Introduction
Histone acetylation is an important step in the
initiation of transcription (1).
The acetylation of lysine residues in histones weakens their
binding to DNA and leads to euchromatin structure, which induces
transcriptional factors to bind to promoter regions of genes
(2). Two opposing enzyme
activities of histone acetyltransferases (HATs) and histone
deacetylases (HDACs) determine the acetylation status of histones,
respectively, acetylating or deacetylating the epsilon-amino groups
of lysine residues located in the amino-terminal tails of the
histones (2). It was reported that
dysregulation of HDAC activity can cause the silence of tumor
suppressor genes such as p53 and contribute to cancer initiation
(3,4). Previous studies have demonstrated
that HDAC activity and expression are increased in many human
cancers including prostate and pancreatic cancer (5,6).
Therefore, HDAC inhibitors have been considered as novel anticancer
drugs. In particular, the reversible HDAC inhibitor, trichostatin A
(TSA) and its hydroxamate analogues can effectively and selectively
induce tumor growth arrest at very low concentrations (nano- to
micromolar range) (7). In fact,
vorinostat (suberoylanilide hydroxamic acid) has been used for the
treatment of cutaneous T-cell lymphoma (8). Other types of HDAC inhibitors such as
romidepsin, panobinostat and valproic acid are clinically evaluated
in cancer therapy (9,10). In general, HDAC inhibitors can
induce cell cycle arrest, cell death and cell differentiation in
various cancer cells (11–13). They have also been shown to
generate reactive oxygen species (ROS) in solid tumor and leukemia
cells (14). Excessive production
of ROS, which is called oxidative stress, has been recognized to
induce cell death.
Cervical cancer is a major cause of cancer-related
death in women worldwide and the occurrence of this cancer is
ascribed to the changes of genetic and epigenetic events.
Epigenetic alterations such as global DNA hypomethylation,
hypermethylation of tumor suppressor genes and histone
modifications take place during cervical carcinogenesis (15). It was reported that phosphorylated
and acetylated forms of histone H3 in cytologic smears are related
to the progression of cervical cancer (16). Furthermore, the overexpression of
HDAC2 is observed in cervical cancer (17). It has been reported that TSA has
anticancer effect in liver, colorectal and breast cancer cells
in vitro and in vivo(18–20).
However, little is known about the anticancer effect of TSA in
cervical cancer cells in view of ROS and GSH levels. Therefore, in
the present study, we investigated the effects of TSA on cell
growth and death in human cervical HeLa cells in relation to ROS
and GSH levels.
Materials and methods
Cell culture
Human cervical adenocarcinoma HeLa cells were
obtained from the American Type Culture Collection (ATCC, Manassas,
VA) and maintained in a humidified incubator containing 5%
CO2 at 37°C. HeLa cells were cultured in RPMI-1640
(Sigma-Aldrich Chemical Co., St. Louis, MO) supplemented with 10%
fetal bovine serum (FBS; Sigma-Aldrich) and 1%
penicillin-streptomycin (Gibco BRL, Grand Island, NY). Cells were
routinely grown in 100-mm plastic tissue culture dishes (Nunc,
Roskilde, Denmark) and harvested with a solution of trypsin-EDTA
while in a logarithmic phase of growth.
Reagents
TSA was purchased from Cayman Chemical Co. (Ann
Arbor, MI) and was dissolved in dimethl sulfoxide (DMSO;
Sigma-Aldrich) at 100 mM as a stock solution. The pan-caspase
inhibitor (Z-VAD-FMK;
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone), caspase-3
inhibitor (Z-DEVD-FMK;
benzyloxycarbonyl-Asp-Glu-Val-Aspfluoromethylketone), caspase-8
inhibitor (Z-IETD-FMK;
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone) and caspase-9
inhibitor (Z-LEHD-FMK;
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone) were obtained
from R&D Systems, Inc. (Minneapolis, MN) and were dissolved in
DMSO at 10 mM to act as stock solutions. NAC was also obtained from
Sigma-Aldrich. NAC was dissolved in the buffer [20 mM HEPES (pH
7.0)] at 100 mM as a stock solution. Cells were pretreated with
each caspase inhibitor or NAC for 1 h prior to TSA treatment. DMSO
(0.05%) was used as a control vehicle.
Growth inhibition assay
Cell growth changes were determined by measuring
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide dye
(MTT; Sigma-Aldrich) absorbance in living cells. In brief,
5×103 cells were seeded in 96-well microtiter plates
(Nunc) for MTT assays. After exposure to the designated doses of
TSA with or without 15 μM of a given caspase inhibitor or 2
mM NAC for the indicated times, 20 μl of MTT solution [2
mg/ml in phosphate-buffered saline (PBS)] were added to each well
of 96-well plates. The plates were incubated for 4 additional hours
at 37°C. Medium in plates was withdrawn using pipetting and 200
μl DMSO was added to each well to solubilize the formazan
crystals. Optical density was measured at 570 nm using a microplate
reader (Synergy™ 2, BioTek Instruments Inc., Winooski, VT).
Measurement of HDAC activity
HDAC activity was assessed using the HDAC Assay kit
(Millipore, Billerica, MA), according to the manufacturer’s
instructions. In brief, 1×106 cells in 60-mm culture
dish (Nunc) were incubated with the indicated doses of TSA for 72
h. The cells were then washed in PBS and suspended in 5 volumes of
lysis buffer (R&D Systems, Inc.). Protein concentrations were
determined using the Bradford method. Supernatant samples
containing 20 μg of total protein were used for
determination of HDAC activity. These samples were added to each
well in 96-well microtiter plates (Nunc) with HDAC substrate
provided by the assay kit at 37°C for 1 h. The optical density of
each well was measured at 405 nm using a microplate reader (Synergy
2, BioTek Instruments).
Western blot analysis
The expression levels of proteins were evaluated
using western blot analysis. In brief, 1×106 cells in
60-mm culture dish (Nunc) were incubated with the designated doses
of TSA for 72 h. The cells were then washed in PBS and suspended in
5 vol of lysis buffer (20 mM HEPES. pH 7.9, 20% glycerol, 200 mM
KCl, 0.5 mM EDTA, 0.5% NP40, 0.5 mM DTT, 1% protease inhibitor
cocktail). Supernatant protein concentrations were determined using
the Bradford method. Supernatant samples containing 30 μg
total protein were resolved by 10 or 15% SDS-PAGE gels depending on
the sizes of target proteins, transferred to Immobilon-P PVDF
membranes (Millipore) by electroblotting and then probed with
anti-acetylated H3, anti-acetylated H4 (Millipore), anti-Bax (Cell
Signaling, Beverly, MA), anti-Bcl-2, anti-PARP, anti-Trx1,
anti-Trx2, anti-TrxR1, anti-Cu/Zn SOD, anti-Mn SOD and anti-β-actin
antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). Membranes
were incubated with horseradish peroxidaseconjugated secondary
antibodies. Blots were developed using an ECL kit (Amersham,
Arlington Heights, IL). Quantitative data were obtained using an
imaging densitometer (ImageJ version 1.33 software, NIH).
Sub-G1 cell analysis
Sub-G1 cells were determined by prop-idium iodide
(PI, Sigma-Aldrich; Ex/Em=488/617 nm) staining. In brief,
1×106 cells in 60-mm culture dish (Nunc) were incubated
with the designated doses of TSA for 72 h. Cells were then washed
with PBS and fixed in 70% ethanol. Cells were washed again with PBS
and then incubated with PI (10 μg/ml) with RNase at 37°C for
30 min. The sub-G1 DNA content cells were measured with a FACStar
flow cytometer (Becton-Dickinson, San Jose, CA).
Annexin V staining
Apoptotic cell death was determined by staining
cells with annexin V-fluorescein isothiocyanate (FITC, Invitrogen
Molecular Probes, OR; Ex/Em = 488/519 nm). In brief,
1×106 cells in 60-mm culture dish (Nunc) were incubated
with the designated doses of TSA with or without 15 μM of a
given caspase inhibitor or 2 mM NAC for 72 h. Cells were washed
twice with cold PBS and then resuspended in 500 μl of
binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM
CaCl2) at a concentration of 1×106 cells/ml.
Annexin V-FITC (5 μl) and PI (1 μg/ml) were then
added to these cells. Stained cells with annexin V-FITC were
analyzed with a FACStar flow cytometer (Becton-Dickinson).
Quantification of caspase-3 activity
The activity of caspase-3 was assessed using the
caspase-3 colorimetric assay kit (R&D Systems). In brief,
1×106 cells in 60 mm culture dish (Nunc) were incubated
with 50 nM TSA for 72 h. The cells were then washed in PBS and
suspended in 5 vol of lysis buffer provided by the kit. Protein
concentrations were determined using the Bradford method.
Supernatant samples containing 50 μg of total protein were
used for determination of caspase-3 activity. These were added to
each well in 96-well microtiter plates (Nunc) with DEVD-pNA as a
caspase-3 substrate at 37°C for 1 h. The optical density of each
well was measured at 405 nm using a microplate reader (Synergy 2,
BioTek Instruments). Caspase-3 activity was expressed in arbitrary
absorbance units.
Measurement of MMP (ΔΨm)
MMP (ΔΨm) levels were measured using a
rhodamine 123 fluorescent dye (Sigma-Aldrich; Ex/Em=485/535 nm). In
brief, 1×106 cells in 60-mm culture dish (Nunc) were
incubated with the designated doses of TSA with or without 15
μM of a given caspase inhibitor or 2 mM NAC for 72 h. Cells
were washed twice with PBS and incubated with a rhodamine 123 (0.1
μg/ml) at 37°C for 30 min. The cells were washed again twice
with PBS and then resuspended in 500 μl of PBS buffer.
Rhodamine 123 staining intensity was determined by the flow
cytometry (Becton-Dickinson). An absence (−) of rhodamine 123
fluorescence in cells was expressed as the loss of MMP
(ΔΨm) in the cells.
Transfection of cells with Bax and Bcl-2
siRNAs
Gene silencing of Bax and Bcl-2 was performed using
a siRNA knockdown system. A non-specific control siRNA duplex
[5′-CCUACGCC ACCAAUUUCGU(dTdT)-3′], Trx1 siRNA duplex [5′-GCAU
GCCAACAUUCCAGUU(dTdT)-3′], Bax siRNA duplex
[5′-GCUGGACAUUGGACUUCCU(dTdT)-3′] and Bcl-2 siRNA duplex
[5′-CAGAAGUCUGGGAAUCGAU(dTdT)-3′] were purchased from the Bioneer
Corp. (Daejeon, South Korea). In brief, 2.5×105 cells in
6-well plates (Nunc) were incubated in RPMI-1640 supplemented with
10% FBS. The next day, cells (∼30–40% confluence) in each well were
transfected with the control, Bax or Bcl-2 siRNA [80 pmol in
Opti-MEM (Gibco BRL)] using Lipofectamine 2000, according to the
manufacturer’s instructions (Invitrogen, Brandford, CT). One day
later, cells were treated with or without 100 nM TSA for additional
24 h. The transfected cells were collected and used for western
blot analysis, growth inhibition assay, annexin-FITC staining,
O2•− and GSH level measurements.
Detection of intracellular
O2•− levels
Intracellular O2•− levels were
detected by means of an oxidation-sensitive fluorescent probe dye,
dihydroethidium (DHE, Invitrogen Molecular Probes; Ex/Em = 518/605
nm). In brief, 1×106 cells in 60-mm culture dish (Nunc)
were incubated with the designated doses of TSA with or without 15
μM of a given caspase inhibitor or 2 mM NAC for 72 h. Cells
were then washed in PBS and incubated with 20 μM DHE at 37°C
for 30 min. The intensity of DHE fluorescence was detected using a
FACStar flow cytometer (Becton-Dickinson).
O2•− levels were expressed as mean
fluorescence intensity (MFI), which was calculated by CellQuest
software (Becton-Dickinson).
Detection of intracellular glutathione
(GSH) levels
Cellular GSH levels were analyzed using a
5-chloromethylfluorescein diacetate dye (CMFDA, Invitrogen
Molecular Probes; Ex/Em = 522/595 nm). In brief, 1×106
cells in 60 mm culture dish (Nunc) were incubated with the
designated doses of TSA with or without 15 μM of a given
caspase inhibitor or 2 mM NAC for 72 h. Cells were then washed with
PBS and incubated with 5 μM CMFDA at 37°C for 30 min. CMF
fluorescence intensity was determined using a FACStar flow
cytometer (Becton-Dickinson). Negative CMF staining cells
indicating GSH depletion were expressed as the percents of (−) CMF
cells.
Statistical analysis
The results represent the mean of at least three
independent experiments (mean ± SD). The data were analyzed using
Instat software (GraphPad Prism4, San Diego, CA). The Student’s
t-test or one-way analysis of variance (ANOVA) with post hoc
analysis using the Tukey multiple comparison test was used for
parametric data. Statistical significance was defined as
p<0.05.
Results
Effects of TSA on the growth and HDAC
activity in HeLa cells
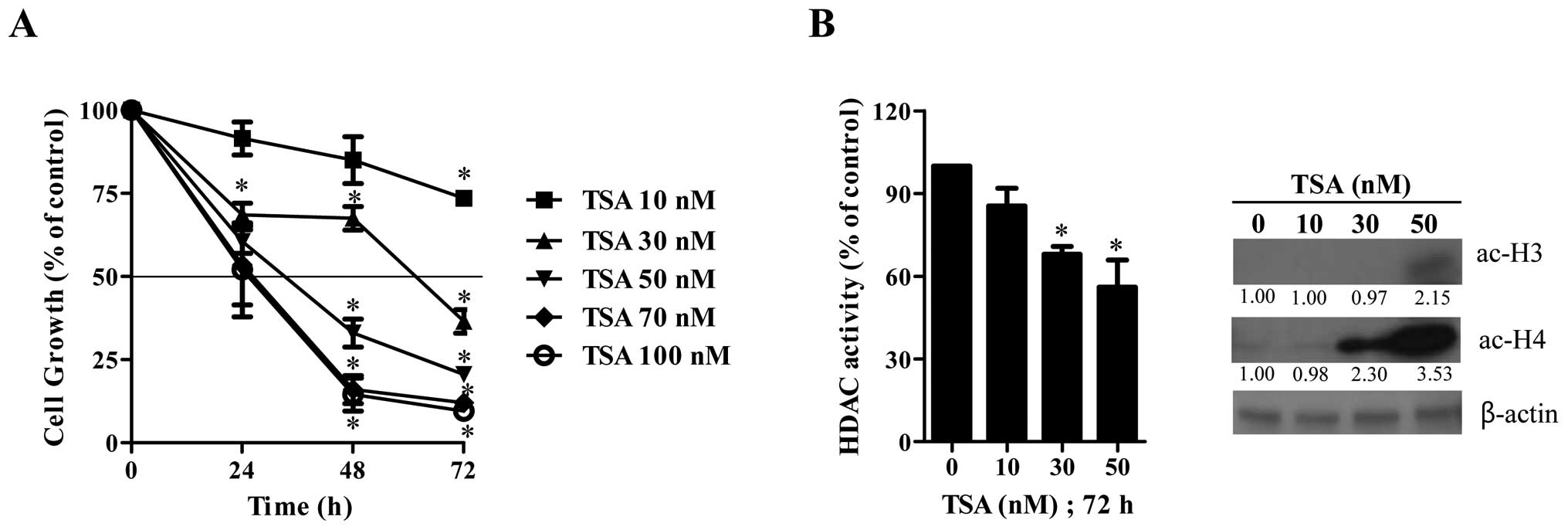
We first examined the effect of TSA on the growth
inhibition of HeLa cells using MTT assays. After exposure to the
various concentrations of TSA, HeLa cell growth was dose- and
time-dependently decreased with an IC50 of ∼100, 40 and
20 nM at 24, 48 and 72 h, respectively (Fig. 1A). When tested whether TSA as an
HDAC inhibitor would inhibit HDAC activity, TSA significantly
attenuated the HDAC activity at 72 h (Fig. 1B). Furthermore, it was observed
that TSA increased the forms of acetylated histone 3 and 4 in HeLa
cells (Fig. 1B).
Effects of TSA on cell death and MMP
(ΔΨm) in HeLa cells
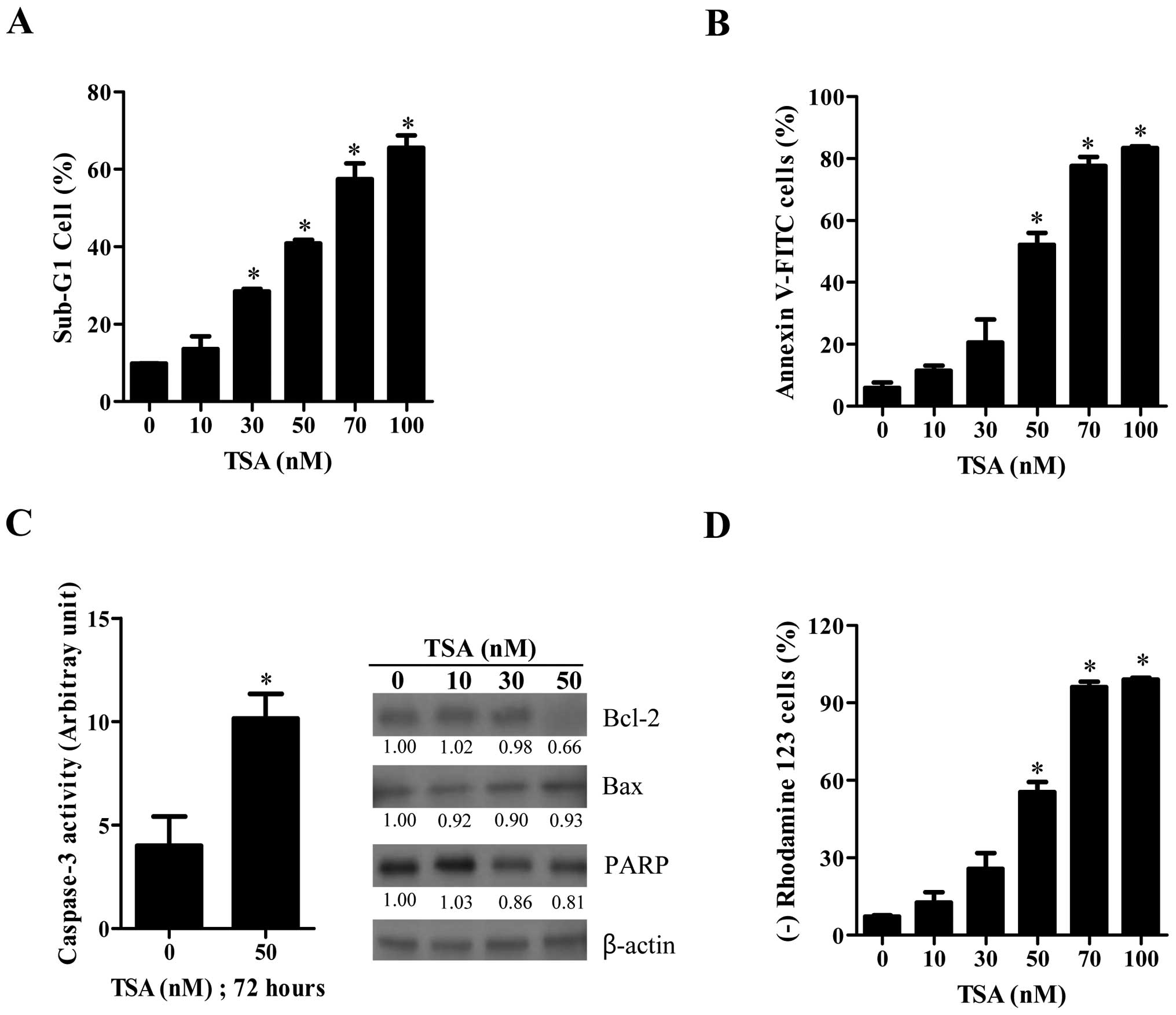
As shown in Fig.
2A, TSA increased the number of sub-G1 cells in HeLa cells in a
dose-dependent manner at 72 h. When HeLa cells were stained with
annexin V-FITC to evaluate the induction of apoptosis, the number
of annexin V-staining cells in TSA-treated cells was
dose-dependently increased (Fig.
2B). In addition, caspase-3 activity was increased in 50 nM
TSA-treated HeLa cells (Fig. 2C).
Examination of apoptosis-related protein changes during TSA-induced
cell death revealed that the levels of Bcl-2 and the intact 116 kDa
form of poly(ADP-ribose) polymerase (PARP) were decreased by TSA
whereas the level of Bax protein was not strongly altered (Fig. 2C). Moreover, TSA increased the
number of MMP (ΔΨm) loss cells in HeLa cells at 72 h in
a dose-dependent manner (Fig.
2D).
Effects of Bax and Bcl-2 siRNAs on cell
growth and death in TSA-treated HeLa cells
To investigate the effects of Bax and Bcl-2 on HeLa
cell growth and death, HeLa cells were transfected with either
non-target control siRNA, Bax or Bcl-2 siRNA. As shown in Fig. 3A, the expressions of Bax and Bcl-2
were clearly decreased by each siRNA as compared with cells
transfected with control siRNA. When we observed the effect of Bax
or Bcl-2 siRNA on cell growth and death in TSA-treated HeLa cells,
Bax siRNA significantly attenuated cell growth inhibition and death
by TSA (Fig. 3B and C). On the
other hand, Bcl-2 siRNA markedly intensified cell growth inhibition
and death in TSA-treated HeLa cells (Fig. 3B and C). In addition, the
administration of Bcl-2 siRNA alone induced cell growth inhibition
and death in the control HeLa cells (Fig. 3B and C).
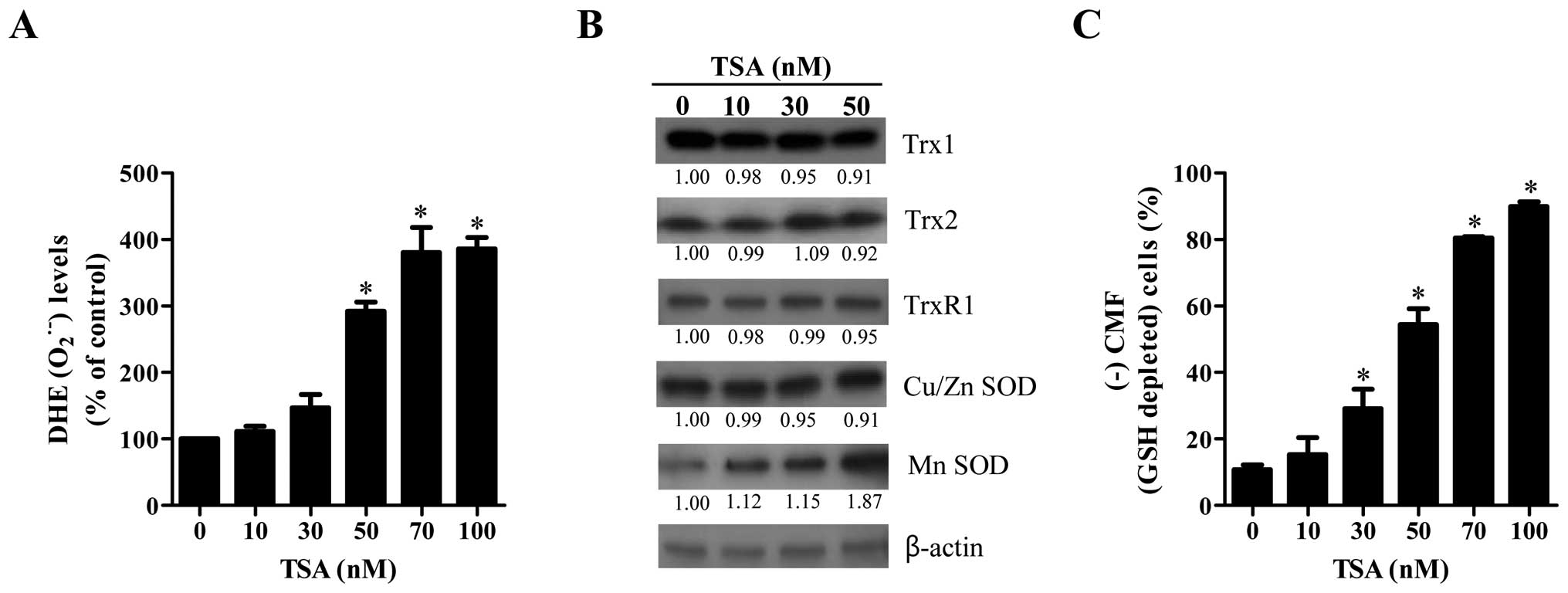
Effects of TSA on intracellular
O2•− and GSH levels in HeLa cells
The intracellular O2•− levels
were measured in TSA-treated HeLa cells using a DHE fluorescence
dye. As shown in Fig. 4A, the
O2•− level was significantly increased in
TSA-treated HeLa cells at 72 h in a dose-dependent manner. In
addition, the level of Mn SOD was upregulated by TSA (Fig. 4B). However, this agent did not
strongly influence the levels of other tested antioxidant proteins;
Trx1, Trx2, TrxR1 and Cu/Zn SOD (Fig.
4B). In relation to GSH level in TSA-treated HeLa cells, TSA
significantly increased GSH depleted cell number at 72 h in a
dose-dependent manner (Fig.
4C).
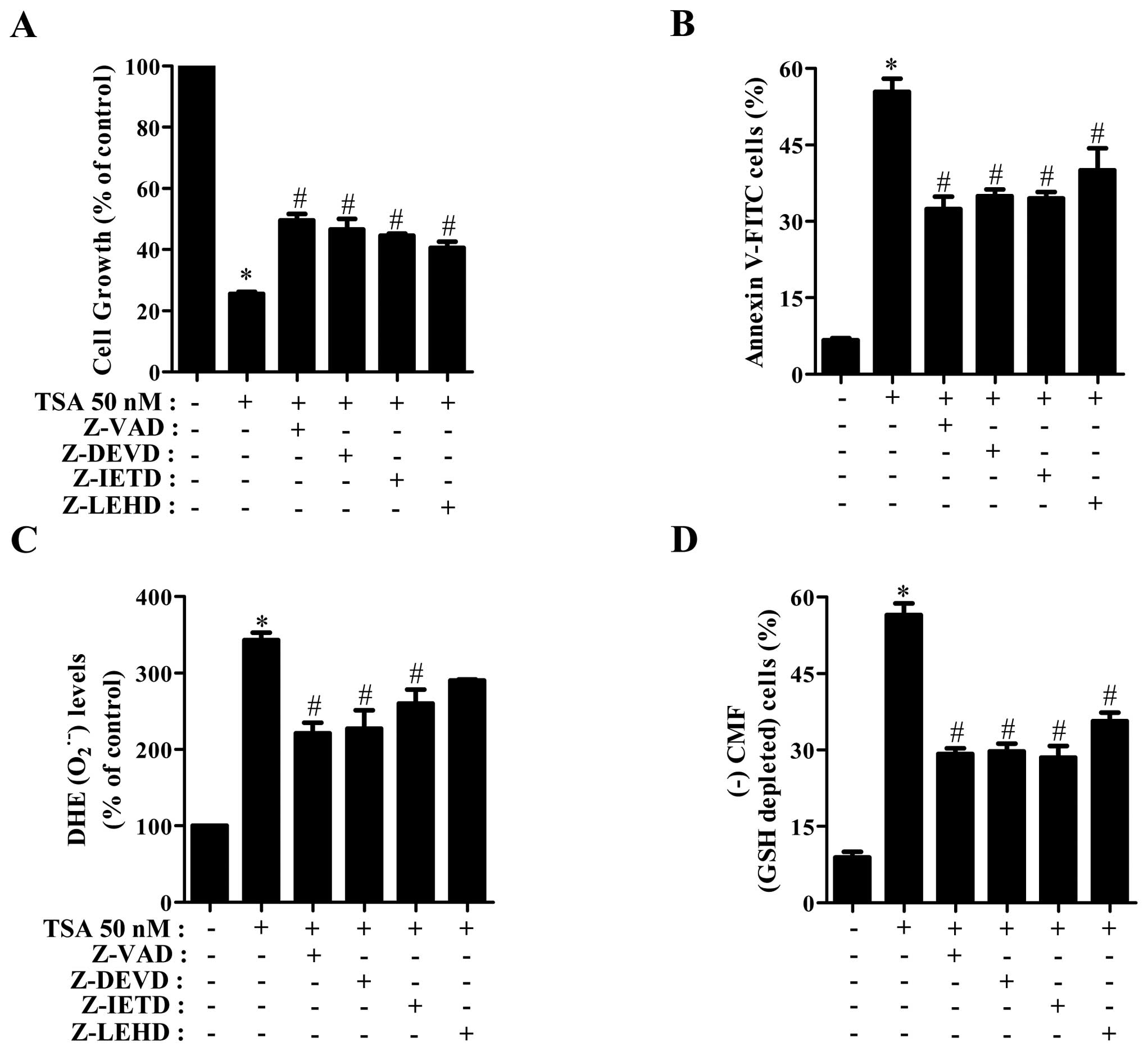
Effects of caspase inhibitors on cell
growth, death, O2•− and GSH levels in
TSA-treated HeLa cells
We determined which caspases were involved in the
cell growth inhibition and death of TSA-treated HeLa cells. For
this experiment, we chose 50 nM TSA as a suitable dose to
differentiate the level of cell growth and death in the presence or
absence of each caspase inhibitor; [pan-caspase inhibitor (Z-VAD),
caspase-3 inhibitor (Z-DEVD), caspase-8 inhibitor (Z-IETD), or
caspase-9 inhibitor (Z-LEHD)]. A concentration of 15 μM
caspase inhibitor was used as an optimal dose in this study, this
dose did not significantly affect cell death in HeLa control cells.
All the caspase inhibitors attenuated cell growth inhibition and
death in TSA-treated HeLa cells (Fig.
5A and B). In relation to O2•− and GSH
levels, all caspase inhibitors, especially Z-VAD, Z-DEVD and Z-IETD
significantly reduced O2•− level in
TSA-treated HeLa cells (Fig. 5C).
Furthermore, all the caspase inhibitors markedly prevented GSH
depletion in TSA-treated HeLa cells (Fig. 5D).
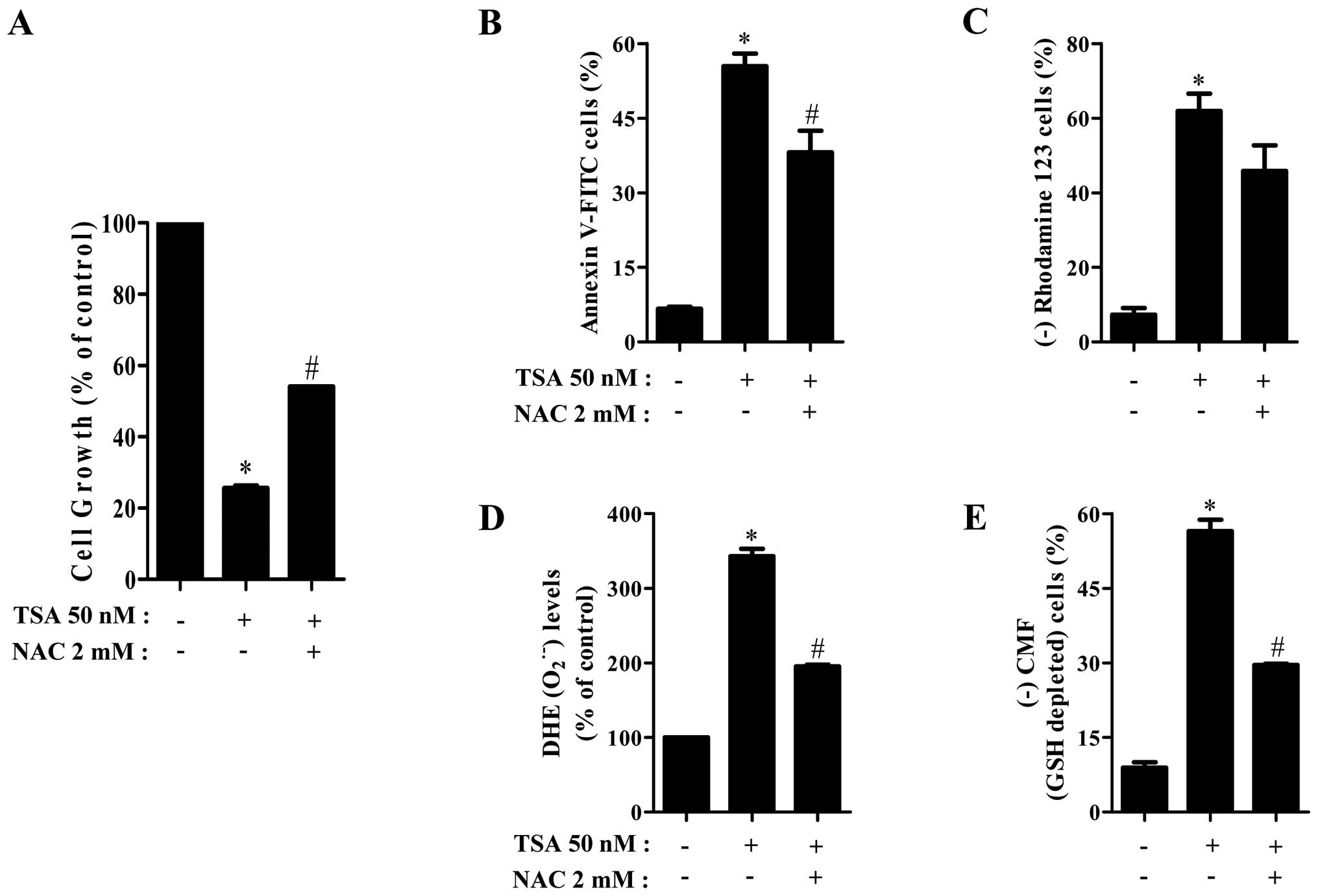
Effects of NAC on cell growth, death,
O2•− and GSH levels in TSA-treated HeLa
cells
To investigate the involvement of
O2•− level increase in TSA-induced HeLa cell
growth inhibition and death, HeLa cells were pretreated with 2 mM
NAC as an antioxidant before the treatment of TSA. As shown in
Fig. 6A and B, NAC significantly
recovered cell growth inhibition and death in TSA-treated HeLa
cells. In addition, NAC attenuated the proportion of MMP
(ΔΨm) loss cells in TSA-treated HeLa cells (Fig. 6C). When assessed whether NAC
influences O2•− and GSH levels, NAC markedly
reduced O2•− level and GSH depletion in
TSA-treated HeLa cells (Fig. 6D and
E).
Discussion
In the present study, we focused on assessing the
effects of TSA on cell growth inhibition and death in HeLa cervical
cancer cells in relation to ROS and GSH levels. Because TSA
decreased the level of HDAC activity and increased the levels of
acetylated histones in HeLa cells, TSA seemed to act as an HDAC
inhibitor in HeLa cells. This agent remarkably induced the
acetylation of histone 4 compared with that of histone 3. However,
another hydroxamic acid-derived HDAC inhibitor, SBHA strongly
induced the acetylation of histone 3 rather than that of histone 4
in HeLa cells (unpublished data). The different effects of TSA and
SBHA on the histone acetylation is probably due to the different
functional bioavailability of these hydroxamic acid-derived HDAC
inhibitors through various biochemical modifications such as
sulfation, hydroxylation, oxidation and methylation in cells. TSA
inhibited the growth of HeLa cells in a dose- and time-dependent
manner and also induced apoptosis. However, this agent did not
significantly induce any specific phase arrest of the cell cycle at
24 and 72 h (data not shown). The growth inhibition in TSA-treated
HeLa cells was due to apoptotic cell death rather than a specific
cell cycle arrest. TSA dose-dependently triggered the loss of MMP
(ΔΨm) and reduced MMP (ΔΨm) levels in HeLa
cells (data not shown). The levels of MMP (ΔΨm) loss
cells were similar to those of annexin V staining cells (Fig. 2), implying that apoptotic cell
death by TSA was tightly correlated with the collapse of MMP
(ΔΨm).
A high ratio of Bax to Bcl-2 is known to be the main
trigger in the collapse of MMP (ΔΨm) and apoptosis in
cells (21). It is reported that
HDAC inhibitors downregulate Bcl-2 expression and induce apoptosis
in many cancer cells (22,23). Likewise, the level of Bcl-2 protein
was downregulated in TSA-treated HeLa cells. Moreover, the
administration of Bcl-2 siRNA enhanced the growth inhibition and
death of TSA-treated HeLa cells. Therefore, TSA seemed to induce
apoptosis in HeLa cells depending on the downregulation of Bcl-2
protein. In relation to Bax protein, TSA did not strongly alter the
expression level of Bax protein. However, the administration of Bax
siRNA attenuated the growth inhibition and death of TSA-treated
HeLa cells. Therefore, these results support the notion that the
relatively high ratio of Bax to Bcl-2 can trigger apoptosis in
cells. In particular, Bcl-2 siRNA alone induced the growth
inhibition and death in HeLa control cells, implying that Bcl-2
protein is a crucial regulator in the survival of HeLa cells. When
determined which caspases were involved in apoptosis in TSA-treated
HeLa cells, all the tested caspase inhibitors prevented TSA-induced
HeLa cell death. In addition, TSA increased the activity of
caspase-3 in HeLa cells. Therefore, TSA-induced HeLa apoptosis is
mediated by the activation of various caspase cascades. In
particular, both cell death receptor pathway of caspase-8 and the
mitochondrial pathway of caspase-9 were involved in the induction
of apoptosis in HeLa cells.
It is reported that HDAC inhibitor increases ROS
levels in solid tumor and leukemia cells (24). Furthermore, oxidative stress seems
to be involved in HDAC inhibitor-induced cell death (25). Similarly, the level of
O2•− was significantly increased in
TSA-treated HeLa cells. Probably, the increased
O2•− level mainly resulted from the damage of
mitochondria by TSA. Importantly, NAC, which strongly suppressed
O2•− levels in TSA-treated HeLa cells,
significantly prevented HeLa cell growth inhibition and death by
TSA and it also attenuated the collapse of MMP (ΔΨm). In
addition, all caspase inhibitors showing the anti-apoptotic effects
decreased O2•− levels in these cells.
Treatment with Bcl-2 siRNA increased O2•−
level in TSA-treated HeLa cells whereas treatment with Bax siRNA
decreased the O2•− level in these cells (data
not shown). These results suggested that TSA-induced HeLa cell
death is mediated by the oxidative stress derived from
O2•− level changes.
TSA increased the level of Mn SOD among the various
antioxidant proteins in the present study. Mn SOD which is located
in mitochondria catalyzes the dismutation of
O2•− into oxygen and hydrogen peroxide
(26). It is possible that an
increase in O2•− level in TSA-treated HeLa
cells leads to upregulation of the expression of Mn SOD in a
compensatory mechanism. However, the expression level of
thioredoxin 2 (Trx2), which is another antioxidant enzyme in
mitochondria, was not altered by TSA in HeLa cells. Because it has
been reported that DNA methylation and histone modification
regulated Mn SOD expression in breast cancer cells (27), it is plausible that the specific
upregulation of Mn SOD can be transcriptionally regulated by the
inhibition of HDAC by TSA.
GSH as the main cellular non-protein antioxidant can
eliminate ROS including O2•−. It is known
that the intracellular GSH content has a decisive effect on
anticancer drug-induced apoptosis (28,29).
According to our current data, TSA increased the number of GSH
depleted cells in HeLa cells. All the tested caspase inhibitors and
NAC prevented the GSH depletion by TSA. In addition, treatment with
Bcl-2 siRNA increased GSH depletion in TSA-treated HeLa cells
whereas treatment with Bax siRNA decreased the GSH depletion in
these cells (data not shown). Therefore, these results support the
notion that apoptotic effects are inversely comparative to GSH
content in the cell.
In conclusion, as depicted in Fig. 7, TSA inhibited the growth of HeLa
cervical cancer cells via Bcl-2-mediated and caspase-dependent
apoptosis, which was closely related to O2•−
and GSH content levels.
Abbreviations:
|
TSA
|
trichostatin A
|
|
HAT
|
histone acetyltransferase
|
|
HDAC
|
histone deacetylase
|
|
ROS
|
reactive oxygen species
|
|
MMP (ΔΨm)
|
mitochondrial membrane potential
|
|
FBS
|
fetal bovine serum
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
PI
|
propidium iodide
|
|
FITC
|
fluorescein isothiocyanate
|
|
Z-VAD-FMK
|
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
|
|
Z-DEVD-FMK
|
benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone
|
|
Z-IETD-FMK
|
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone
|
|
Z-LEHD-FMK
|
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone
|
|
DHE
|
dihydroethidium
|
|
GSH
|
glutathione
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
|
Trx
|
thioredoxin
|
|
TrxR
|
thioredoxin reductase
|
|
Cu/Zn SOD
|
copper zinc superoxide dismutase
|
|
Mn SOD
|
manganese superoxide dismutase
|
|
NAC
|
N-acetyl cysteine
|
Acknowledgements
This study was supported by the Basic
Science Research Program through the National Research Foundation
of Korea (NRF) funded by the Ministry of Education, Science and
Technology (2010-007059) and by a grant from the Ministry of
Science & Technology (MoST)/Korea Science & Engineering
Foundation (KOSEF) through the Diabetes Research Center at Chonbuk
National University (2012-0009323).
References
|
1.
|
Burgess DJ: Histone modification at the
gene level. Nat Rev Cancer. 12:1562012. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Ganesan A, Nolan L, Crabb SJ and Packham
G: Epigenetic therapy: histone acetylation, DNA methylation and
anti-cancer drug discovery. Curr Cancer Drug Targets. 9:963–981.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Lu Z, Luo RZ, Peng H, Huang M, Nishmoto A,
Hunt KK, Helin K, Liao WS and Yu Y: E2F-HDAC complexes negatively
regulate the tumor suppressor gene ARHI in breast cancer. Oncogene.
25:230–239. 2006.PubMed/NCBI
|
|
4.
|
Khan O and La Thangue NB: HDAC inhibitors
in cancer biology: emerging mechanisms and clinical applications.
Immunol Cell Biol. 90:85–94. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lehmann A, Denkert C, Budczies J,
Buckendahl AC, Darb-Esfahani S, Noske A, Muller BM, Bahra M,
Neuhaus P, Dietel M, et al: High class I HDAC activity and
expression are associated with RelA/p65 activation in pancreatic
cancer in vitro and in vivo. BMC Cancer. 9:3952009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Wang L, Zou X, Berger AD, Twiss C, Peng Y,
Li Y, Chiu J, Guo H, Satagopan J, Wilton A, et al: Increased
expression of histone deacetylaces (HDACs) and inhibition of
prostate cancer growth and invasion by HDAC inhibitor SAHA. Am J
Transl Res. 1:62–71. 2009.PubMed/NCBI
|
|
7.
|
Vanhaecke T, Papeleu P, Elaut G and
Rogiers V: Trichostatin A-like hydroxamate histone deacetylase
inhibitors as therapeutic agents: toxicological point of view. Curr
Med Chem. 11:1629–1643. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Marks PA: Discovery and development of
SAHA as an anticancer agent. Oncogene. 26:1351–1356. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Lane AA and Chabner BA: Histone
deacetylase inhibitors in cancer therapy. J Clin Oncol.
27:5459–5468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Campas-Moya C: Romidepsin for the
treatment of cutaneous T-cell lymphoma. Drugs Today. 45:787–795.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Pettazzoni P, Pizzimenti S, Toaldo C,
Sotomayor P, Tagliavacca L, Liu S, Wang D, Minelli R, Ellis L,
Atadja P, et al: Induction of cell cycle arrest and DNA damage by
the HDAC inhibitor panobinostat (LBH589) and the lipid peroxidation
end product 4-hydroxynonenal in prostate cancer cells. Free Radic
Biol Med. 50:313–322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Phillip CJ, Giardina CK, Bilir B, Cutler
DJ, Lai YH, Kucuk O and Moreno CS: Genistein cooperates with the
histone deacetylase inhibitor vorinostat to induce cell death in
prostate cancer cells. BMC Cancer. 12:1452012. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Sweet MJ, Shakespear MR, Kamal NA and
Fairlie DP: HDAC inhibitors: modulating leukocyte differentiation,
survival, proliferation and inflammation. Immunol Cell Biol.
90:14–22. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Gong K, Xie J, Yi H and Li W: CS055
(Chidamide/HBI-8000), a novel histone deacetylase inhibitor,
induces G1 arrest, ROS-dependent apoptosis and differentiation in
human leukaemia cells. Biochem J. 443:735–746. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Duenas-Gonzalez A, Lizano M, Candelaria M,
Cetina L, Arce C and Cervera E: Epigenetics of cervical cancer. An
overview and therapeutic perspectives. Mol Cancer. 4:382005.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Anton M, Horky M, Kuchtickova S, Vojtesek
B and Blaha O: Immunohistochemical detection of acetylation and
phosphorylation of histone H3 in cervical smears. Ceska Gynekol.
69:3–6. 2004.PubMed/NCBI
|
|
17.
|
Huang BH, Laban M, Leung CH, Lee L, Lee
CK, Salto-Tellez M, Raju GC and Hooi SC: Inhibition of histone
deacetylase 2 increases apoptosis and p21Cip1/WAF1
expression, independent of histone deacetylase 1. Cell Death
Differ. 12:395–404. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Vigushin DM, Ali S, Pace PE, Mirsaidi N,
Ito K, Adcock I and Coombes RC: Trichostatin A is a histone
deacetylase inhibitor with potent antitumor activity against breast
cancer in vivo. Clin Cancer Res. 7:971–976. 2001.PubMed/NCBI
|
|
19.
|
Zhang CZ, Zhang HT, Chen GG and Lai PB:
Trichostatin A sensitizes HBx-expressing liver cancer cells to
etoposide treatment. Apoptosis. 16:683–695. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Meng J, Zhang HH, Zhou CX, Li C, Zhang F
and Mei QB: The histone deacetylase inhibitor trichostatin A
induces cell cycle arrest and apoptosis in colorectal cancer cells
via p53-dependent and -independent pathways. Oncol Rep. 28:384–388.
2012.PubMed/NCBI
|
|
21.
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Duan H, Heckman CA and Boxer LM: Histone
deacetylase inhibitors down-regulate bcl-2 expression and induce
apoptosis in t(14;18) lymphomas. Mol Cell Biol. 25:1608–1619. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ellis L and Pili R: Histone deacetylase
inhibitors: advancing therapeutic strategies in hematological and
solid malignancies. Pharmaceuticals. 3:2411–2469. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Eot-Houllier G, Fulcrand G,
Magnaghi-Jaulin L and Jaulin C: Histone deacetylase inhibitors and
genomic instability. Cancer Lett. 274:169–176. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Ungerstedt JS, Sowa Y, Xu WS, Shao Y,
Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X and Marks PA:
Role of thioredoxin in the response of normal and transformed cells
to histone deacetylase inhibitors. Proc Natl Acad Sci USA.
102:673–678. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Miriyala S, Spasojevic I, Tovmasyan A,
Salvemini D, Vujaskovic Z, St Clair D and Batinic-Haberle I:
Manganese superoxide dismutase, MnSOD and its mimics. Biochim
Biophys Acta. 1822:794–814. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Hitchler MJ, Wikainapakul K, Yu L, Powers
K, Attatippaholkun W and Domann FE: Epigenetic regulation of
manganese superoxide dismutase expression in human breast cancer
cells. Epigenetics. 1:163–171. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Brechbuhl HM, Kachadourian R, Min E, Chan
D and Day BJ: Chrysin enhances doxorubicin-induced cytotoxicity in
human lung epithelial cancer cell lines: the role of glutathione.
Toxicol Appl Pharmacol. 258:1–9. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Fath MA, Ahmad IM, Smith CJ, Spence J and
Spitz DR: Enhancement of carboplatin-mediated lung cancer cell
killing by simultaneous disruption of glutathione and thioredoxin
metabolism. Clin Cancer Res. 17:6206–6217. 2011. View Article : Google Scholar : PubMed/NCBI
|