% paraformaldehyde in PBS
buffer for 15 min at room temperature, and permeabilized with 0.2%
Triton X-100 in PBS buffer for 5 min at room temperature. The
permeabilized cells were blocked in block buffer (0.1% Tween-20, 5%
goat normal serum in PBS buffer) for 2 h at room temperature and
incubated with primary anti-AR antibody (100X diluted in block
buffer) overnight at 4°C. After washing, the cells were incubated
with a secondary antibody (goat anti-rabbit IgG-FITC, 40X diluted
in 1% BSA, PBS buffer) for 2 h at room temperature, counterstained
with Hoechst 33342, and mounted in Vectashield® Mounting
Medium (Vector Laboratories, Inc., Burlingame, CA, USA) for
confocal microscopy.
Affinity imaging of PSMAs with
Cy5.5-CTT-54.2
The cells cultured on the 2-well slides were washed
twice with warm medium (37°C) A (phosphate-free RPMI-1640
containing 1% FBS), then incubated with 1 ml of Cy5.5-CTT-54.2 (10
μM) in warm medium A for 1 h in a humidified incubator at
37°C and 5% CO2. The above treated cells were washed
three times with cold-KRB buffer pH 7.4 (mmol/l: NaCl 154.0, KCl
5.0, CaCl2 2.0, MgCl2 1.0, HEPES 5.0,
D-glucose 5.0) and fixed with 4% paraformaldehyde in KRB for 15 min
at room temperature. The cellular nuclei were counterstained with
Hoechst 33342, and then mounted in Vectashield Mounting Medium for
microscopy.
Confocal laser scanning microscopy
Cells were visualized under 40× oil immersion
objective using an LSM 510 META laser scanning microscope. Hoechst
33342 was excited with a Diode laser (405 nm), and the emission was
collected with a BP420-480-nm filter. AR immunofluorescence (with
goat anti-rabbit IgG-FITC) was excited at 488 nm using an Argon
laser, and the emission was collected with a LP505-nm filter.
PSMA-targeted imaging with Cy5.5-CTT-54.2 was excited using 633 nm
from a HeNe laser, and the emission collected with an LP 650-nm
filter. To reduce interchannel crosstalk, a multi-tracking
technique was used, and images were taken at a resolution of
1,024×1,024 pixels. Confocal scanning parameters were set up so
that the control cells without treatment did not have background
fluorescence. The imaging colors of the fluorescent dyes, Hoechst
33342 and FITC, were defined as blue and green, respectively. As
the emission wavelength of the near-infrared fluorescent dye Cy5.5
was beyond visible ranges, fluorescence pseudocolor of Cy5.5 was
assigned as red. The images were edited by National Institutes of
Health (NIH) Image J software (http://rsb.info.nih.gov/ij) and Adobe Photoshop
CS2.
Whole cell lysate extraction and western
blot analysis
The controls: PC-3 and LNCaP cells (cultured in
normal growth media) and LNCaP cells under androgen deprivation
over time (2 days, 5, 10 or 20 passages) were collected by
scraping, washed once in ice-cold PBS, re-suspended in 3-fold cell
pellet volumes of lysis buffer (1% NP-40, 20 mM Tris pH 8.0, 137 mM
NaCl, 10% glycerol) (16,17) supplemented with 1X Halt Protease
Inhibitor Cocktail for 15 min on ice, then transferred to Eppendorf
tubes for centrifugation at 10,000 g for 15 min at 4°C, the
supernatant was saved as whole-cell protein extracts. Protein
concentrations were determined using Non-Interfering Protein Assay
(G-Biosciences, St. Louis, MO, USA). Western blotting was performed
as described previously with only minor modifications (17,18).
In brief, detergent soluble proteins (30 μg) were loaded and
separated on a NuPAGE™ 4–12% Bis-Tris Gel (Invitrogen, Carlsbad,
CA, USA) by electrophoresis for 40 min at a constant 200 V under
reducing conditions, and then transferred to a 0.45 μm PVDF
Immobilon-P Transfer Membrane (Millipore Corp., Bedford, MA, USA)
at 400 mA for 100 min in a transfer apparatus-Owl Bandit VEP-2
(Owl, Portsmouth, NH, USA) according to the manufacturer’s
instructions. Membranes were incubated with primary antibody at
corresponding dilution overnight at 4°C and then with horseradish
peroxidase conjugated-second antibody for 1 h at room temperature.
The immunoreactive bands were visualized using Protein Detector TMB
Western Blot kit (KPL, Gaithersburg, MD, USA) following the
manufacturer’s instructions.
Evaluation of PSMA relative enzymatic
activity
HPLC-based PSMA enzymatic activity analysis was
performed in triplicate as described previously with only minor
modifications (19,20). Working solutions of the substrate
(N-[4-(phenylazo)-benzoyl]-glutamyl-γ-glutamic acid, PABGgG) were
made at 10 μM in Tris-buffer (50 mM, pH 7.4). Working
solutions of each protein sample were diluted at 30 μg/ml in
Tris-buffer (50 mM, pH 7.4 containing 1% Triton X-100). A typical
incubation mixture (final volume 250 μl) was prepared by the
addition of 200 μl Tris buffer (50 mM, pH 7.4) and
PAB-Glu-γ-Glu (25 μl, 10 μM) in a test tube. The
enzymatic reaction was initiated by the addition of 25 μl of
the PSMA working solution. The reaction was allowed to proceed for
15 min with constant shaking at 37°C and terminated by the addition
of 25 μl methanolic TFA (2.5% trifluoroacetic acid by volume
in methanol) followed by vortexing. The quenched incubation mixture
was quickly buffered by the addition of 25 μl
K2HPO4 (0.1 M), vortexed, iced for 15 min,
followed by centrifugation (10 min at 7,000 g). An 85-μl
aliquot of the resulting supernatant was subsequently quantified
for the contents of substrate and product by HPLC as previously
described (21,22). Enzymatic activity for each protein
sample was calculated from HPLC data. Activities were compared with
normally-cultured LNCaP cells to evaluate relative enzymatic
activity (REA).
Results
Downregulation of AR protein expression
induced by androgen deprivation
In a positive control, a strong immunofluorescence
signal for AR was observed throughout the nuclei of normal growth
medium-cultured LNCaP cells (Fig.
1). In contrast, there is no signal for AR in AR-negative PC-3
cells (Fig. 1). Compared to the
positive control, androgen deprivation induced downregulation of AR
protein expression in LNCaP cells over time; AR was not detectable
after 10 passages (Fig. 1). Based
on these results, it is clear that downregulation of AR protein
expression is a cell-adaptable response to prolonged androgen
deprivation. Although the presented results were not consistent
with the previous report which demonstrated that the administration
of androgens resulted in downregulated AR mRNA levels in LNCaP
cells (23), our data matched with
androgen-independent LNCaP subline (4).
Affinity labeling of PSMA
Cy5.5-CTT-54.2, a specific PSMA fluorescent
inhibitor (IC50 = 0.55 nM) was designed and evaluated
for PSMA-targeted fluorescence imaging of LNCaP cells in our
previous published study (15). In
this study, Cy5.5-CTT-54.2 was employed to detect the change of
active PSMAs on the cellular surface by fluorescence imaging of
androgen-deprived LNCaP cells. Consistent with the results for AR
immunofluorescence study above, the greatest cell labeling by
Cy5.5-CTT-54.2 was observed for LNCaP cells cultured in normal
growth media (Fig. 2A), with
decreased signals through 5 passages under androgen-deprived
conditions (Fig. 2B and C), and no
detectable signals by 10 passages under the same conditions
(Fig. 2D and E), similar to that
of PSMA-negative PC-3 cells (Fig.
2F).
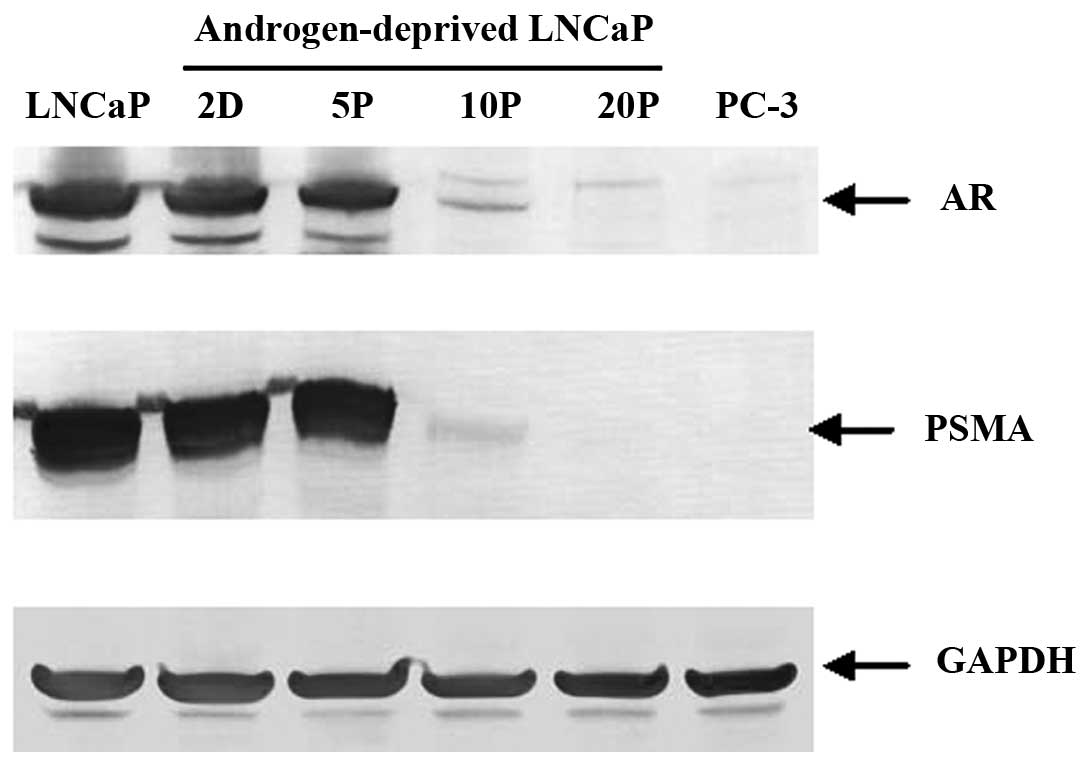
Western blot analysis
Western blot analysis further confirmed that the
total amount of AR and PSMA decreased over time with a dramatic
loss by 10 passages and absent after 20 passages in
androgen-deprived conditions; GAPDH served as a protein loading
controls (Fig. 3). These data
suggest that downregulation of both AR and PSMA expression is
dependent on the androgen levels and the length of time of androgen
deprivation during cell growth.
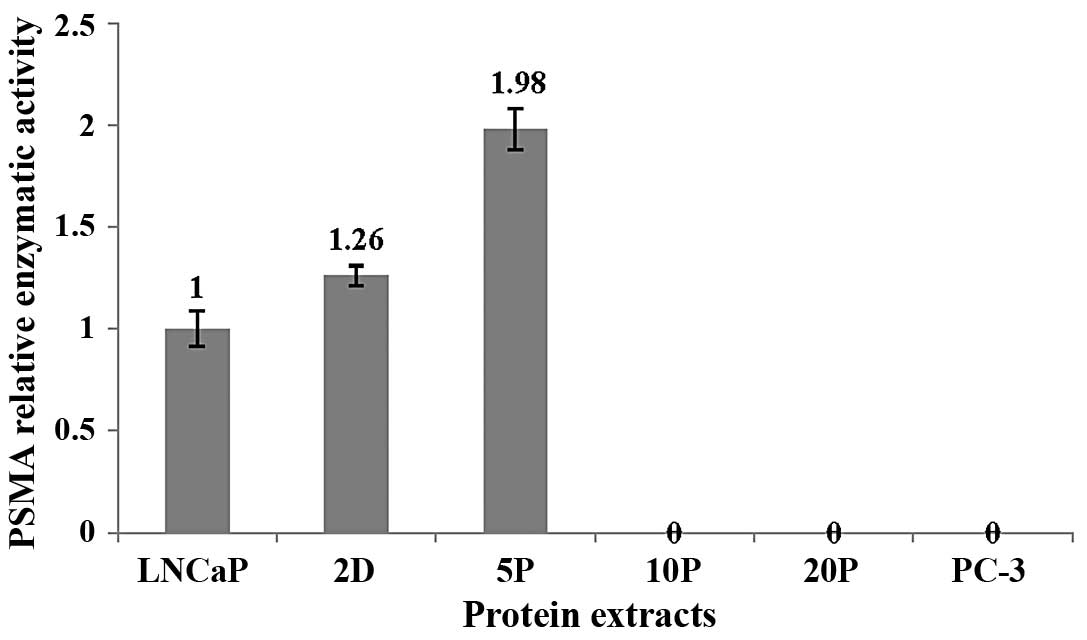
PSMA relative enzymatic activity
(REA)
Analysis of PSMA relative enzymatic activity
revealed that there was an apparent increase for whole-cell protein
samples for 2 days (REA = 1.26) and 5 passages (REA = 1.98),
compared to the membrane-protein sample from normally-cultured
LNCaP cells (Fig. 4). However,
there was no detectable PSMA enzymatic activity after 10 passages
in prolonged androgen-deprived conditions, similar to that of
PSMA-negative PC-3 cells (Fig. 4),
and consistent with the PSMA expression (Figs. 2 and 3).
Discussion
Our data suggest that long-term androgen-depletion
may induce downregulation of AR and PSMA which may lead to a
diagnostically and therapeutically elusive androgen-independent
disease-state. These new data provide additional knowledge of
androgen-independent prostate cancer progression compared to
previous studies, which primarily focused on changes in AR ligand
binding specificity that may result from gene structure changes
(e.g., mutation, amplification, or spliced variant expression) or
AR ligand-independent activation arising from alternative signal
pathways that activate AR activity at the castration level of
androgen (2,24). With respect to PSMA, it is thought
that increased expression is correlated with prostate cancer
progression, especially in recurrent, metastatic cancers after
androgen deprivation therapy (25,26).
While increased or consistent AR levels have been
reported in established androgen-independent LNCaP sublines
(27,28), loss of AR expression has been
detected in other LNCaP sublines (4). Although all androgen-independent
LNCaP sublines have been derived from a parent androgen-sensitive
LNCaP cell line, they have been established in different labs under
various conditions such as passage number of starting parent LNCaP
cells, culture conditions, and duration of propagation under
androgen-deprived growth conditions, which may result in different
characteristics. In addition, the parent androgen-sensitive LNCaP
cell line is not a homogeneous population, but is a multiple
hypotetraploid mixture including cells with 84, 86, 87 or more
chromosomes (product description of CRL-1740, http://www.atcc.org/). Single or a combination of
multiple factors may contribute to these contradictory observations
on AR expression in these established LNCaP sublines. A recent
microarray analysis on patient tissues revealed that strong AR
expression was detected in benign prostate (83%), localized
prostate cancer (100%), and lymph node metastasis (80%), but less
(40%) in metastatic hormone-resistant prostate cancer (3). It was also noticed that two highly
aggressive androgen-independent prostate cancer cell lines: DU145
and PC-3 are double-negative of AR and PSMA (29) and the loss of their AR and PSMA
expression is due to epigenetic silencing by CpG island
hypermethylation of their promoter regions (29–31).
The DU145 and PC-3 lines were derived from brain and bone
metastases of prostate cancer, whereas the AR(+), PSMA(+) LNCaP
cell line was derived from a lymph node metastasis. The attributes
of these three cell lines are consistent with the above clinical
analysis for AR expression. Although the molecular basis for
downregulation of AR and PSMA expression still remains unclear, the
present study reveals a dynamic progression in the loss of AR and
PSMA expression during androgen deprivation. We also observed that
LNCaP cells gained the capability of suspension growth and more
rapid proliferation after 10 passages in culture with 10%
charcoal-stripped fetal bovine serum and RPMI-1640 media (data not
shown). Combined, the data suggest that the protein expression
levels of AR and PSMA may correlate to the progression and
metastatic sites of prostate cancer. Also, highly metastatic,
aggressive, and androgen-independent prostate cancer cells are more
likely to exhibit an AR(−) and PSMA(−) genotype. While some
evidence is suggestive of novel roles for AR and PSMA as
tumor-suppressor activity in androgen-independent prostate cancer
cells (2,32), the loss of AR and PSMA expression
may be a consequence of dedifferentiation or a stem cell-like
transition of prostate cancer cells (33) induced by androgen deprivation.
It was observed that short-term androgen deprivation
(up to 5 passages) lead to an apparent increase of PSMA enzymatic
activity (Fig. 4) while cell
labeling with a fluorescent PSMA inhibitor decreased for these
time-points (Fig 2). This
inconsistency may implicate a possible specific post-translational
modification change (e.g., N-glycosylation pattern), which
occurs during short-term androgen deprivation resulting in a
modified PSMA with improved enzymatic activity and a relative loss
of either affinity for inhibitors or cell-surface expression. In
previous studies with cell lines and patient samples, different
N-glycosylation patterns for PSMA have been detected
(34,35). Furthermore, it has been confirmed
that the different N-glycosylation patterns can strongly affect
PSMA enzymatic activity (34,36).
In addition, changes in substrate specificity and enzymatic
activity were exhibited by brain GCP II (37,38),
which shares sequence homology with PSMA (39), but is expressed in a different
tissue.
In conclusion, our in vitro data suggest that
the continuous, long-term androgen deprivation induces
downregulation or loss of AR and PSMA expression in prostate cancer
cells, which may have significance in the progression toward a more
aggressive, metastatic prostate cancer disease-state during
androgen deprivation therapy. The implication of these observations
is that it may be critical to identify AR(+) or PSMA(+) tumors
early to ensure the therapeutic efficacy of novel AR- or
PSMA-targeted agents to treat recurrent, metastatic, and
hormone-refractory prostate cancer patients. However, an unsolved
question remains whether these androgen-independent LNCaP cells
originate from the minor population of stem-like cancer cells or
result from the accumulation of the changes in genetic structures,
gene expression profiles, or alternative signal pathways in
androgen-sensitive LNCaP cells.
Acknowledgements
The authors thank Cytogen Corp.
(Princeton, NJ, USA) for the gift of the mouse monoclonal antibody
7E11, and extend their gratitude for technical assistance from C.
Davitt and V. Lynch-Holm at the WSU Franceschi Microscopy and
Imaging Center. This study was supported in part by the National
Institutes of Health (R01CA140617).
References
|
1.
|
H GustavssonK WelenJE DamberTransition of
an androgen-dependent human prostate cancer cell line into an
androgen-independent subline is associated with increased
angiogenesisProstate62364373200510.1002/pros.2014515389782
|
|
2.
|
Y NiuS AltuwaijriKP LaiAndrogen receptor
is a tumor suppressor and proliferator in prostate cancerProc Natl
Acad Sci USA1051218212187200810.1073/pnas.080470010518723679
|
|
3.
|
JN DavisKJ WojnoS DaignaultElevated E2F1
inhibits transcription of the androgen receptor in metastatic
hormone-resistant prostate cancerCancer
Res661189711906200610.1158/0008-5472.CAN-06-249717178887
|
|
4.
|
SO LeeSS DuttN NadimintyE PinderH LiaoAC
GaoDevelopment of an androgen-deprivation induced and androgen
suppressed human prostate cancer cell
lineProstate6712931300200710.1002/pros.2062117626246
|
|
5.
|
P SaraonK JarviEP DiamandisMolecular
alterations during progression of prostate cancer to androgen
independenceClin
Chem5713661375201110.1373/clinchem.2011.16597721956922
|
|
6.
|
HL DevlinM MudryjProgression of prostate
cancer: multiple pathways to androgen independenceCancer
Lett274177186200910.1016/j.canlet.2008.06.00718657355
|
|
7.
|
PA WatsonYF ChenMD BalbasConstitutively
active androgen receptor splice variants expressed in
castration-resistant prostate cancer require full-length androgen
receptorProc Natl Acad Sci
USA1071675916765201010.1073/pnas.1012443107
|
|
8.
|
TC YuanS VeeramaniFF LinAndrogen
deprivation induces human prostate epithelial neuroendocrine
differentiation of androgen-sensitive LNCaP cellsEndocr Relat
Cancer13151167200610.1677/erc.1.01043
|
|
9.
|
K ZhangDJ WaxmanPC3 prostate
tumor-initiating cells with molecular profile
FAM65Bhigh/MFI2low/LEF1low increase tumor angiogenesisMol
Cancer9319201010.1186/1476-4598-9-31921190562
|
|
10.
|
KM BaeZ SuC FryeExpression of pluripotent
stem cell reprogramming factors by prostate tumor initiating cellsJ
Urol18320452053201010.1016/j.juro.2009.12.09220303530
|
|
11.
|
AK RajasekaranG AnilkumarJJ ChristiansenIs
prostate-specific membrane antigen a multifunctional protein?Am J
Physiol Cell
Physiol288C975C981200510.1152/ajpcell.00506.200415840561
|
|
12.
|
SJ LeeK LeeX YangNFATc1 with AP-3 site
binding specificity mediates gene expression of
prostate-specific-membrane-antigenJ Mol
Biol330749760200310.1016/S0022-2836(03)00640-512850144
|
|
13.
|
A GhoshWD HestonTumor target prostate
specific membrane antigen (PSMA) and its regulation in prostate
cancerJ Cell Biochem91528539200410.1002/jcb.10661
|
|
14.
|
RS IsraeliCT PowellJG CorrWR FairWD
HestonExpression of the prostate-specific membrane antigenCancer
Res541807181119947511053
|
|
15.
|
T LiuLY WuMR HopkinsJK ChoiCE BerkmanA
targeted low molecular weight near-infrared fluorescent probe for
prostate cancerBioorg Med Chem
Lett2071247126201010.1016/j.bmcl.2010.09.05720947349
|
|
16.
|
JY MatrouleCM CarthyDJ GranvilleO JoloisDW
HuntJ PietteMechanism of colon cancer cell apoptosis mediated by
pyropheophorbide-a methylester
photosensitizationOncogene2040704084200110.1038/sj.onc.120454611494135
|
|
17.
|
T LiuLY WuCE BerkmanProstate-specific
membrane antigen-targeted photodynamic therapy induces rapid
cytoskeletal disruptionCancer
Lett296106112201010.1016/j.canlet.2010.04.00320452720
|
|
18.
|
T LiuY ToriyabeCE BerkmanPurification of
prostate-specific membrane antigen using conformational
epitope-specific antibody-affinity chromatographyProtein Expr
Purif49251255200610.1016/j.pep.2006.05.008
|
|
19.
|
T LiuLY WuM KazakCE BerkmanCell-surface
labeling and internalization by a fluorescent inhibitor of
prostate-specific membrane
antigenProstate68955964200810.1002/pros.2075318361407
|
|
20.
|
LY WuMO AndersonY ToriyabeThe molecular
pruning of a phosphoramidate peptidomimetic inhibitor of
prostate-specific membrane antigenBioorg Med
Chem1574347443200710.1016/j.bmc.2007.07.02817869524
|
|
21.
|
MO AndersonLY WuNM SantiagoSubstrate
specificity of prostate-specific membrane antigenBioorg Med
Chem1566786686200710.1016/j.bmc.2007.08.00617764959
|
|
22.
|
J MaungJP MallariTA GirtsmanProbing for a
hydrophobic a binding register in prostate-specific membrane
antigen with phenylalkylphosphonamidatesBioorg Med
Chem1249694979200410.1016/j.bmc.2004.06.031
|
|
23.
|
DA WolfT HerzingerH HermekingD BlaschkeW
HorzTranscriptional and posttranscriptional regulation of human
androgen receptor expression by androgenMol
Endocrinol792493619938413317
|
|
24.
|
Y NiuTM ChangS YehWL MaYZ WangC
ChangDifferential androgen receptor signals in different cells
explain why androgen-deprivation therapy of prostate cancer
failsOncogene2935933604201010.1038/onc.2010.12120440270
|
|
25.
|
JS RossCE SheehanHA FisherCorrelation of
primary tumor prostate-specific membrane antigen expression with
disease recurrence in prostate cancerClin Cancer
Res963576362200314695135
|
|
26.
|
S PernerMD HoferR KimProstate-specific
membrane antigen expression as a predictor of prostate cancer
progressionHum
Pathol38696701200710.1016/j.humpath.2006.11.01217320151
|
|
27.
|
JM KokontisN HayS LiaoProgression of LNCaP
prostate tumor cells during androgen deprivation:
hormone-independent growth, repression of proliferation by
androgen, and role for p27Kip1 in androgen-induced cell
cycle arrestMol
Endocrinol12941953199810.1210/mend.12.7.01369658399
|
|
28.
|
N IshikuraH KawataA NishimotoR NakamuraN
IshiiY AokiEstablishment and characterization of an androgen
receptor-dependent, androgen-independent human prostate cancer cell
line, LNCaP-CS10Prostate70457466201019902465
|
|
29.
|
H SuzukiT UedaT IchikawaH ItoAndrogen
receptor involvement in the progression of prostate cancerEndocr
Relat Cancer10209216200310.1677/erc.0.010020912790784
|
|
30.
|
A ChlenskiK NakashiroKV KetelsGI
KorovaitsevaR OyasuAndrogen receptor expression in
androgen-independent prostate cancer cell
linesProstate476675200110.1002/pros.104811304731
|
|
31.
|
P Mhawech-FaucegliaDJ SmiragliaW
BsharaProstate-specific membrane antigen expression is a potential
prognostic marker in endometrial adenocarcinomaCancer Epidemiol
Biomarkers Prev17571577200810.1158/1055-9965.EPI-07-0511
|
|
32.
|
A GhoshX WangE KleinWD HestonNovel role of
prostate-specific membrane antigen in suppressing prostate cancer
invasivenessCancer Res65727731200515705868
|
|
33.
|
Y TangAW HamburgerL WangMA KhanA
HussainAndrogen deprivation and stem cell markers in prostate
cancersInt J Clin Exp Pathol3128138200920126580
|
|
34.
|
A GhoshWD HestonEffect of carbohydrate
moieties on the folate hydrolysis activity of the prostate specific
membrane antigenProstate57140151200310.1002/pros.1028912949938
|
|
35.
|
EH HolmesTG GreeneWT TinoAnalysis of
glycosylation of prostate-specific membrane antigen derived from
LNCaP cells, prostatic carcinoma tumors, and serum from prostate
cancer
patientsProstateSuppl72529199610.1002/(SICI)1097-0045(1996)7+%3C25::AID-PROS3%3E3.0.CO;2-I
|
|
36.
|
C BarinkaP SachaJ SklenarIdentification of
the N-glycosylation sites on glutamate carboxypeptidase II
necessary for proteolytic activityProtein
Sci1316271635200410.1110/ps.0462210415152093
|
|
37.
|
R Luthi-CarterAK BarczakH SpenoJT
CoyleHydrolysis of the neuropeptide N-acetylaspartylglutamate
(NAAG) by cloned human glutamate carboxypeptidase IIBrain
Res795341348199810.1016/S0006-8993(98)00244-39622670
|
|
38.
|
CW TiffanyRG LapidusA MerionDC CalvinBS
SlusherCharacterization of the enzymatic activity of PSM:
comparison with brain
NAALADaseProstate392835199910.1002/(SICI)1097-0045(19990401)39:1%3C28::AID-PROS5%3E3.0.CO;2-A10221263
|
|
39.
|
R Luthi-CarterAK BarczakH SpenoJT
CoyleMolecular characterization of human brain N-acetylated
alpha-linked acidic dipeptidase (NAALADase)J Pharmacol Exp
Ther286102010251998
|