Introduction
Metastasis is a multi-step cascade involving the
migration of tumour cells from their site of origin, evasion from
host defence systems, subsequent seeding at distant organs and
growth of secondary tumours. The first step of metastasis is
migration of the tumour cells from the primary tumour nest. In this
process, tumour cells are required to loosen their homophilic cell
adhesion, enabling tumour cells to escape from the tumour nest.
Classic cadherins interact homophilically with cadherins of
neighbouring cells to form adherens junctions, which serve both as
mechanical linkages between cells and signalling hubs that relay
information from the extracellular environment (1–5).
Epithelial cadherin, or E-cadherin, is thought to be a
tumour-suppressor molecule largely because it is frequently
downregulated in carcinomas (6–8).
Loss of E-cadherin has also been shown to be a hallmark of
epithelial-mesenchymal transition (EMT) in cancer cells and to
directly correlate with malignant phenotype and poor prognosis
(9–11).
The cellular distribution of β-catenin has a major
influence on the control of the malignant phenotype of cancer
cells. Accumulation of β-catenin in the nucleus correlates with
poor prognosis in many types of cancer. Upon translocation into the
nucleus, β-catenin forms complexes with members of the T cell
factor (TCF)/lymphoid enhancer factor (LEF) family of transcription
factors (12–14), leading to the activation of
responsive genes involved in cell proliferation, differentiation
and other malignant phenotypes (15,16).
Cytosolic β-catenin is the principal mediator of canonical Wnt
signalling (17,18). In the absence of an extracellular
Wnt ligand, cytosolic β-catenin is phosphorylated at Ser45 by the
priming kinase casein kinase 1α (CK1α) and incorporated into a
cytosolic protein complex containing Axin, the adenomatous
polyposis coli gene product (APC) and glycogen synthase kinase-3β
(GSK-3β) (19). Axin and APC serve
as scaffolding proteins that enable GSK-3β to phosphorylate
β-catenin at residues Ser33, Ser37 and Thr41 (20), thereby targeting it for
ubiquitination by β-TrCP (β-transducin repeat-containing homologue
protein) and subsequent degradation in the proteasome. Cytosolic
β-catenin protein levels are thus kept low in the absence of Wnt
ligand stimulation. Binding of a Wnt ligand to its co-receptors
Frizzled (Fzd) and low-density lipoprotein (LDL) receptor-related
protein (LRP) 5/6 results in the activation of the Dishevelled
(Dvl) protein, which then inhibits GSK-3β-mediated phosphorylation
of β-catenin. Cytosolic β-catenin is thus stabilized and is able to
accumulate. This pool of β-catenin translocates to the nucleus and
promotes malignancy by binding to TCF/LEF (18,19).
In addition to its function in Wnt signalling, β-catenin is a
component of the cadherin-based adherens junction complexes formed
at cell-cell adhesion sites. β-catenin binds the cytoplasmic domain
of cadherin and acts as a structural protein by linking
cell-surface cadherins to the actin cytoskeleton (21). By sequestering β-catenin at the
membrane, cadherins modulate the signalling properties of cytosolic
β-catenin, creating a finely tuned balance between Wnt signalling
and cell-cell adhesion (22–24).
Tetraspanins, or TM4SF (transmembrane 4 superfamily)
proteins, compose a large group of cell-surface transmembrane
proteins, some of which can form complexes with integrins. Several
tetraspanins appear to be particularly relevant to tumour cell
metastasis (25,26). CD82 (CD82/KAI-1), a member of the
tetraspanin superfamily, was originally identified as an accessory
molecule in T cell activation (27). The role played by CD82 in cancer
progression was discovered during a genetic screen to identify
metastasis-suppressor genes (28).
Ample evidence suggests that CD82 acts as a broad-spectrum
suppressor of invasion and metastasis during the progression of
various solid tumours. In malignant solid tumours, the detection of
CD82 expression indicates a better prognosis for cancer patients,
whereas the downregulation or loss of CD82 expression is commonly
associated with clinically advanced cancers (29,30).
Our previous studies indicate that CD82 negatively controls cancer
cell migration and proteinase secretion by regulating cell
signalling events, particularly those mediated by receptor tyrosine
kinase (RTK) and phosphoinositide 3-kinase (PI3K) (25,31,32).
These results suggest that the chief function of CD82 involves the
normalization of uncontrolled malignant phenotypes in cancer cells
by regulating the expression of cell-surface molecules. Recently,
we have unravelled a novel function for CD82 in E-cadherin-mediated
cellular adhesion (33). CD82
inhibits β-catenin tyrosine phosphorylation and stabilizes
E-cadherin-β-catenin complexes at the cell membrane. CD82 favours
homocellular adhesion and controls the cellular distribution of
β-catenin. This function inhibits cancer cell dissociation from the
primary cancer nest and limits metastasis (33).
In this study, we investigated the effect of CD82 on
the canonical Wnt pathway (also important in the control of
β-catenin cellular distribution) and showed that CD82 inhibits Wnt
signalling in a multifunctional way.
Materials and methods
Antibodies
Mouse monoclonal antibodies against KAI-1 (G-2),
E-cadherin (H-106) and rabbit polyclonal antibodies against KAI-1
(C-16) were purchased from Santa Cruz Biotechnology (Santa Cruz,
CA, USA). Rabbit polyclonal and mouse monoclonal antibodies against
β-catenin were purchased from Upstate Laboratories (Temecula, CA,
USA). The rabbit monoclonal antibody against phospho-β-catenin
(pSer33/pSer37) was purchased from Upstate Laboratories. Mouse
monoclonal antibodies against phospho-β-catenin (pThr41 and pSer45)
were purchased from Sigma-Aldrich (St. Louis, MO, USA). The sheep
polyclonal antibody against CK1α was purchased from R&D Systems
(Minneapolis, MN, USA) and the rabbit monoclonal antibody against
GSK-3β, from Cell Signalling Technology (Danvers, MA, USA).
The anti-Wnt protein antibodies used in this study
were as follows: anti-Wnt1 (rabbit polyclonal; GeneTex, Inc.,
Irvine, CA, USA), anti-Wnt2 (rabbit polyclonal; ProteinTech Group,
Chicago, IL, USA), anti-Wnt2B (rabbit polyclonal; AVIVA Systems
Biology, San Diego, CA, USA), anti-Wnt3 (mouse monoclonal; LifeSpan
Biosciences, Seattle, WA, USA), anti-Wnt3a (rabbit polyclonal;
Acris Antibodies, Herford, Germany), anti-Wnt4 (rat monoclonal;
Acris Antibodies), anti-Wnt5a (rabbit polyclonal; LifeSpan
Biosciences), anti-Wnt5b (rabbit polyclonal; Novus Biologicals,
Littleton, CO, USA), anti-Wnt6 (rabbit polyclonal; Novus
Biologicals), anti-Wnt7a and anti-Wnt7b (goat polyclonal; R&D
Systems), anti-Wnt8a (rabbit polyclonal; Novus Biologicals),
anti-Wnt8b (rat monoclonal; Acris Antibodies), anti-Wnt9a (goat
polyclonal; R&D Systems), anti-Wnt9b (rabbit polyclonal;
GeneWay Biotech, Inc., San Diego, CA, USA), anti-Wnt10a (rabbit
polyclonal; Novus Biologicals) and anti-Wnt10b (Wnt12) (rabbit
polyclonal; LifeSpan Biosciences).
The anti-Fzd protein antibodies used in this study
were as follows: rabbit polyclonal antibodies against Fzd1, Fzd3,
Fzd4, Fzd6, Fzd7, Fzd8, Fzd9 and Fzd10 (GenWay Biotech, Inc.) and
rabbit polyclonal antibodies against Fzd2 and Fzd5 (GeneTex,
Inc.).
Cell cultures
The human cell line h1299 (non-small-cell lung
carcinoma) and its transfectant derivatives (h1299/zeo and
h1299/CD82) were established in our laboratory by transfection of a
control vector or CD82 cDNA and cell sorting-based clone selection,
as described previously (50).
Further, h1299/zeo is a mock transfectant with weak CD82 expression
and h1299/CD82 overexpresses CD82. The protein levels of CD82 in
h1299/CD82 cells, as assayed by immunoblotting, are 20 times higher
but its cell surface expression, as assessed by flow cytometry, is
only ∼9-fold higher than that in the wild-type or h1299/zeo cells.
The cell lines used in this study were maintained in Dulbecco’s
modified Eagle’s medium (DMEM; Sigma) supplemented with 10% fetal
bovine serum (FBS; ICN Biomedicals, Aurora, OH, USA) and 2 mM
L-glutamine at 37°C and in an atmosphere of 5% CO2.
Transfection of short hairpin RNA
(shRNA)
The h1299/CD82-sh.control and h1299/CD82-sh.CD82
cell lines were generated by Lipofectamine (Invitrogen Life
Technologies, Carlsbad, CA, USA) transfection of h1299/CD82 cells
with pLKO.1-puro Control Vector (Sigma) and pLKO.1-puro/sh.CD82
(NM_002231; Sigma), respectively. Colonies that showed resistance
to puromycin (Sigma) were pooled from the individual transfection
experiments. The expression levels of CD82 in shRNA-transfected
h1299 cells were monitored by reverse transcriptase-polymerase
chain reaction (RT-PCR) and immunoblotting. The
h1299/CD82-sh.control and h1299/CD82-sh.CD82 cells were maintained
in DMEM containing 10% FBS and 2 μg/ml puromycin.
Immunoblot analysis
Cell lysates for immunoblotting were prepared in
cell lysis buffer [1% Triton X-100, 2 mM sodium orthovanadate, 500
mM NaCl, 10 mM MgCl2, 10 μg/ml leupeptin, 10
μg/ml aprotinin, 1 mM PMSF, 50 mM Tris-HCl (pH 7.2)].
Subcellular fractionation was performed using the
ProteoExtract® Subcellular Proteome Extraction kit from
Merck (Darmstadt, Germany) according to the manufacturer’s
instructions.
The samples were resolved by sodium dodecyl
sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred
to a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA) and
incubated with specific primary antibodies. Protein bands were
visualized using horseradish peroxidise (HRP)-conjugated secondary
antibodies and Enhanced Chemiluminescence Reagent (Amersham
Pharmacia Biotech, Piscataway, NJ, USA). The bands were scanned by
computer-assisted densitometry (ChemiDoc XRS-J; Bio-Rad) and
analysed using the Quantity One software (Bio-Rad).
Real-time RT-PCR
Total RNA was extracted from h1299 cells by using
TRIzol (Invitrogen, Carlsbad, CA, USA) and used for first-strand
cDNA synthesis. The mRNA levels were measured in triplicate using a
real-time PCR system with the Brilliant SYBR Green qPCR kit
(Stratagene, La Jolla, CA, USA). Specific primers for GSK-3β and
CK1α were as follows: GSK-3β (F: 5′-GG TCTATCTTAATCTGGTGCTGG-3′ and
R: 5′-AGGTTCTGC GGTTTAATATCCC-3′) and CK1α (F: 5′-F:GGAAAAGAAGC
ATGACTGTTAG-3′ and R: 5′-TCTGTATGGTATGTGTTGCC TT-3′). PCR cycling
conditions were 10 min at 95°C for 1 cycle followed by 45 cycles at
95°C for 30 sec, 60°C for 30 sec and 72°C for 60 sec. Dissociation
curve analyses confirmed that signals corresponded to unique
amplicons. Expression levels were normalized to the
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA level of each
sample, obtained from parallel assays.
Results
CD82 does not influence Wnt protein
expression
The cellular distribution of β-catenin is regulated
by the Wnt/β-catenin (canonical) signalling pathway. This pathway
is initiated by binding of Wnt ligands to their Fzd receptor
proteins. To evaluate the effect of CD82 on Wnt signalling, we
first analysed the protein expression levels of Wnt ligands on
h1299 cells by immunoblotting. As shown in Fig. 1, h1299 cells expressed all classes
of Wnt ligands (1, 2, 2b, 3a, 5a, 5b, 6, 7a, 7b, 8a, 8b, 9a, 9b,
10a and 10b). The expression of Wnt ligands was not significantly
altered after ectopic expression of CD82 at the protein level.
CD82 inhibits the expression of specific
Fzd proteins
Next, we examined the expression of Fzd proteins,
which are the transmembrane receptors for Wnt ligands (34). Binding of Wnt proteins to their Fzd
receptors transduces Wnt signalling via inactivation of GSK-3β and
consequent nuclear translocation of unphosphorylated β-catenin.
Fzd1-Fzd10 were expressed on h1299 cells. In our
model, the expression of Fzd1, Fzd4, Fzd6, Fzd8 and Fzd10 was not
markedly affected by CD82. In contrast, CD82 significantly
downregulated the expressions of Fzd2, Fzd3, Fzd5, Fzd7 and Fzd9.
Knocking down of CD82 mRNA expression by shRNA in h1299/CD82 cells
resulted in recovery of the expression of those downregulated Fzd
proteins, suggesting a specific effect of CD82 on Fzd2, Fzd3, Fzd5,
Fzd7 and Fzd9 expression (Fig.
2).
This result suggests that CD82 reduces receptor
binding of Wnt ligands by inhibiting Fzd expression, thereby
reducing Wnt signalling and β-catenin translocation to the
nucleus.
CD82 controls β-catenin cellular
distribution and inhibits β-catenin phosphorylation
In the absence of Wnt ligands or in the event of
impaired Wnt signalling, cytosolic β-catenin is phosphorylated at
Ser45 by CK1α. Consequently, GSK-3β phosphorylates β-catenin at
Thr41, Ser37 and Ser33 (20).
Ser33/Ser37 double-phosphorylated β-catenin is specifically
recognized by β-TrCP (35) and
rapidly degraded.
Therefore, we next examined the effect of CD82 on
the cellular distribution and phosphorylation of β-catenin. We
performed subcellular fractionation and determined the amount of
total and phosphorylated β-catenin in h1299 cells by
immunoblotting.
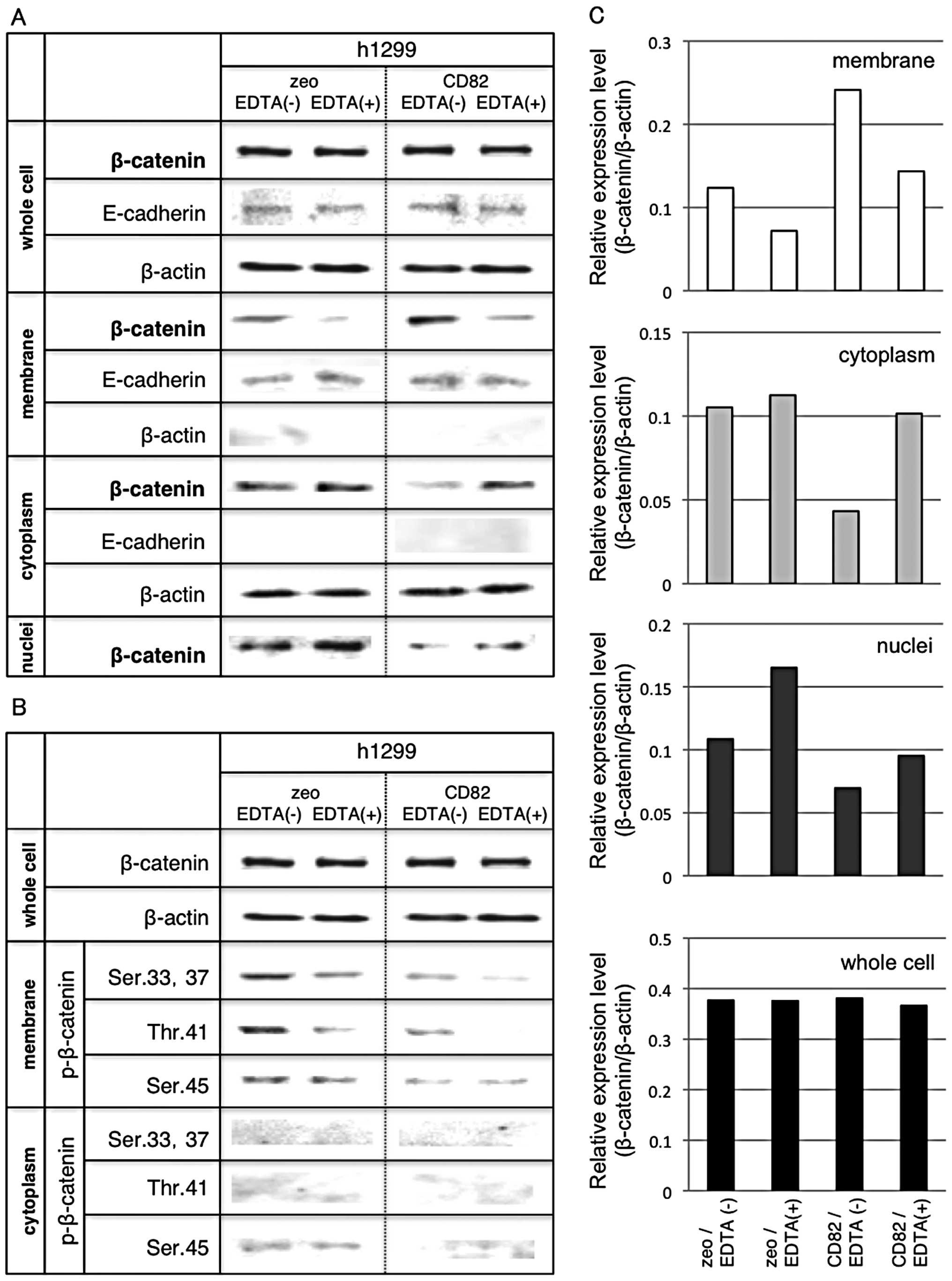
Fig. 3A shows the
cellular distribution of β-catenin in h1299 cells. The quantitated
data are shown in Fig. 3C. We
found significant accumulation of β-catenin at the cell membrane of
h1299/CD82 cells. From our previous studies that showed increase of
the E-cadherin-β-catenin complex in h1299/CD82 cells (33), we used EDTA in serum-free medium in
order to destabilize E-cadherin. Low Ca2+ treatment
enhances E-cadherin internalization (which is independent of
tyrosine phosphorylation and ubiquitination) and E-cadherin is
recycled back to the plasma membrane (36). EDTA treatment reduced β-catenin at
the cell membrane in both h1299/zeo and h1299/CD82 cells. The
amount of cytosolic β-catenin in h1299/zeo cells was only mildly
increased by EDTA treatment, whereas that in h1299/CD82 cells was
remarkably increased. Nuclear β-catenin was basically decreased in
h1299/CD82 cells. Interestingly, EDTA treatment significantly
increased β-catenin levels in h1299/zeo cells but not significantly
so in h1299/CD82 cells (Fig. 3A and
C). These results support our previously reported results
(33) and suggest that CD82
controls the distribution of cytoplasmic β-catenin to the membrane
rather than into the nucleus.
Fig. 3B shows the
levels of phosphorylated β-catenin in h1299 cells. The amount of
phosphorylated β-catenin (Ser33, Ser37, Thr41 and Ser45) at the
cell membrane was reduced by overexpression of CD82. In addition,
EDTA treatment inhibited β-catenin phosphorylation (at Ser33,
Ser37, Thr41 and Ser45) in both cell lines.
These data suggest that CD82 inhibits the
cytoplasmic translocation and phosphorylation of β-catenin (Ser33,
Ser37, Thr41 and Ser45) at the cell membrane. Moreover, CD82
reduces nuclear translocation of β-catenin even when E-cadherin is
destabilized.
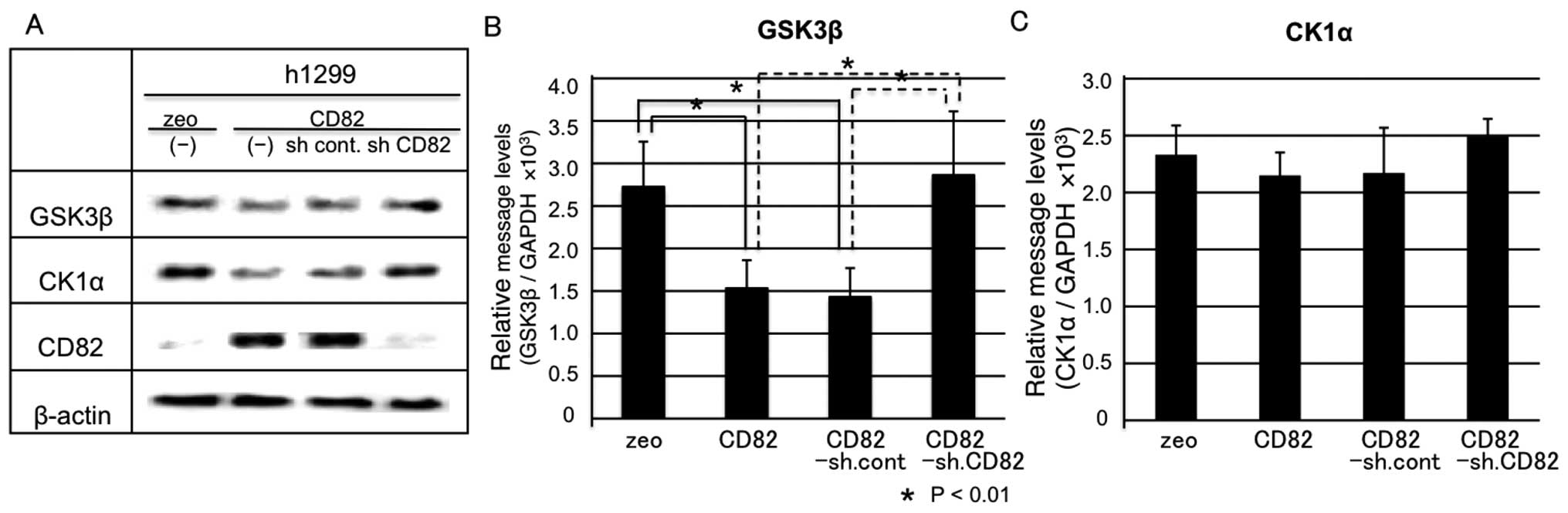
CD82 inhibits GSK-3β and CK1α
expression
Inhibition of β-catenin phosphorylation (Ser33,
Ser37, Thr41 and Ser45) by CD82 also suggests that β-catenin may be
differently phosphorylated by GSK-3β and CK1α.
In a first step, we examined the protein expression
levels of GSK-3β and CK1α by immunoblotting. The protein levels of
GSK-3β and CK1α were downregulated by CD82 and this effect was
reverted by knocking down CD82. These results indicate a specific
effect of CD82 on GSK-3β and CK1α (Fig. 4A).
Next, we examined the mRNA levels of GSK-3β and CK1α
by real-time RT-PCR. GSK-3β mRNA levels were significantly
downregulated by CD82 and this downregulation reverted by knocking
down CD82 (Fig. 4B). In contrast,
CK1α mRNA levels were not significantly downregulated by CD82, but
the slight downregulation was still reverted after knocking down
CD82 (Fig. 4C). This result
suggests that CK1α protein levels are inhibited by CD82
post-transcriptionally; however, the underlying mechanisms remain
unknown.
Discussion
In our previous research, we showed that CD82
inhibits tyrosine phosphorylation of β-catenin and stabilizes
E-cadherin-β-catenin complexes at the cell membrane (33). CD82 induces homocellular adhesion
and controls cellular distribution of β-catenin to the cell
membrane rather than to the nucleus. It has been shown that
cellular β-catenin levels are regulated at 3 different levels: the
first is regulation by RTK (37,38);
the second, regulation by the Wnt/β-catenin (canonical) pathway
(18); and the last, control by
endosomes and exosomes (39–41).
We have also shown that CD82 inhibits β-catenin phosphorylation via
the epidermal growth factor receptor (EGFR) and c-Met (33). This is a novel function of CD82 in
E-cadherin-mediated homocellular adhesion and one of the most
important functions of CD82 in inhibiting cancer metastasis. In
this study, we further found evidence that CD82 regulates β-catenin
cell distribution by inhibiting Wnt signalling at multiple
molecular levels.
All members of the Wnt protein family are
extracellular secreted proteins. Binding of a Wnt ligand to its
co-receptors Fzd and LRP5/6 results in the activation of Dvl, which
in turn inhibits GSK-3β-mediated phosphorylation of β-catenin
(19). We could not find any
differences in the protein expression of Wnt proteins after ectopic
expression of CD82. On the other hand, we found significant
downregulation of Fzd2, Fzd3, Fzd5, Fzd7 and Fzd9 after CD82
ectopic expression. This effect was reverted by shRNA knock-down of
CD82, suggesting the specific effect of CD82. Although the
specificity of the Wnt signalling is determined by the interaction
of specific pairs of Wnt and Fzd proteins, the mechanisms remain to
be elucidated. However, in various types of cancer cells, it has
been reported that particular Wnt-Fzd interactions are important in
tumour progression (18). In
particular, binding of Wnt5a to Fzd2 or Fzd7 controls
metalloproteinase production (42,43)
and focal adhesion dynamics via the Wnt signalling pathway
(44). It has also been reported
that Wnt5a expression is of clinical relevance in prostate cancer
(42). These reports support the
idea that Fzd2, Fzd3, Fzd5, Fzd7 and Fzd9 (down-regulated by
overexpression of CD82) are key players in many types of cancer
cells. Therefore, specific downregulation of Fzd receptors by CD82
may reduce Wnt/β-catenin signalling. Furthermore, Wnt target genes
(Wnt3a, Fzd7, Axin, TCF/LEF, among others) are all molecules
related to Wnt signalling itself (45–49).
Inhibition of Wnt signalling pathway leads to further negative
regulation of the pathway. This results in inhibition of β-catenin
nuclear translocation and consequent accumulation in the
cytoplasm.
Accumulated cytoplasmic β-catenin is phosphorylated
at Ser45 by CK1α and incorporated into a cytosolic protein complex
containing Axin, APC and GSK-3β. GSK-3β phosphorylates β-catenin at
residues Ser33, Ser37 and Thr41 (20), thereby targeting it for
ubiquitination by β-TrCP and subsequent degradation in the
proteasome. Ectopic expression of CD82 inhibits phosphorylation of
β-catenin at Ser45, Ser33, Ser37 and Thr41 by downregulation of
GSK-3β and CK1α, thereby favouring the escape of β-catenin from the
ubiquitination and degradation process. Therefore, downregulation
of GSK-3β and CK1α by CD82 ultimately leads to accumulation of
β-catenin in the cytoplasm. Contradictorily, we found a decrease in
cytoplasmic β-catenin and a significant increase in β-catenin
accumulation at the cell membrane of CD82-transfected cells
(Fig. 3A). We also found that
EDTA-destabilized E-cadherin inhibits the translocation of
β-catenin to the cell membrane (Fig.
3A). This result suggests that β-catenin, when at the cell
membrane, is associated with E-cadherin. We have already shown that
overexpression of CD82 induces interaction between E-cadherin and
β-catenin and one possible mechanism is the inhibition of RTK
signalling pathway by CD82. CD82 inhibits EGFR tyrosine
phosphorylation (32) and Ras- and
PI3K-dependent c-Met signalling (50). Direct association of CD82 with
these growth factor receptors inhibits receptor signal
transduction, which will result in inhibition of β-catenin tyrosine
phosphorylation (50). Several
studies have shown that either increased kinase activity through
stimulation with EGF or decreased phosphatase activity using
pervanadate or phosphatase mutants leads to decreased interaction
between cadherin-catenin complexes and the cytoskeleton. CK1α also
destabilizes E-cadherin by phosphorylation of the cytoplasmic
domain of E-cadherin. Downregulation of CK1α by CD82 overexpression
will result in E-cadherin stabilization (51). Therefore, CD82 strengthens the
interaction between E-cadherin and β-catenin by multiple pathways
and results in translocation of accumulated cytoplasmic β-catenin
to the membrane.
Furthermore, this result also serves as evidence to
show that CD82 inhibits the nuclear translocation of β-catenin.
Accumulation of β-catenin in the cytoplasm will result in nuclear
translocation of β-catenin, as observed in the EDTA-treated
h1299/zeo cells (Fig. 3A). In
contrast, EDTA treatment actually increased β-catenin in the
cytoplasm while impairing its translocation to the nucleus. These
findings highlight an important function of CD82 in inhibiting
β-catenin translocation to the nucleus. However, the mechanism
remains to be elucidated.
Recently, a novel inhibitory mechanism of Wnt
signalling by CD82 was described. CD82 and other tetraspanins are
organized in a signalling complex with E-cadherin at the plasma
membrane. This signalling complex, including tetraspanins,
E-cadherin and β-catenin, is internalized and delivered to early
endosomes (41). Exosome
biogenesis begins with outward vesicle budding at the limiting
membrane of endosomes, generating intraluminal vesicles (ILVs).
These exosome-containing endosomes eventually mature into late
endosomes, also known as multivesicular bodies (MVBs). These MVBs
then fuse with the plasma membrane and release their intraluminal
vesicles, referred to as exosomes, which contain β-catenin.
Exosomal targeting of β-catenin causes a reduction in the
intracellular pool of β-catenin and therefore reduces Wnt/β-catenin
signalling. This mechanism is thought to occur after CD82-induced
β-catenin membrane translocation, as shown in this study.
Therefore, CD82 recruits β-catenin and E-cadherin to the plasma
membrane, thereby contributing to the formation of large signalling
complexes, which are in turn incorporated into β-catenin-containing
exosomes, whose contents are released outside the cells.
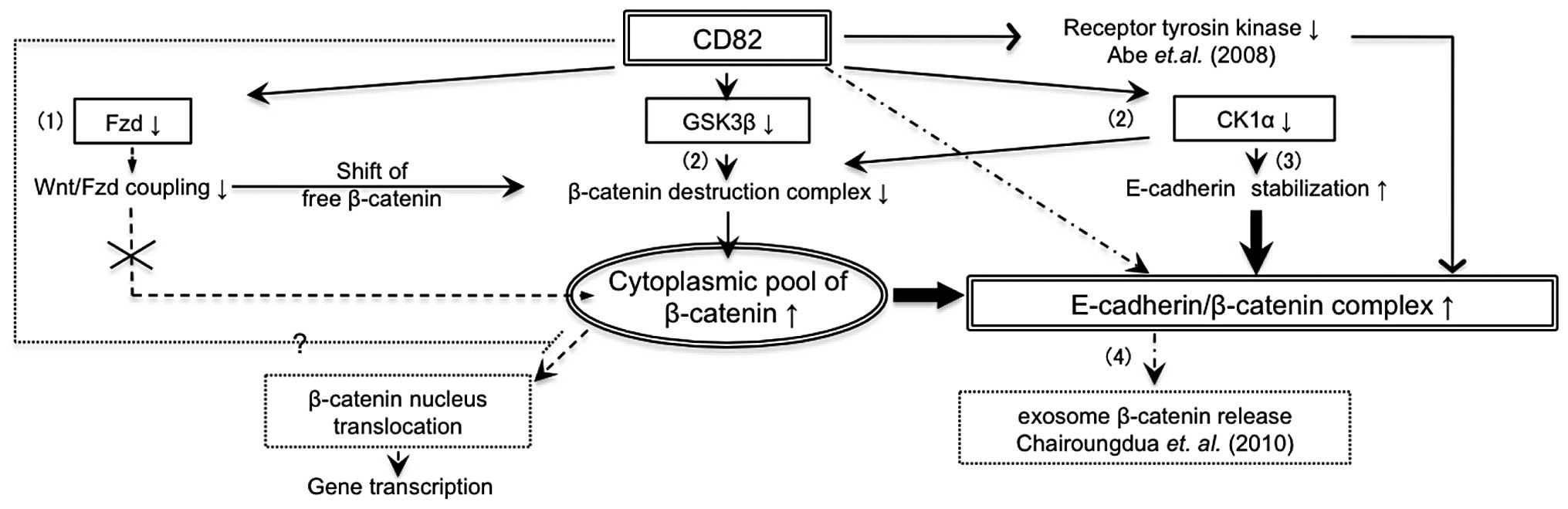
Altogether, CD82 attenuates Wnt signalling in
multiple ways (Fig. 5): i)
inhibition of β-catenin nuclear translocation by downregulation of
Fzd receptors or other mechanism; ii) accumulation of β-catenin at
the cell membrane by downregulation of GSK-3β and CK1α; iii)
stabilization of the E-cadherin-β-catenin complex by inhibition of
RTK and downregulation of CK1α; and iv) induction of exosomal
release of β-catenin. In the first step, (i) and (ii) will increase
the cytoplasmic pool of β-catenin, whereas in the second step,
(iii) and (iv) will reduce the cytoplasmic pool of β-catenin.
Inhibition of Wnt signalling by β-catenin translocation but not by
β-catenin degradation leads to E-cadherin stabilization at the cell
surface and strengthens homophilic adhesions between cancer
cells.
In conlusion, CD82 suppresses cancer metastasis
through the canonical Wnt pathway via multifunctional ways.
Down-regulation of Fzd receptors also suggests inhibition of the
non-canonical Wnt pathway. However, this mechanism remains to be
elucidated. Overall, accumulating evidence of the anti-metastatic
effect of CD82 suggests CD82 as a novel therapeutic target for
anti-metastasis cancer therapy.
Acknowledgements
This study was supported by
Grants-in-Aid (no. 20390517 to K.S. and T.S., no. 23390465 to T.S.)
from the Ministry of Education, Culture, Sports, Science and
Technology of Japan.
References
|
1.
|
M TakeichiCadherins: a molecular family
important in selective cell-cell adhesionAnnu Rev
Biochem59237252199010.1146/annurev.bi.59.070190.0013212197976
|
|
2.
|
R KemlerFrom cadherins to catenins:
cytoplasmic protein interactions and regulation of cell
adhesionTrends
Genet9317321199310.1016/0168-9525(93)90250-L8236461
|
|
3.
|
PA PiepenhagenWJ NelsonDefining
E-cadherin-associated protein complexes in epithelial cells:
plakoglobin, beta- and gamma-catenin are distinct componentsJ Cell
Sci1047517621993
|
|
4.
|
A NagafuchiS TsukitaM
TakeichiTransmembrane control of cadherin-mediated cell-cell
adhesionSemin Cell Biol4175181199310.1006/scel.1993.10218347834
|
|
5.
|
M TakeichiThe cadherin cell adhesion
receptor family: roles in multicellular organization and
neurogenesisProg Clin Biol Res39014515319947724643
|
|
6.
|
H SembG ChristoforiThe tumor-suppressor
function of E-cadherinAm J Hum
Genet6315881593199810.1086/3021739837810
|
|
7.
|
Y DokiH ShiozakiH TaharaCorrelation
between E-cadherin expression and invasiveness in vitro in a human
esophageal cancer cell lineCancer Res533421342619938324752
|
|
8.
|
H OkaH ShiozakiK KobayashiExpression of
E-cadherin cell adhesion molecules in human breast cancer tissues
and its relationship to metastasisCancer
Res531696170119938453644
|
|
9.
|
MH YangCL ChenGY ChauComprehensive
analysis of the independent effect of twist and snail in promoting
metastasis of hepatocellular
carcinomaHepatology5014641474200910.1002/hep.2322119821482
|
|
10.
|
DI BellovinRC BatesA MuzikanskyDL RimmAM
MercurioAltered localization of p120 catenin during epithelial to
mesenchymal transition of colon carcinoma is prognostic for
aggressive diseaseCancer
Res651093810945200510.1158/0008-5472.CAN-05-194716322241
|
|
11.
|
G HanSL LuAG LiW HeCL CorlessM
Kulesz-MartinXJ WangDistinct mechanisms of TGF-beta1-mediated
epithelial-to-mesenchymal transition and metastasis during skin
carcinogenesisJ Clin
Invest11517141723200510.1172/JCI2439915937546
|
|
12.
|
J BehrensJP von KriesM KuhlL BruhnD
WedlichR GrosschedlW BirchmeierFunctional interaction of
beta-catenin with the transcription factor
LEF-1Nature382638642199610.1038/382638a08757136
|
|
13.
|
O HuberR KornJ McLaughlinM OhsugiBG
HerrmannR KemlerNuclear localization of beta-catenin by interaction
with transcription factor LEF-1Mech
Dev59310199610.1016/0925-4773(96)00597-78892228
|
|
14.
|
M MolenaarM van de WeteringM
OosterwegelXTcf-3 transcription factor mediates
beta-catenin-induced axis formation in Xenopus
embryosCell86391399199610.1016/S0092-8674(00)80112-98756721
|
|
15.
|
TC HeAB SparksC RagoIdentification of
c-MYC as a target of the APC
pathwayScience2811509151219989727977
|
|
16.
|
O TetsuF McCormickBeta-catenin regulates
expression of cyclin D1 in colon carcinoma
cellsNature398422426199910.1038/1888410201372
|
|
17.
|
PJ Morinbeta-catenin signaling and
cancerBioessays2110211030199910.1002/(SICI)1521-1878(199912)22:1%3C1021::AID-BIES6%3E3.0.CO;2-P10580987
|
|
18.
|
P PolakisWnt signaling and cancerGenes
Dev14183718512000
|
|
19.
|
D WuW PanGSK3: a multifaceted kinase in
Wnt signalingTrends Biochem
Sci35161168201010.1016/j.tibs.2009.10.00219884009
|
|
20.
|
C LiuY LiM SemenovControl of beta-catenin
phosphorylation/degradation by a dual-kinase
mechanismCell108837847200210.1016/S0092-8674(02)00685-211955436
|
|
21.
|
RL DaughertyCJ GottardiPhospho-regulation
of Beta-catenin adhesion and signaling
functionsPhysiology22303309200710.1152/physiol.00020.200717928543
|
|
22.
|
J HeasmanA CrawfordK
GoldstoneOverexpression of cadherins and underexpression of
beta-catenin inhibit dorsal mesoderm induction in early Xenopus
embryosCell79791803199410.1016/0092-8674(94)90069-87528101
|
|
23.
|
RT CoxC KirkpatrickM PeiferArmadillo is
required for adherens junction assembly, cell polarity, and
morphogenesis during Drosophila embryogenesisJ Cell
Biol134133148199610.1083/jcb.134.1.1338698810
|
|
24.
|
F FagottoN FunayamaU GluckBM
GumbinerBinding to cadherins antagonizes the signaling activity of
beta-catenin during axis formation in XenopusJ Cell
Biol13211051114199610.1083/jcb.132.6.11058601588
|
|
25.
|
T SugiuraF BerditchevskiFunction of
alpha3beta1-tetraspanin protein complexes in tumor cell invasion.
Evidence for the role of the complexes in production of matrix
metalloproteinase 2 (MMP–2)J Cell Biol14613751389199610491398
|
|
26.
|
F BerditchevskiE OdintsovaCharacterization
of integrintetraspanin adhesion complexes: role of tetraspanins in
integrin signalingJ Cell
Biol146477492199910.1083/jcb.146.2.47710427099
|
|
27.
|
S Lebel-BinayML GilC LagaudriereFurther
characterization of CD82/IA4 antigen (type III surface protein): an
activation/differentiation marker of mononuclear cellsCell
Immunol154468483199410.1006/cimm.1994.1092
|
|
28.
|
JT DongPW LambCW Rinker-SchaefferJ
VukanovicT IchikawaJT IsaacsJC BarrettKAI1, a metastasis suppressor
gene for prostate cancer on human chromosome
11p11.2Science268884886199510.1126/science.77543747754374
|
|
29.
|
M AdachiT TakiT KonishiCI HuangM
HigashiyamaM MiyakeNovel staging protocol for non-small-cell lung
cancers according to MRP-1/CD9 and KAI1/CD82 gene expressionJ Clin
Oncol161397140619989552043
|
|
30.
|
X GuoH FriessHU GraberM KashiwagiA
ZimmermannM KorcMW BuchlerKAI1 expression is up-regulated in early
pancreatic cancer and decreased in the presence of metastasesCancer
Res564876488019968895737
|
|
31.
|
M TakahashiT SugiuraK ShirasunaTetraspanin
CD82/KAI-1 regulates growth factor induced cancer cell migration by
forming complexes with growth factor receptorsInt J Oral Maxillofac
Surg34146200510.1016/S0901-5027(05)81459-6
|
|
32.
|
E OdintsovaT SugiuraF
BerditchevskiAttenuation of EGF receptor signaling by a metastasis
suppressor, the tetraspanin CD82/KAI-1Curr
Biol1010091012200010.1016/S0960-9822(00)00652-710985391
|
|
33.
|
M AbeT SugiuraM TakahashiK IshiiM ShimodaK
ShirasunaA novel function of CD82/KAI-1 on E-cadherin-mediated
homophilic cellular adhesion of cancer cellsCancer
Lett266163170200810.1016/j.canlet.2008.02.05818395972
|
|
34.
|
P BhanotM BrinkCH SamosA new member of the
frizzled family from Drosophila functions as a Wingless
receptorNature382225230199610.1038/382225a08717036
|
|
35.
|
JT WinstonP StrackP Beer-RomeroCY ChuSJ
ElledgeJW HarperThe SCFbeta-TRCP-ubiquitin ligase complex
associates specifically with phosphorylated destruction motifs in
IkappaBalpha and beta-catenin and stimulates IkappaBalpha
ubiquitination in vitroGenes
Dev13270283199910.1101/gad.13.3.270
|
|
36.
|
F PalaciosJS TushirY FujitaC
D’Souza-SchoreyLysosomal targeting of E-cadherin: a unique
mechanism for the down-regulation of cell-cell adhesion during
epithelial to mesenchymal transitionsMol Cell
Biol25389402200510.1128/MCB.25.1.389-402.200515601859
|
|
37.
|
CH LeeHW HungPH HungYS ShiehEpidermal
growth factor receptor regulates beta-catenin location, stability,
and transcriptional activity in oral cancerMol
Cancer964201010.1186/1476-4598-9-6420302655
|
|
38.
|
F VerkaarGJ ZamanA model for signaling
specificity of Wnt/Frizzled combinations through co-receptor
recruitmentFEBS
Lett58438503854201010.1016/j.febslet.2010.08.03020800062
|
|
39.
|
TL LeAS YapJL StowRecycling of E-cadherin:
a potential mechanism for regulating cadherin dynamicsJ Cell
Biol146219232199910.1083/jcb.146.1.21910402472
|
|
40.
|
AI IvanovA NusratCA ParkosEndocytosis of
epithelial apical junctional proteins by a clathrin-mediated
pathway into a unique storage compartmentMol Biol
Cell15176188200410.1091/mbc.E03-05-031914528017
|
|
41.
|
A ChairoungduaDL SmithP PochardM HullMJ
CaplanExosome release of beta-catenin: a novel mechanism that
antagonizes Wnt signalingJ Cell
Biol19010791091201010.1083/jcb.20100204920837771
|
|
42.
|
H YamamotoN OueA SatoWnt5a signaling is
involved in the aggressiveness of prostate cancer and expression of
metalloproteinaseOncogene2920362046201010.1038/onc.2009.49620101234
|
|
43.
|
A SatoH YamamotoH SakaneH KoyamaA
KikuchiWnt5a regulates distinct signalling pathways by binding to
Frizzled2EMBO J294154201010.1038/emboj.2009.32219910923
|
|
44.
|
S MatsumotoK FumotoT OkamotoK KaibuchiA
KikuchiBinding of APC and dishevelled mediates Wnt5a-regulated
focal adhesion dynamics in migrating cellsEMBO
J2911921204201010.1038/emboj.2010.2620224554
|
|
45.
|
YW ZhangYF MiaoJ YiJ GengR WangLB
ChenTranscriptional inactivation of secreted frizzled-related
protein 1 by promoter hypermethylation as a potential biomarker for
non-small cell lung
cancerNeoplasma57228233201010.4149/neo_2010_03_22820353273
|
|
46.
|
J WillertM EppingJR PollackPO BrownR
NusseA transcriptional response to Wnt protein in human embryonic
carcinoma cellsBMC Dev Biol28200210.1186/1471-213X-2-812095419
|
|
47.
|
B LustigB JerchowM SachsNegative feedback
loop of Wnt signaling through upregulation of conductin/axin2 in
colorectal and liver tumorsMol Cell
Biol2211841193200210.1128/MCB.22.4.1184-1193.200211809809
|
|
48.
|
J RooseG HulsM van BeestSynergy between
tumor suppressor APC and the beta-catenin-Tcf4 target
Tcf1Science28519231926199910.1126/science.285.5435.192310489374
|
|
49.
|
M FilaliN ChengD AbbottV LeontievJF
EngelhardtWnt-3A/beta-catenin signaling induces transcription from
the LEF-1 promoterJ Biol
Chem2773339833410200210.1074/jbc.M10797720012052822
|
|
50.
|
M TakahashiT SugiuraM AbeK IshiiK
ShirasunaRegulation of c-Met signaling by the tetraspanin
KAI-1/CD82 affects cancer cell migrationInt J
Cancer12119191929200710.1002/ijc.2288717621632
|
|
51.
|
S Dupre-CrochetA FigueroaC HoganCasein
kinase 1 is a novel negative regulator of E-cadherin-based
cell-cell contactsMol Cell
Biol2738043816200710.1128/MCB.01590-0617353278
|