Introduction
Prostate cancer is the second leading cause of male
cancer-related mortality in the United States (1). A distinctive characteristic of this
cancer type is that prostate tumors are critically dependent on
androgen for development, growth and survival (2,3).
Androgen ablation therapy is the foundation of current prostate
cancer treatment for patients that present with locally advanced or
metastatic disease. This is typically achieved through chemical
castration using selective agents that reduce levels of circulating
androgens, such as luteinizing hormone-releasing hormone (LHRH)
agonists or androgen receptor (AR) antagonists such as bicalutamide
(4). Although this approach
initially induces clinical remissions, most patients ultimately
relapse and progress to castration-resistant disease within a
median of 18–24 months (5).
It is now clear that these advanced tumors continue
to rely on AR signaling, and a number of mechanisms have been
proposed for reactivation of AR in the castrate environment
(3,5). Novel endocrine treatments targeting
the AR signaling axis, including abiraterone acetate and MDV3100,
have recently shown clinical promise for advanced prostate cancer,
particularly in the second-line therapeutic setting (6,7).
Reports however suggest that resistance to these new agents, linked
to continued hormone-driven oncogenesis, can develop (8). Thus the incomplete efficacy of
androgen deprivation therapy highlights an urgent need for
alternative treatment strategies to improve patient outcomes.
In this regard, targeting heat shock protein 90
(Hsp90) has emerged as a potential avenue for therapeutic
intervention. Hsp90 is a molecular chaperone required for the
post-translational stability and function of numerous key signal
transduction proteins, termed ‘client’ proteins (9,10).
Of note, a number of these clients have been causally implicated in
the pathogenesis of prostate cancer, including AR, HER2, AKT and
RAF1 (11–13). Interaction with Hsp90 regulates the
half-life of these proteins and the AR is particularly reliant on
Hsp90 function for its activity. Within the cytoplasm, the receptor
is maintained in a multichaperone complex with Hsp90 that is
essential for stabilizing the protein in a conformation receptive
to ligand binding (14).
Importantly, inhibition of Hsp90 activity targets its clients for
proteasomal destruction. Thus pharmacological blockade of Hsp90 can
overcome signaling redundancies and mechanisms of drug resistance
commonly observed in many cancers (15–17)
because of its coordinate and simultaneous impact on multiple
signaling cascades. For these reasons, Hsp90 represents an
attractive molecular target for the development of new anticancer
agents (18,19).
A number of preclinical studies have provided
compelling evidence supporting the potential utility of Hsp90
inhibitors in prostate cancer (20–23).
Unfortunately, the clinical experience using such compounds in the
single-agent setting has been disappointing, with minimal effects
on PSA levels or tumor burden being observed along with
unacceptable toxicities (24,25).
Ganetespib (formerly STA-9090) is a new small molecule inhibitor of
Hsp90 with superior pharmacologic and biologic properties that
distinguish it from other first- and second-generation inhibitors
in terms of antitumor activity, potency and safety (26). In light of these considerations,
here we have undertaken a comprehensive evaluation of ganetespib
activity in prostate cancer cell lines both in vitro and
in vivo.
Materials and methods
Cell lines, antibodies and reagents
The LNCaP, VCaP, 22Rv1, DU145 and PC3 human prostate
cancer cell lines and HeLa cells were all purchased from the
American Type Culture Collection (Manassas, VA, USA). Cells were
maintained and cultured according to standard techniques at 37°C in
5% (v/v) CO2 using culture medium recommended by the
supplier. All primary antibodies were purchased from Cell Signaling
Technology (Beverly, MA, USA) with the exception of RAF1 (Santa
Cruz Biotechnology, Santa Cruz, CA, USA), p-EGFR (Tyr1068)
(Invitrogen, Carlsbad, CA, USA), actin (GE Healthcare, UK) and the
AR mouse monoclonal antibody AR441 (27), which was prepared by the antibody
core of the Dan L. Duncan Cancer Center at Baylor College of
Medicine. The Hsp90 inhibitors ganetespib and 17-AAG were
synthesized at Synta Pharmaceuticals Corp. Methyltrienolone (R1881)
was purchased from Perkin-Elmer (Boston, MA, USA).
Cell viability assays
Cellular viability was assessed using the
CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison,
WI, USA) according to the manufacturer’s protocol. Twenty-four
hours after plating at 5×103 cells/well in triplicate in
96-well plates, cells were dosed with graded concentrations of
ganetespib or 17-AAG for 72 h. CellTiter-Glo was added (50% v/v) to
the cells, and the plates incubated for 10 min prior to luminescent
detection in a SpectraMax Plus 384 microplate reader (Molecular
Devices, Sunnyvale, CA, USA). Data were normalized to percent of
control and IC50 values used to determine the
sensitivity of each line.
Western blotting
Prostate cancer cell lines were lysed in RIPA buffer
(Cell Signaling Technology) and HeLa lysed by four rounds of
freeze/thawing using 1X Reporter Lysis Buffer (Promega) containing
0.4 M NaCl. Lysates were clarified by centrifugation and equal
amounts of protein resolved by SDS-PAGE before transfer to
nitrocellulose membranes. Membranes were blocked with 5% skim milk
in TBS with 0.5% Tween and immunoblotted with indicated antibodies.
Antigen-antibody complexes were visualized using an Odyssey system
(LI-COR, Lincoln, NE, USA) or using ECL reagents.
Quantitative RT-PCR
LNCaP cells were cultured in charcoal-stripped
medium for 24 h and then treated with 250 nM ganetespib, 1
μM geldanamycin, or vehicle for 24 h in the absence or
presence of 10 nM methyltrienolone (R1881). RNA was prepared from
the LNCaP cells post-treatment using TRIzol reagent (Invitrogen,
Grand Island, NY, USA). Previously reported prostate specific
antigen (PSA), transmembrane protease, serine 2 (TMPRSS2), and 18S
primer sets (28) were used for
target gene expression and were analyzed using SYBR green PCR
Master mix in an ABI 7500 Fast sequence detection system. PSA and
TMPRSS2 mRNA levels were normalized to 18S mRNA values.
Transient transfection of HeLa cells
HeLa cells were transiently transfected using a
poly-L-lysine coupled adenoviral-mediated DNA transfer technique as
previously described (29). The
plasmid constructs used were pCR3.1-AR (encoding full-length AR)
and pCR3.1-V7 (encoding the V7 truncated AR isoform, a gift from
Manjula Nakka and William Krause, Baylor College of Medicine). For
the expression study, HeLa cells were transfected with 3 ng of
pCR3.1-AR or 0.5 ng of pCR3.1-V7 for 24 h. Cells were treated with
R1881 (10 nM), GA (1 μM), and/or ganetespib (250 nM) or
vehicle (ethanol and DMSO) for 24 h prior to lysis and
immunoblotting. To determine the effect of Hsp90 inhibitors on AR
and variant activity, HeLa cells were transiently transfected with
250 ng of GRE-luciferase reporter, 30 ng of pCR3.1 β-galactosidase,
3 ng of pCR3.1-AR, or 0.03 ng of pCR3.1-V7 and treated as above
except that inhibitors were added immediately after the completion
of the transfection procedure. Luciferase and β-galactosidase
activities were measured and luciferase levels normalized to
β-galactosidase levels as previously described (30).
Flow cytometry
For cell cycle analysis, PC3 and DU145 cells were
seeded overnight at 0.3×106 cells/5 ml in a 6-well plate
and then exposed to increasing concentrations of ganetespib (0–500
nM) for 24 h. Cells were harvested and stained with propidium
iodide using the BD Cycle Test Plus Reagent Kit (BD Biosciences,
San Jose, CA, USA) according to the manufacturer’s instructions.
Twenty thousand cells were analyzed for their DNA content using a
FACS Calibur cytometer (BD Biosciences, Billerica, MA, USA). For
the apoptosis assay in the DU145 cell line, cells were treated with
ganetespib (10, 100 or 500 nM), 17-AAG (500 or 1000 nM) or control
(DMSO) for 24 h. Following treatment cells were harvested and
stained using a fluorescein-conjugated anti-Annexin V antibody (BD
Biosciences) and apoptosis assessed by flow cytometry.
In vivo prostate xenograft model
Eight-week-old female immunodeficient nude and CB-17
severe combined immunodeficient (SCID) mice (Charles River
Laboratories, Wilmington, MA, USA) were maintained in a
pathogen-free environment, and all in vivo procedures were
approved by the Synta Pharmaceuticals Corp. Institutional Animal
Care and Use Committee in accordance with the Guide for Care and
Use of Laboratory Animals. PC3 tumor cells (5×106) were
subcutaneously implanted into nude mice and 22Rv1 cells
(5×106) into SCID mice. Animals bearing established
tumors (100–200 mm3) were randomized into treatment
groups of 8 and i.v. dosed via the tail vein with either vehicle or
ganetespib formulated in 10/18 DRD (10% DMSO, 18% Cremophor RH 40,
3.6% dextrose, 68.4% water). Tumor volumes (V) were calculated by
caliper measurements of the width (W), length (L), and thickness
(T) of each tumor using the formula: V = 0.5236 (LWT). Tumor growth
inhibition was determined as described previously (31).
Results
Ganetespib potently induces cell death in
prostate cancer cells irrespective of androgen receptor status
We initially examined the growth inhibitory effects
of ganetespib in vitro using a panel of prostate cancer cell
lines. In all cases, ganetespib reduced cell viability in a
dose-dependent manner and was more potent than the first-generation
ansamycin Hsp90 inhibitor 17-AAG (Table I). In the AR-negative cell lines
DU145 and PC3 the cytotoxicity IC50 values at 72 h were
12 and 77 nM, respectively. The AR-positive, androgen-dependent
cell lines LNCaP and VCaP were more sensitive to ganetespib
exposure (IC50 values of 8 and 7 nM). The 22Rv1 cell
line, which while AR-positive is only weakly androgen responsive,
was also highly sensitive to ganetespib (IC50, 20 nM).
These data demonstrate that Hsp90 inhibition by ganetespib results
in potent cytotoxic effects in prostate cancer lines regardless of
their AR status or androgen sensitivity.
 | Table I.Comparison of ganetespib and 17-AAG
in vitro cytotoxicity in a panel of prostate cancer cell
lines. |
Table I.
Comparison of ganetespib and 17-AAG
in vitro cytotoxicity in a panel of prostate cancer cell
lines.
| Cell line | AR
expression/androgen sensitivity | Ganetespib
(nM) | 17-AAG (nM) |
|---|
| LNCaP | +/Dependent | 8 | 266 |
| VCaP | +/Dependent | 7 | 2645 |
| 22Rv1 | +/Partial | 20 | 1270 |
| DU145 | −/Independent | 12 | 36 |
| PC3 | −/Independent | 77 | 246 |
Coordinate inhibition of AR activity and
multiple oncogenic signaling pathways in prostate cancer cells by
ganetespib
Targeted degradation of client proteins is a feature
of Hsp90 inhibition. We therefore examined expression changes in
Hsp90 clients known to be associated with prostate tumor
progression. AR-positive LNCaP cells were treated with ganetespib
or 17-AAG for 24 h and protein levels determined by western blot
analysis (Fig. 1A). Ganetespib
treatment resulted in a potent and dose-dependent decrease in AR
levels. Hsp90-directed loss of AR receptor expression resulted in
consequent suppression of AR-directed gene regulation. To show
this, LNCaP cells were cultured in charcoal-stripped medium for 24
h and then treated with ganetespib, geldanamycin (GA, the parent
compound from which 17-AAG is derived), or vehicle for 24 h in the
absence or presence of androgen (R1881). As a read-out of
AR-specific transcriptional activity, PSA and TMPRSS2 mRNA levels
were measured and normalized to 18S mRNA values (Fig. 1B). In accordance with the
androgen-inducible expression of both genes, R1881 exposure
increased PSA and TMPRSS2 levels in control cells. This induction
was significantly inhibited in the presence of either Hsp90
inhibitor (*P<0.001) (Fig. 1B).
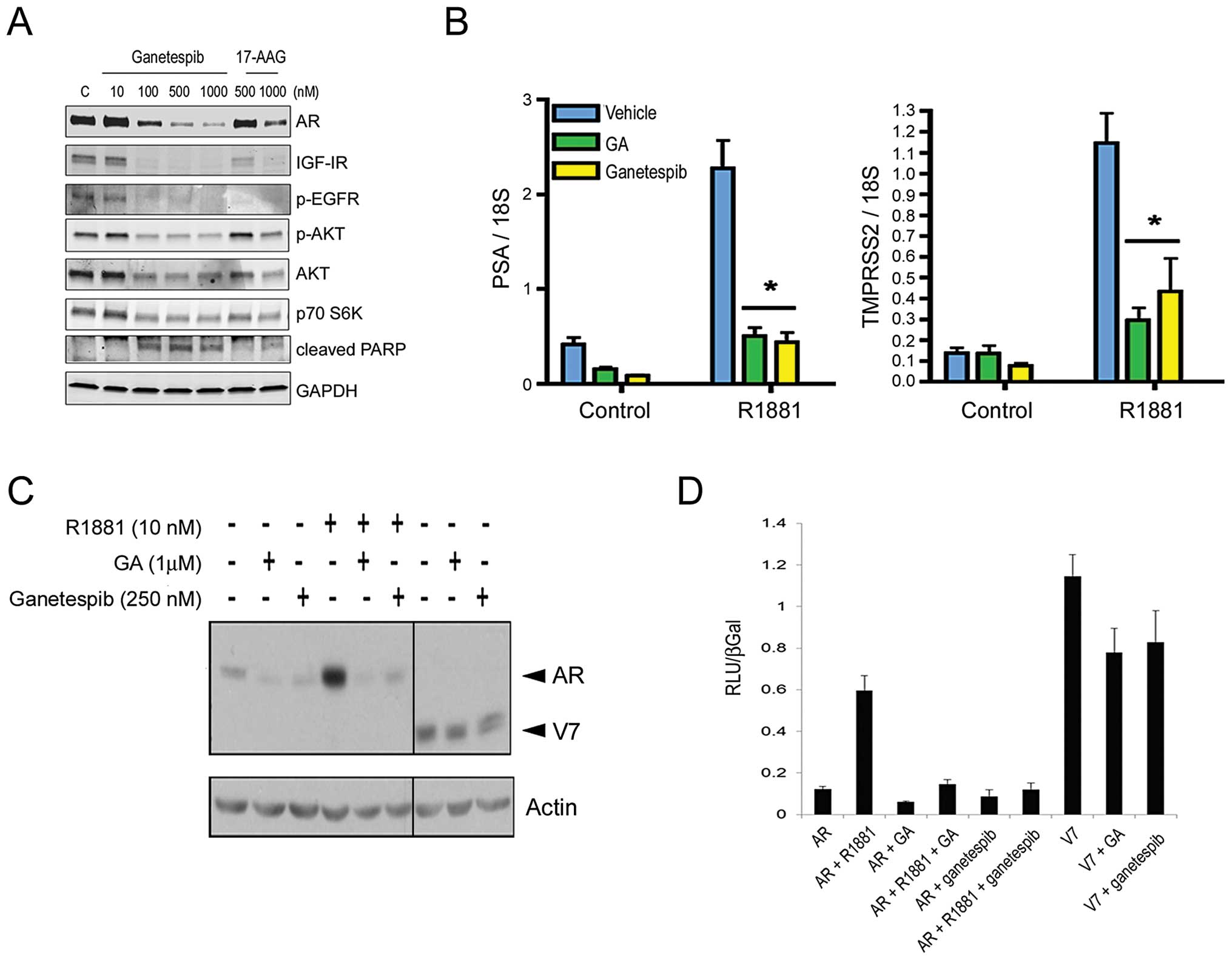 | Figure 1.Ganetespib treatment destabilizes
full-length AR receptor expression and activity, as well as
multiple client proteins, in AR-positive cancer cell lines. (A),
LNCaP cells were exposed to increasing concentrations of ganetespib
or 17-AAG as indicated for 24 h. Cell lysates were immunoblotted
using antibodies against AR, IGF-IR, phosphorylated EGFR (p-EGFR),
phosphorylated AKT (p-AKT), total AKT and p70 S6K as shown. Cleaved
PARP expression was included as a marker of apoptosis. Total
protein levels were determined using GAPDH. (B), LNCaP cells were
cultured in charcoal-stripped medium for 24 h and then treated with
250 nM ganetespib, 1 μM geldanamycin (GA), or vehicle for 24
h in the absence or presence of 10 nM androgen (R1881). Prostate
specific antigen (PSA) and transmembrane protease, serine 2
(TMPRSS2) mRNA levels were measured and normalized to 18S mRNA
values. Experiments were performed in triplicate.
Androgen-inducible transcriptional activation was significantly
inhibited in the presence of either Hsp90 inhibitor
(*P<0.001). (C), HeLa cells were transiently
transfected with 3 ng of pCR3.1-AR or 0.5 ng of pCR3.1-ARV7 plasmid
to induce expression of the full-length and V7 truncated AR
proteins, respectively (arrowheads). Twenty-four hours following
infection, cells were treated with 10 nM R1881, 1 μM GA, or
250 nM ganetespib as indicated. Cell lysates were resolved by
SDS-PAGE and immunoblotted with an anti-AR antibody. Total protein
levels were determined using an anti-actin antibody. (D), To
determine the effect of Hsp90 inhibitors on AR and variant
activity, HeLa cells were transiently transfected with 250 ng of
GRE-luciferase reporter, 30 ng of pCR3.1 β-galactosidase, 3 ng of
pCR3.1-AR, or 0.03 ng of pCR3.1-V7 and treated with vehicle
(ethanol and DMSO), R1881 (10 nM), GA (1 μM), and/or
ganetespib (250 nM) for 24 h. Luciferase and β-galactosidase
activities were measured and luciferase levels were normalized to
β-galactosidase levels. Experiments were performed in
triplicate. |
Importantly, ganetespib also induced degradation of
IGF-IR and phosphorylated EGFR receptors, previously implicated in
the pathogenesis of prostate cancer, as well as the downstream
effectors AKT and p70 S6K, in LNCaP cells (Fig. 1A). Moreover a concomitant increase
in PARP cleavage, a marker of apoptosis, accompanied the reductions
in these protein levels. Consistent with the differences in
sensitivity shown in Table I,
ganetespib was comparatively more potent than 17-AAG at inducing
targeted loss of these oncogenic proteins and signaling
pathways.
Constitutively active AR variant
expression does not confer resistance to ganetespib
The expression of alternatively spliced,
terminally-truncated AR isoforms is one potential mechanism for the
development of a castration-resistant phenotype (32). For example, the 22Rv1 cell line
expresses the full-length AR protein as well as constitutively
active variants that lack the carboxyl-terminal ligand-binding
domain (33,34), thereby reducing its dependence on
exogenous androgen. Of note, we found that 22Rv1 cells were acutely
sensitive to the effects of ganetespib treatment (Table I), although loss of the truncated
receptor appeared less pronounced than that of full-length AR
following treatment (data not shown). To directly examine the
effects of Hsp90 inhibition on alternate receptor proteins, we
transiently transfected plasmids encoding full-length AR as well as
the truncated isoform corresponding to the known V7 variant
(33) into HeLa cells (Fig. 1C). Androgen treatment increased
full-length AR expression at 24 h and this response was completely
abrogated in the presence of either 1 μM GA or 250 nM
ganetespib. Both Hsp90 inhibitors were also effective at targeted
degradation of AR in the absence of androgen stimulation; however
neither inhibitor significantly altered expression of the variant
receptor (Fig. 1C). Similarly, GA
and ganetespib strongly inhibited full-length AR activity but were
less effective against constitutive V7 activity measured using an
AR responsive luciferase reporter assay (Fig. 1D). Although the truncated V7
isoform appears less sensitive to Hsp90 inhibition, the potent
activity of ganetespib in 22Rv1 cells suggests that its concomitant
impacts on multiple signaling pathways can overcome any potential
selective advantages provided by constitutively active variant
expression.
Ganetespib inhibits multiple oncogenic
Hsp90 client proteins in AR-negative prostate cancer cells to
induce cell death
The DU145 prostate cancer cell line lacks AR
receptor expression. However, the growth and survival of these
cells has been reported to be regulated through autocrine
activation of EGFR by its ligands (35), in turn leading to oncogenic STAT
activation. Further, this line also expresses an autocrine IL-6
cytokine signaling loop that results in persistent activation of
the JAK/STAT signaling pathway (36). Ganetespib effectively targeted EGFR
and completely abrogated STAT3 signaling in these cells in a
dose-dependent manner (Fig. 2A).
In addition, IGF-IR and downstream signaling pathways mediated
through p-AKT, RAF1, and p-ERK1/2 were also destabilized following
ganetespib exposure, similar to that observed in LNCaP cells
(Fig. 1A). The correlative
increase in cleaved PARP expression indicated that simultaneous
blockade of these signaling pathways triggered apoptosis and this
was further supported by Annexin V staining (Fig. 2B). Cells were treated with
escalating doses of ganetespib or 17-AAG for 24 h and then analyzed
by flow cytometry. Ganetespib treatment resulted in a
dose-dependent increase in apoptotic cells. A comparable proportion
of apoptotic cells was seen following high doses of 17-AAG, a
response that was saturated by the 500 nM exposure level.
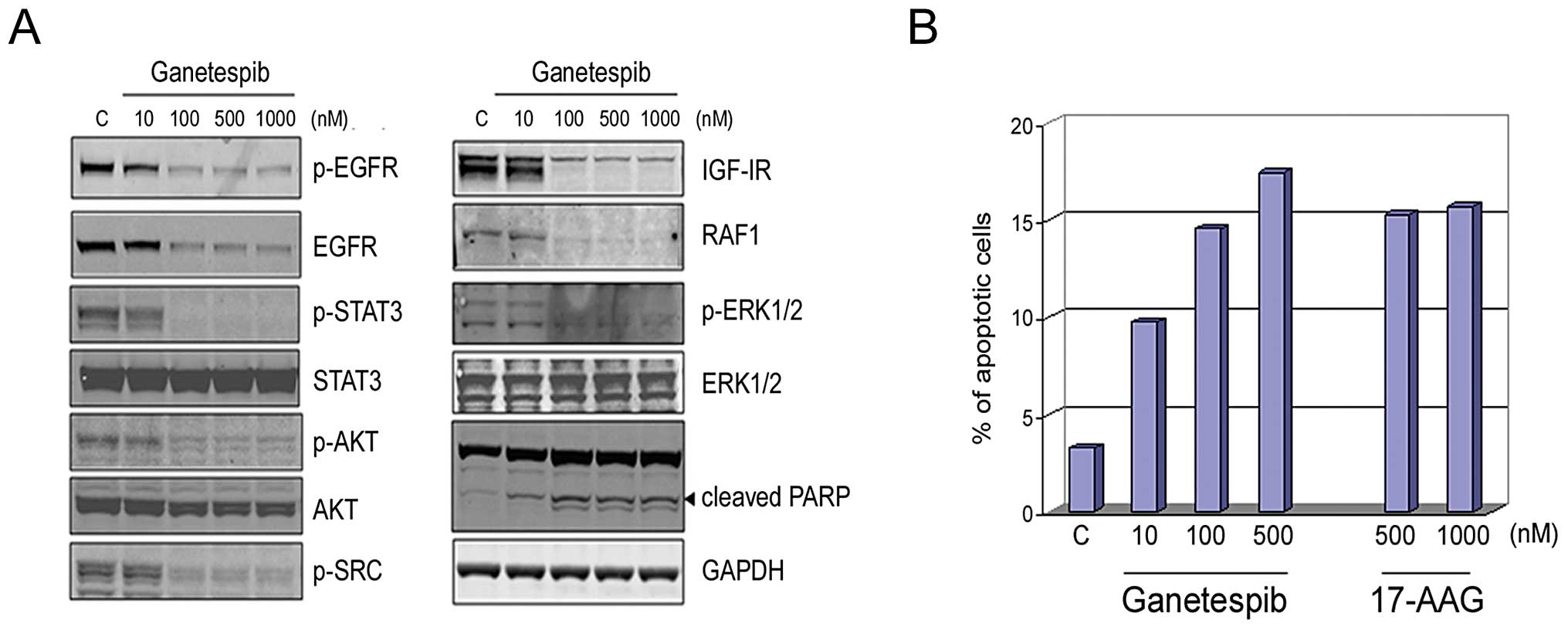 | Figure 2.Ganetespib inhibits multiple
Hsp90-dependent signaling pathways in AR-negative DU145 prostate
cells to induce apoptosis. (A), DU145 cells were exposed to graded
concentrations of ganetespib as indicated for 24 h. Cell lysates
were immunoblotted using antibodies against phosphorylated EGFR
(p-EGFR), total EGFR, phosphorylated STAT3 (p-STAT3), total STAT3,
phosphorylated AKT (p-AKT), total AKT, phosphorylated SRC (p-SRC),
IGF-IR, RAF1, phosphorylated ERK1/2 (p-ERK1/2) and total ERK1/2 as
shown. Cleaved PARP expression is included as a marker of
apoptosis. Total protein levels were determined using GAPDH. (B),
DU145 cells were treated with ganetespib (10, 100 or 500 nM),
17-AAG (500 or 1000 nM) or control (DMSO) for 24 h. Cells were
harvested, stained with a fluorescent conjugated anti-Annexin V
antibody and apoptosis measured by flow cytometry. |
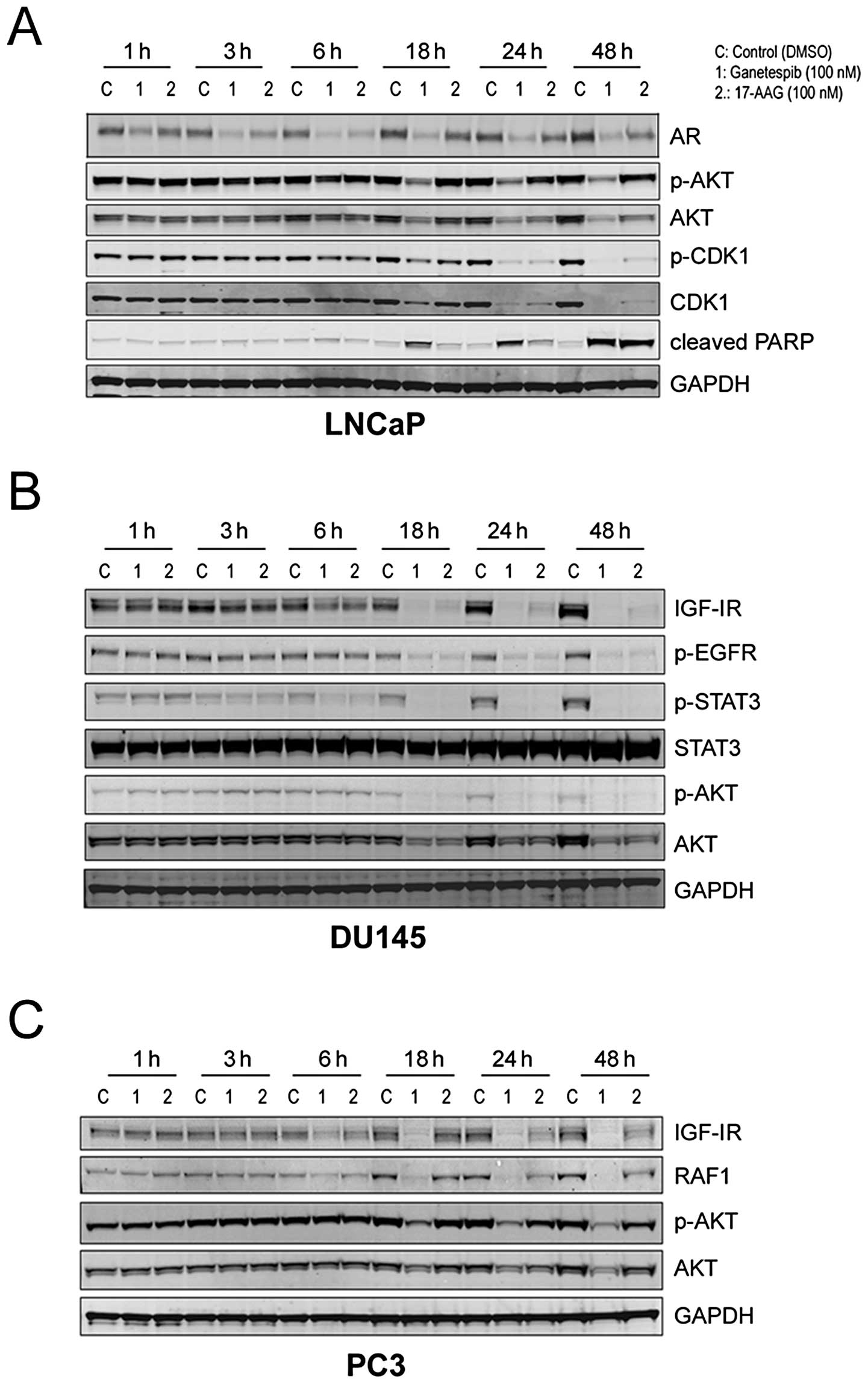
Kinetics of Hsp90 client protein
degradation by ganetespib
We next examined the kinetics of client protein loss
in response to Hsp90 inhibition. In LNCaP cells, 100 nM ganetespib
treatment rapidly (within 3 h) resulted in a measurable reduction
in AR expression and this effect was sustained over a 48-h time
course (Fig. 3A). Destabilization
of p-AKT/AKT was a relatively later event occurring at 18 h; these
kinetics matched those observed for the elevation of cleaved PARP.
Interestingly, ganetespib also induced a loss of both the total and
phosphorylated forms of cyclin dependent kinase 1 (CDK1), a key
regulator of the G2/M checkpoint, by 24 h and this
effect persisted until at least 48 h (Fig. 3A).
The kinetics of targeted AKT degradation were
similar in the AR-negative prostate cell lines DU145 and PC3
(Fig. 3B and C, respectively). In
DU145 cells, significant reductions in p-EGFR expression also
required an 18-h exposure to either ganetespib or 17-AAG, whereas
destabilization of IGF-IR and p-STAT3 was evident by 6 h (Fig. 3B). Like LNCaP cells, PC3 prostate
cells were significantly more sensitive to the effects of
ganetespib treatment compared to an equivalent dose of 17-AAG
(Fig. 3C). Consistent with the
DU145 results, ganetespib reduced IGF-1R levels in this line by 6 h
and sustained loss of the receptor was observed over the 48-h time
course. In addition, a potent and time-dependent reduction in RAF1
protein expression which also preceded AKT modulation was observed
(Fig. 3C).
Modulation of cell cycle protein
expression by ganetespib induces growth arrest and apoptosis
We have previously reported that ganetespib
treatment can exert profound effects on cell cycle regulatory
proteins, in addition to oncogenic signaling pathways, that
contribute to its antitumor activity (31). Cell cycle analysis revealed that
ganetespib exposure led to a dose-dependent accumulation of cells
in the G2/M phase in both DU145 and PC3 cells, with a
concomitant loss of S phase (Fig.
4A). In both cell lines, we observed a corresponding reduction
in protein expression of CDK1 as well as CHK1, another kinase that
plays an essential role in the integrity of the G2/M
checkpoint (Fig. 4B). Next, more
extensive characterization of the concomitant impact of ganetespib
on both oncogenic and cell cycle signaling was performed in
androgen-dependent VCaP prostate cells. As seen in the LNCaP line
(Fig. 1A), ganetespib treatment of
these cells induced AR and IGF-IR degradation and reduced p-AKT/AKT
levels in a dose-dependent manner (Fig. 4C). In agreement with recent
findings (37), ablation of AR/AKT
signaling resulted in accumulation of the cyclin-dependent kinase
inhibitor p27Kip1. In addition, loss of both the total
and phosphorylated forms of CDK1 was observed as a function of
dose. Taken together, these data suggest that loss of checkpoint
control and G2/M arrest accompanies blockade of
oncogenic signaling in prostate cancer cells as a result of Hsp90
inhibition by ganetespib. Moreover, we observed concomitant
elevations in phosphorylated histone H2AX and PARP cleavage
(Fig. 4C). Since the
phosphorylated form of H2AX is a sensitive indicator of DNA double
strand break formation, these data suggest that G2/M
arrest leads to subsequent apoptosis.
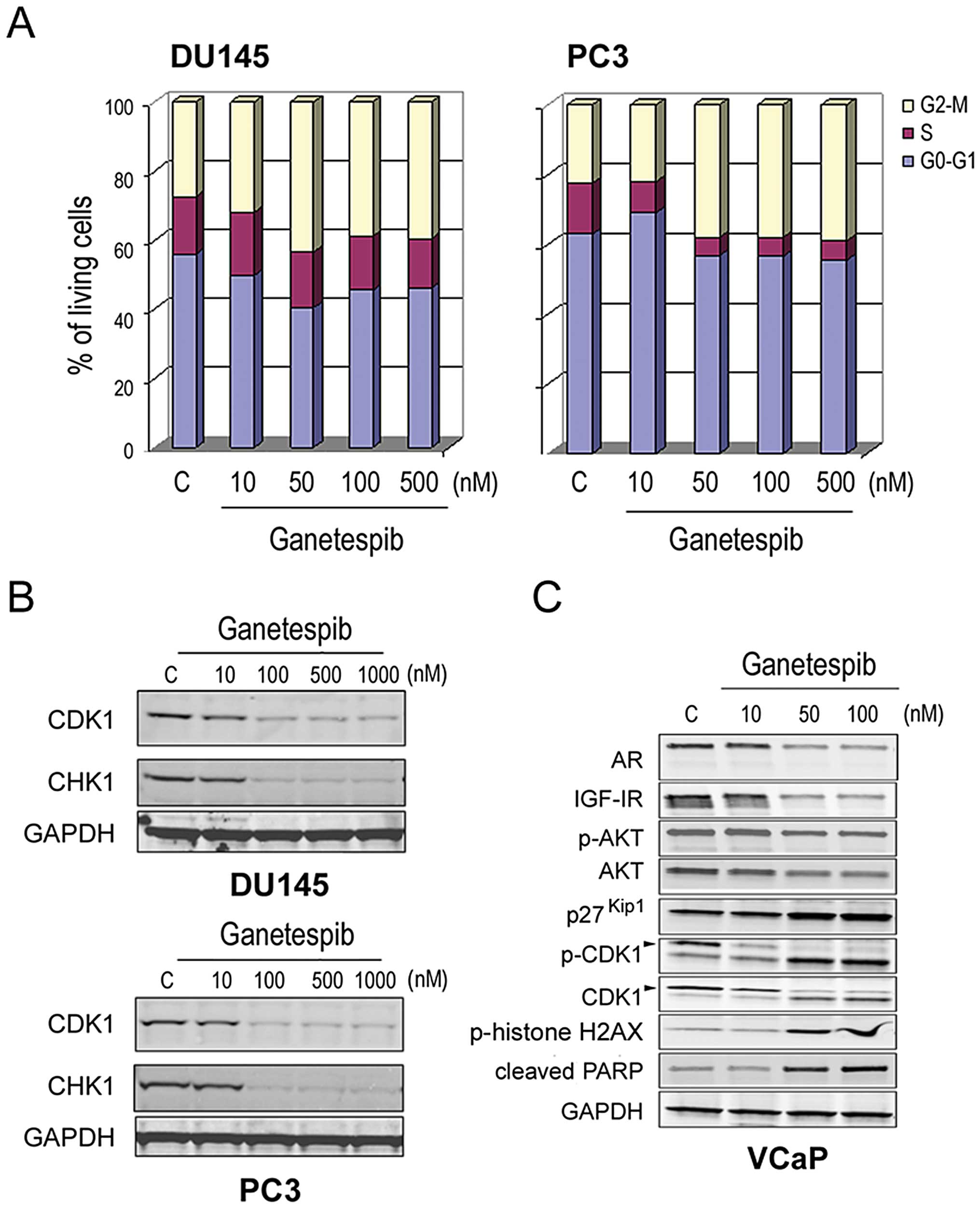 | Figure 4.Ganetespib modulates cell cycle
protein expression and induces growth arrest in prostate cancer
cells. (A), DU145 and PC3 cells were treated with increasing
concentrations of ganetespib as indicated. Cell cycle distribution
was determined in each line by flow cytometry 24-h post-treatment.
(B), DU145 and PC3 cells were treated with increasing
concentrations of ganetespib for 24 h as in (A). Cell lysates were
immunoblotted using antibodies against CDK1, CHK1 and GAPDH. (C),
VCaP cells were treated with ganetespib at 0, 10, 50 and 100 nM for
24 h. Cell lysates were immunoblotted using antibodies against AR,
IGF-IR, phosphorylated AKT (p-AKT), total AKT, p27Kip1,
phosphorylated-CDK1 (p-CDK1), total CDK1, phosphorylated
(p-)histone H2AX, PARP and GAPDH. |
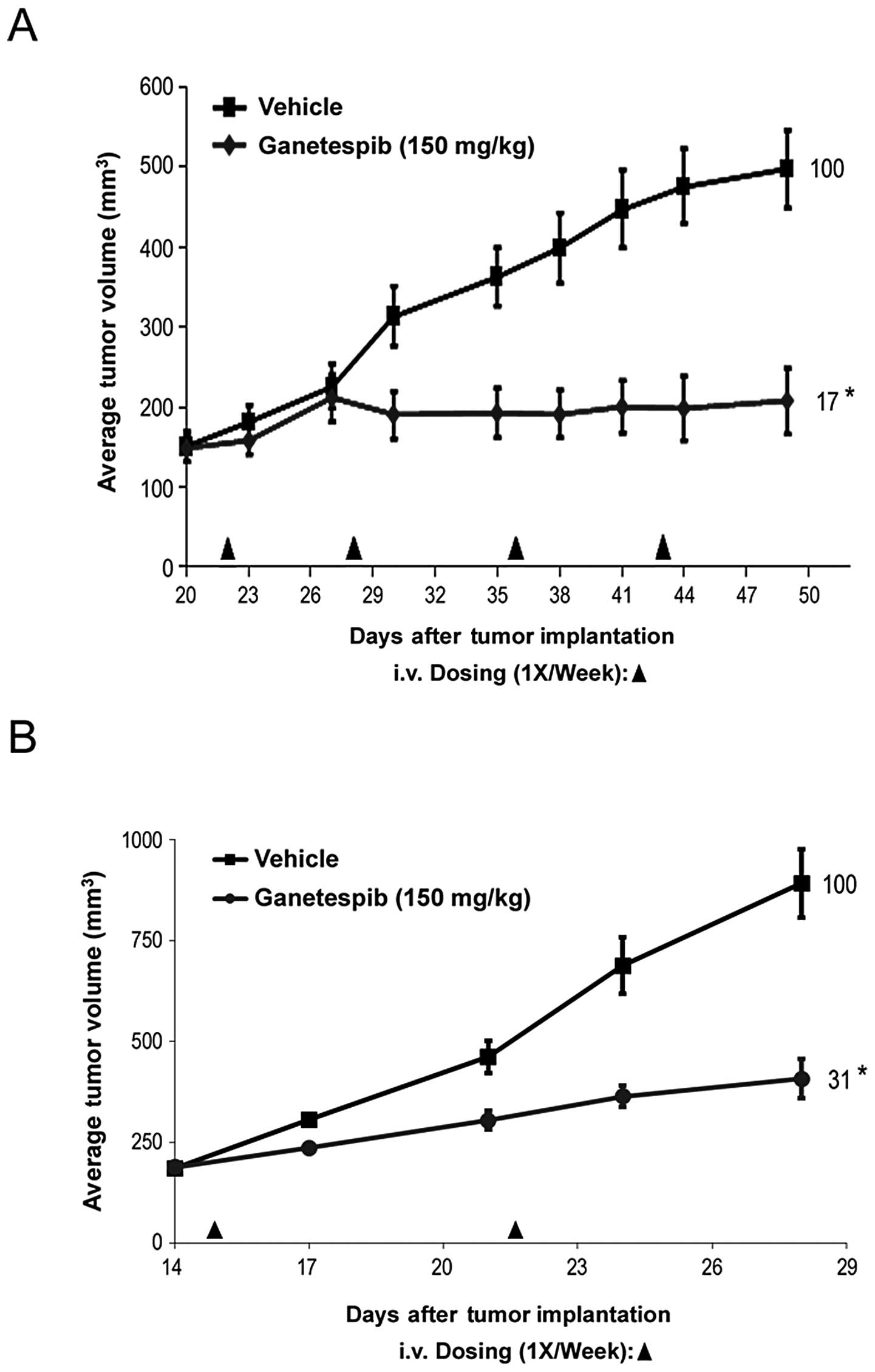
Ganetespib inhibits AR-dependent and
AR-independent tumor growth in vivo
Finally, to determine whether the potent in
vitro effects of ganetespib translated to in vivo
antitumor activity, we studied the efficacy of single-agent
ganetespib treatment on the growth of prostate cancer xenografts.
We have previously determined that the highest non-severely toxic
dose of ganetespib on a weekly dosing regimen is 150 mg/kg
(26). As shown in Fig. 5A, mice bearing AR-independent PC3
xenograft tumors treated on this schedule exhibited a significant
decrease in tumor volume compared to control animals (T/C value
17%). Even at this dose the regimen was well tolerated. Minor body
weight losses occurred post-administration but were rapidly
recovered between dosing points and no net loss of body weight was
observed over the duration of the study (data not shown).
Ganetespib treatment was also highly efficacious in rapidly growing
xenografts derived from the AR-dependent 22Rv1 cell line (Fig. 5B), consistent with the sensitivity
data obtained in vitro. Together, these data show that
ganetespib treatment can significantly inhibit prostate tumor
growth, again irrespective of androgen receptor status.
Discussion
Androgen ablation therapy has been a mainstay of
prostate cancer treatment since the concept was first introduced
over 70 years ago (38). For
advanced disease, however, this approach has not proven curative
since durable tumor suppression is typically not achieved and
patients invariably progress to a castrate-resistant phenotype.
Until the introduction of docetaxel as a standard of care in 2010,
first-line chemotherapeutics such as estramustine and mitoxantrone
failed to provide overall survival benefit to patients with
advanced or recurrent tumors (39). More recently, an improved
understanding of the underlying biology of prostate cancer has led
to major clinical and translational advances, particularly in the
development of novel androgen-ablative and AR antagonist strategies
(6,7). Even with such progress, an urgent
need for more effective and alternative approaches to combat the
disease remains. In this era of molecularly targeted therapies
Hsp90 inhibition has emerged as an exciting potential avenue of
therapeutic intervention in a variety of human malignancies,
including prostate tumors (15,40).
To date, however, the clinical experience with first-generation
ansamycin inhibitors of Hsp90 in prostate cancer has been
disappointing, hampered by poor single-agent activity and adverse
toxicity profiles (24,25).
Ganetespib is a unique resorcinolic triazolone small
molecule inhibitor of Hsp90, structurally unrelated to the
ansamycin class, which exhibits potent activity in a broad range of
preclinical models of human malignancies (26,41).
Moreover, ganetespib displays superior pharmacological and safety
properties compared to other Hsp90 inhibitors and is currently
undergoing clinical evaluation in multiple phase I and II trials.
Here we examined the effects of ganetespib in a panel of prostate
tumor lines and in both AR-dependent and independent xenograft
models. With low nanomolar potency, ganetespib reduced cell
viability in vitro in all the lines examined, irrespective
of their androgen sensitivity and/or AR status. Ganetespib exposure
resulted in a dose-dependent destabilization of multiple Hsp90
client proteins, including AR, EGFR, IGF-IR and AKT. Importantly,
ganetespib demonstrated superior potency and more durable responses
in terms of client protein suppression compared to 17-AAG. Thus,
while ganetespib exerts its pharmacological effects on Hsp90, it
was clear that the downstream consequences involved an array of
client proteins and biochemical pathways. The combinatorial
blockade of multiple key signaling components required for prostate
cancer cell growth and survival, and subsequent induction of
apoptosis, accounted for the potent cytotoxic activity of the
compound.
The AR is an established Hsp90 client, and the
relationship between the chaperoning function of Hsp90 with steroid
receptor stability, conformation and modulation of ligand binding
is well characterized (reviewed in ref. 42). For the AR-expressing cell lines,
abrogation of this critical signaling axis by ganetespib likely
underlies their acute sensitivity. For example, and in agreement
with studies of other Hsp90 inhibitors (21,23),
ganetespib treatment of LNCaP cells promoted the rapid degradation
of AR expression. This was accompanied by inhibition of
AR-transactivation and AR-dependent gene expression. Of note,
destabilization of AR was an early event, and preceded the
downstream loss of AKT signaling and induction of apoptosis by
several hours, highlighting the importance of Hsp90 for stability
and function of the steroid receptor.
In AR-negative cell lines ganetespib exposure
resulted in the simultaneous disruption of signaling networks that
have been implicated in the aberrant growth and survival of
prostate cancer. IGF-IR activation has mitogenic and antiapoptotic
effects in prostate tumor cells and circulating serum levels of
IGF1 have been associated with increased risk of prostate cancer
(43,44). Ganetespib treatment of DU145 and
PC3 cells led to the potent and sustained degradation of IGF-IR and
its downstream effector pathways PI3K/AKT and ERK1/2. In addition
persistent STAT3 activation, through either autocrine cytokine or
EGFR activation, is a feature of DU145 cells (35,36)
and this pathway was also effectively inhibited following
ganetespib exposure. Thus, targeting Hsp90 can overcome the
compensatory signaling pathways present in androgen-insensitive
prostate cancer cells that promote aberrant cell survival. Taken
together, these data suggest that ganetespib may be effective in
controlling castrate-resistant disease. In support of this premise,
we observed encouraging antitumor efficacy of ganetespib as a
single agent in an in vivo study using the PC3 xenograft
model. Additional efficacy and pharmacodynamic studies of
ganetespib in preclinical models of androgen-dependent and
castrate-resistant prostate cancer are underway.
It has been proposed that one mechanism that may
contribute to the development of a castration resistant phenotype
is the expression of truncated, constitutively active AR isoforms,
including the well described V7 variant (32–34).
This variant, expressed by the 22Rv1 cell line, has been shown to
be enriched in xenograft models of androgen-refractory prostate
cancer, to promote the growth of androgen-dependent xenografts in
castrate mice, and to be upregulated in malignant human prostate
tissues compared to their benign counterparts (32,45).
Interestingly, we found that while full-length AR was potently
destabilized following Hsp90 blockade, ganetespib had minimal
effect on V7, either in terms of its targeted degradation or
inhibition of its constitutive transactivation activity. It has
been well established that AR associates with Hsp90, through its
ligand binding domain, in order to adopt a confirmation that is
competent to bind ligand. In the case of the constitutively active
V7, which lacks the ligand binding domain, this interaction is
likely no longer required; a property distinct to most other client
proteins that are generally more reliant on Hsp90 for their
stability and function (10,16).
Despite this apparent lack of direct Hsp90 modulation, the acute
sensitivity of the 22Rv1 line in vitro as well as the
antitumor efficacy observed in vivo indicates that
ganetespibs’ multifaceted mode of action can bypass the selective
advantages provided by truncated AR isoform expression.
We have previously shown that the cellular impact of
Hsp90 inhibition by ganetespib is not restricted to oncogenic
survival signaling but also includes profound effects on the cell
cycle regulatory machinery (31).
In the data presented here, ganetespib exposure resulted in
G2/M accumulation and loss of S phase in prostate cancer
cells, mediated at least in part through loss of the checkpoint
regulatory proteins CDK1 and CHK1. In this regard, it is known that
Hsp90 inhibition can sensitize cancer cells to the effects of
chemotherapy and radiotherapy (46,47),
and modulation of the cell division machinery represents an
important component of this cytotoxic sensitizing property. For
example, depletion of CHK1 and loss of checkpoint control as a
result of Hsp90 inhibition has been reported to enhance the
cytotoxic activity of the chemotherapeutic agents gemcitabine and
irinotecan (48,49). Moreover, we recently showed that
ganetespib synergistically potentiated the cytotoxic effects of
taxanes, a group of microtubule-targeted agents that cause mitotic
catastrophe, in preclinical models of non-small cell lung cancer
(41). Interestingly, mitotic
disruption can also be exacerbated by Hsp90 inhibition in cell
lines with defects in the function of the retinoblastoma (RB) tumor
suppressor protein (50),
presumably linked to interference with Hsp90’s role in centrosome
organization (51,52). RB is a master cell cycle regulator
and key component of the proliferative response to AR which is lost
or inactivated with high frequency (30–60%) in prostatic neoplasms
(2). Taken together, these
findings provide additional evidence for the potential advantages
of Hsp90 inhibitors such as ganetespib, based on their multifaceted
mode of action, to overcome deficiencies of AR-directed
therapeutics.
In conclusion, we have shown that the unique small
molecule Hsp90 inhibitor ganetespib exhibits robust cytotoxic
activity and antitumor efficacy in preclinical models of prostate
cancer. Importantly, due to concomitant effects on oncogenic
survival pathways and cell cycle progression, ganetespib treatment
potently induced cancer cell death irrespective of androgen
sensitivity. Together, the data suggest that ganetespib may serve
as an effective treatment strategy for prostate cancers driven by
AR, truncated forms of the receptor that confer androgen
independence, as well as castrate-resistant tumors no longer
reliant on the receptor itself. In light of these findings, further
evaluation of the therapeutic utility of this agent is
warranted.
Acknowledgements
The authors wish to thank Dr Richard
Bates who provided drafts and editorial assistance during
preparation of this study.
References
|
1.
|
Jemal A, Siegel R, Xu J, et al: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
2.
|
Balk SP and Knudsen KE: AR, the cell
cycle, and prostate cancer. Nucl Recept Signal.
6:e0012008.PubMed/NCBI
|
|
3.
|
Chen Y, Sawyers CL and Scher HI: Targeting
the androgen receptor pathway in prostate cancer. Curr Opin
Pharmacol. 8:440–448. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Scher HI, Buchanan G, Gerald W, et al:
Targeting the androgen receptor: improving outcomes for
castration-resistant prostate cancer. Endocr Relat Cancer.
11:459–476. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lonergan PE and Tindall DJ: Androgen
receptor signaling in prostate cancer development and progression.
J Carcinog. 10:202011. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Yap TA, Zivi A, Omlin A and De Bono JS:
The changing therapeutic landscape of castration-resistant prostate
cancer. Nat Rev Clin Oncol. 8:597–610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Osanto S and van Poppel H: Emerging novel
therapies for advanced prostate cancer. Ther Adv Urol. 4:3–12.
2012. View Article : Google Scholar
|
|
8.
|
Attard G, Cooper CS and De Bono JS:
Steroid hormone receptors in prostate cancer: a hard habit to
break? Cancer Cell. 16:458–462. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Hahn JS: The Hsp90 chaperone machinery:
from structure to drug development. BMB Rep. 42:623–630. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Whitesell L and Lindquist SL: HSP90 and
the chaperoning of cancer. Nat Rev Cancer. 5:761–772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Saporita AJ, Ai J and Wang Z: The Hsp90
inhibitor, 17-AAG, prevents the ligand-independent nuclear
localization of androgen receptor in refractory prostate cancer
cells. Prostate. 67:509–520. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Trepel J, Mollapour M, Giaccone G, et al:
Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer.
10:537–549. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Vanaja DK, Mitchell SH, Toft DO, et al:
Effect of geldanamycin on androgen receptor function and stability.
Cell Stress Chaperones. 7:55–64. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Pratt WB and Toft DO: Steroid receptor
interactions with heat shock protein and immunophilin chaperones.
Endocr Rev. 18:306–360. 1997.PubMed/NCBI
|
|
15.
|
Banerji U: Heat shock protein 90 as a drug
target: some like it hot. Clin Cancer Res. 15:9–14. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Neckers L: Heat shock protein 90: the
cancer chaperone. J Biosci. 32:517–530. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Xu W and Neckers L: Targeting the
molecular chaperone heat shock protein 90 provides a multifaceted
effect on diverse cell signaling pathways of cancer cells. Clin
Cancer Res. 13:1625–1629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Li Y, Zhang T, Schwartz SJ and Sun D: New
developments in Hsp90 inhibitors as anti-cancer therapeutics:
mechanisms, clinical perspective and more potential. Drug Resist
Updat. 12:17–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Taldone T, Gozman A, Maharaj R, et al:
Targeting Hsp90: small-molecule inhibitors and their clinical
development. Curr Opin Pharmacol. 8:370–374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Eskew JD, Sadikot T, Morales P, et al:
Development and characterization of a novel C-terminal inhibitor of
Hsp90 in androgen dependent and independent prostate cancer cells.
BMC Cancer. 11:4682011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Lamoureux F, Thomas C, Yin MJ, et al: A
novel HSP90 inhibitor delays castrate-resistant prostate cancer
without altering serum PSA levels and inhibits osteoclastogenesis.
Clin Cancer Res. 17:2301–2313. 2011. View Article : Google Scholar
|
|
22.
|
O’Malley KJ, Langmann G, Ai J, et al:
Hsp90 inhibitor 17-AAG inhibits progression of LuCaP35 xenograft
prostate tumors to castration resistance. Prostate. 72:1117–1123.
2011.PubMed/NCBI
|
|
23.
|
Solit DB, Zheng FF, Drobnjak M, et al:
17-Allylamino-17-demethoxygeldanamycin induces the degradation of
androgen receptor and HER-2/neu and inhibits the growth of prostate
cancer xenografts. Clin Cancer Res. 8:986–993. 2002.PubMed/NCBI
|
|
24.
|
Heath EI, Hillman DW, Vaishampayan U, et
al: A phase II trial of 17-allylamino-17-demethoxygeldanamycin in
patients with hormone-refractory metastatic prostate cancer. Clin
Cancer Res. 14:7940–7946. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Oh WK, Galsky MD, Stadler WM, et al:
Multicenter phase II trial of the heat shock protein 90 inhibitor,
retaspimycin hydro-chloride (IPI-504), in patients with
castration-resistant prostate cancer. Urology. 78:626–630. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Ying W, Du Z, Sun L, et al: Ganetespib, a
unique triazolone-containing hsp90 inhibitor, exhibits potent
antitumor activity and a superior safety profile for cancer
therapy. Mol Cancer Ther. 11:475–484. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Nazareth LV, Stenoien DL, Bingman WE, et
al: A C619Y mutation in the human androgen receptor causes
inactivation and mislocalization of the receptor with concomitant
sequestration of SRC-1 (steroid receptor coactivator 1). Mol
Endocrinol. 13:2065–2075. 1999. View Article : Google Scholar
|
|
28.
|
Agoulnik IU, Vaid A, Nakka M, et al:
Androgens modulate expression of transcription intermediary factor
2, an androgen receptor coactivator whose expression level
correlates with early biochemical recurrence in prostate cancer.
Cancer Res. 66:10594–10602. 2006. View Article : Google Scholar
|
|
29.
|
Nazareth LV and Weigel NL: Activation of
the human androgen receptor through a protein kinase A signaling
pathway. J Biol Chem. 271:19900–19907. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Agoulnik IU, Krause WC, Bingman WE, et al:
Repressors of androgen and progesterone receptor action. J Biol
Chem. 278:31136–31148. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Proia DA, Foley KP, Korbut T, et al:
Multifaceted intervention by the Hsp90 inhibitor ganetespib
(STA-9090) in cancer cells with activated JAK/STAT signaling. PLoS
One. 6:e185522011. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Dehm SM, Schmidt LJ, Heemers HV, et al:
Splicing of a novel androgen receptor exon generates a
constitutively active androgen receptor that mediates prostate
cancer therapy resistance. Cancer Res. 68:5469–5477. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Hu R, Dunn TA, Wei S, et al:
Ligand-independent androgen receptor variants derived from splicing
of cryptic exons signify hormone-refractory prostate cancer. Cancer
Res. 69:16–22. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Li Y, Alsagabi M, Fan D, et al: Intragenic
rearrangement and altered RNA splicing of the androgen receptor in
a cell-based model of prostate cancer progression. Cancer Res.
71:2108–2117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Connolly JM and Rose DP: Autocrine
regulation of DU145 human prostate cancer cell growth by epidermal
growth factor-related polypeptides. Prostate. 19:173–180. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Okamoto M, Lee C and Oyasu R:
Interleukin-6 as a paracrine and autocrine growth factor in human
prostatic carcinoma cells in vitro. Cancer Res. 57:141–146.
1997.PubMed/NCBI
|
|
37.
|
Fang Z, Zhang T, Dizeyi N, et al: Androgen
receptor enhances p27 degradation in prostate cancer cells through
rapid and selective TRC2 activation. J Biol Chem. 287:2090–2098.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Huggins C, Stevens RE and Hodges CV:
Studies on prostatic cancer. II. The effect of castration on
advanced carcinoma of the prostate gland. Arch Surg. 43:209–228.
1941. View Article : Google Scholar
|
|
39.
|
Sartor O, Michels RM, Massard C, et al:
Novel therapeutic strategies for metastatic prostate cancer in the
post-docetaxel setting. Oncologist. 16:1487–1497. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Kim YS, Alarcon SV, Lee S, et al: Update
on Hsp90 inhibitors in clinical trial. Curr Top Med Chem.
9:1479–1492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Proia DA, Sang J, He S, et al: Synergistic
activity of the Hsp90 inhibitor ganetespib with taxanes in
non-small cell lung cancer models. Invest New Drugs. View Article : Google Scholar : [Epub ahead of
print]. 2012.PubMed/NCBI
|
|
42.
|
Smith DF and Toft DO: Minireview: the
intersection of steroid receptors with molecular chaperones:
observations and questions. Mol Endocrinol. 22:2229–2240. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Monti S, Proietti-Pannunzi L, Sciarra A,
et al: The IGF axis in prostate cancer. Curr Pharm Des. 13:719–727.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Ozkan EE: Plasma and tissue insulin-like
growth factor-I receptor (IGF-IR) as a prognostic marker for
prostate cancer and anti-IGF-IR agents as novel therapeutic
strategy for refractory cases: a review. Mol Cell Endocrinol.
344:1–24. 2011. View Article : Google Scholar
|
|
45.
|
Guo Z, Yang X, Sun F, et al: A novel
androgen receptor splice variant is up-regulated during prostate
cancer progression and promotes androgen depletion-resistant
growth. Cancer Res. 69:2305–2313. 2009. View Article : Google Scholar
|
|
46.
|
Hwang M, Moretti L and Lu B: HSP90
inhibitors: multi-targeted antitumor effects and novel
combinatorial therapeutic approaches in cancer therapy. Curr Med
Chem. 16:3081–3092. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Solit DB and Chiosis G: Development and
application of Hsp90 inhibitors. Drug Discov Today. 13:38–43. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Arlander SJ, Eapen AK, Vroman BT, et al:
Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to
replication stress. J Biol Chem. 278:52572–52577. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Flatten K, Dai NT, Vroman BT, et al: The
role of checkpoint kinase 1 in sensitivity to topoisomerase I
poisons. J Biol Chem. 280:14349–14355. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Munster PN, Basso A, Solit D, et al:
Modulation of Hsp90 function by ansamycins sensitizes breast cancer
cells to chemotherapy-induced apoptosis in an RB- and
schedule-dependent manner. Clin Cancer Res. 7:2228–2236. 2001.
|
|
51.
|
Lange BM, Bachi A, Wilm M, et al: Hsp90 is
a core centrosomal component and is required at different stages of
the centrosome cycle in Drosophila and vertebrates. EMBO J.
19:1252–1262. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
De Carcer G: Heat shock protein 90
regulates the metaphaseanaphase transition in a polo-like
kinase-dependent manner. Cancer Res. 64:5106–5112. 2004.PubMed/NCBI
|