Introduction
Physiological estrogens are a group of steroid
hormones that include estrone (E1), estradiol (E2), and estriol
(E3). Although E3 is the most plentiful among these three factors,
E2, also known as 17β-estradiol, exerts the strongest estrogenic
effect. Estrogens are produced in ovaries, adrenal glands, and fat
tissues, and function as the primary female sex hormones that
promote the development of secondary sexual characteristics and
regulate certain functions of the reproductive system. In addition,
these compounds control various metabolic processes including bone
growth, protein synthesis, and fat deposition. Estrogens have also
been reported to be linked to the pathogenesis of several cancers
in the reproductive organs. Previous studies have shown that
circulating levels of estrogens may be most strongly associated
with the risks of breast (1–4),
ovarian (5–7), endometrial (8), and cervical (9) cancers. These diseases are known as
estrogen-responsive or estrogen receptor (ER)-positive cancers
because the actions of estrogen are mediated by ERs and ER
expression has been observed in these cancers.
Recently, chemical compounds called endocrine
disrupting chemicals (EDCs) are emerging as another risk factor for
hormone-responsive cancers (10).
EDCs are environmental substances that interfere with the
biosynthesis, signaling, or metabolism of natural hormones in the
body, thus having serious detrimental effects on reproductive and
developmental processes (11).
Xenoestrogens are classified as EDCs with estrogenic activity that
disrupt normal estrogen signaling mediated by ERs (12–15).
Bisphenol A (BPA) is a widely used industrial compound and a
typical xenoestrogen (16,17). This chemical has been used for the
manufacturing of polycarbonate plastics and polystyrene resins, and
is commonly found in plastic bottles, plastic food containers,
dental materials, and compounds used to coat containers for canned
food. BPA can leach from these products in appreciable quantities,
and thus humans are easily exposed to it through normal product use
(18–20). After the estrogenic properties of
BPA were discovered in 1930 (16),
many studies published in the following decades have characterized
the hazardous health effects of this compound and identified BPA as
an endocrine disruptor. For instance, perinatal exposure to
environmentally relevant concentrations of BPA causes morphological
and functional alterations of the male and female genital tracts
(21). In so doing, BPA may
predispose the affected individuals to earlier onset of disease and
reduced fertility, and induce neoplastic transformation in human
breast epithelial cells (21–23).
Currently, the connection between perinatal BPA exposure and breast
cancer is being examined (24).
In the present study, we examined the effect of BPA
on the risk of ovarian cancer cell proliferation. Although this
disease is one of the most frequently observed gynecologic cancers
and is an estrogen-responsive disorder (25–27),
the pathogenic actions of BPA on ovarian carcinoma have not been
fully elucidated. Some previous reports suggest that BPA stimulates
the proliferation of OVCAR-3 human ovarian cancer cells by inducing
leptin receptor expression (28)
or decreasing caspase-3 activity (29). To evaluate the effect of BPA on
ovarian cancer development, we used the BG-1 ovarian adenocarcinoma
cell line, an estrogen-dependent cell line that expresses ERs. In a
cell proliferation assay, BPA promoted BG-1 cell growth as did E2,
indicating that BPA acts as a xenoestrogen which has an obvious
estrogenic effect on estrogen-responsive ovarian cancer. To explore
ways to reverse the positive effects of BPA on cancer cell
proliferation, we also examined the suppressive effect of genistein
(GEN) on cell growth promoted by E2 or BPA. GEN is a classical
phytoestrogen that is a plant-derived and naturally occurring
dietary xenoestrogen which influences multiple biochemical
functions (30). Based on
epidemiologic observations indicating that incidences of cancer,
including breast cancer, are much lower in Asian populations that
consume significantly higher amounts of phytoestrogens compared to
Western individuals, the chemoprotective properties of GEN have
been extensively studied (31–33)
although the anticancer effect of GEN remains unclear. Our present
study showed that GEN effectively suppressed BG-1 ovarian cancer
cell proliferation induced by E2 or BPA. These findings may be
considered as an evidence of another chemopreventive activity of
GEN that can nullify the carcinogenic risks associated with BPA, a
potent chemosynthetic EDC, or E2.
Materials and methods
Reagents and chemicals
17β-estradiol (E2), BPA, and ICI 182,780 were
purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). GEN was
obtained from LC Laboratories (Woburn, MA, USA). Propyl pyrazole
triol (PPT) and diarylpropionitrile (DPN) were purchased from
Tocris (Ellisville, MO, USA). All chemicals were dissolved in 100%
dimethyl sulfoxide (DMSO; Junsei Chemical Co., Tokyo, Japan) and
stored as stock solutions at 4°C.
Cell culturing
BG-1 ovarian adenocarcinoma cells were obtained from
Dr K.S. Korach (National Institute of Environmental Health
Sciences, Research Triangle Park, NC, USA). The cells were cultured
in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone Laboratories
Inc., Logan, UT, USA) supplemented with 10% heat-inactivated fetal
bovine serum (FBS; Hyclone Laboratories Inc.), 1% penicillin G and
streptomycin (Cellgro Mediatech, Inc., Manassas, VA, USA), and 1%
anti-fungal HEPES (Invitrogen Life Technologies, Carlsbad, CA, USA)
at 37°C in a humidified atmosphere of 5% CO2-95% air. To
prevent the effects of estrogenic components in the DMEM and FBS,
BG-1 cells were also cultured in phenol red-free DMEM supplemented
with 5% charcoal-dextran treated FBS (CD-FBS) to measure the
estrogenicity of the EDCs. Cells were detached with 0.05%
trypsin/0.02% EDTA in Mg2+/Ca2+-free Hank’s
balanced salt solution (PAA Laboratories, Pasching, Austria).
Cell viability assay
To evaluate the effect of E2 or BPA on BG-1 cell
proliferation, a cell viability assay was conducted as previously
described (34–36). BG-1 cells were seeded at a density
of 4,000 cells/100 μl of phenol red-free DMEM with 5% CD-FBS
medium per well of 96-well plates. After incubating for 48 h, the
cells were washed and treated with various concentrations of E2 or
BPA (E2: 10−10–10−6 M, BPA:
10−10–10−5 M) in phenol red-free DMEM
supplemented with 0.1% DMSO for 5 days. DMSO was used as a vehicle
and a negative control. Cell viability was assessed with the
addition of 3-(4-,5-dimethylthiazol-2-yl)-2,5-dyphenyltetrazolium
bromide (MTT; Sigma-Aldrich) solution. MTT (10 μl of 5-mg/ml
solution) was added to each well and the plates were incubated for
4 h at 37°C. Supernatants were removed and 100 μl of DMSO
was added to each well to dissolve the resultant formazan crystals.
The optical density (OD) of each well was measured at 540 nm using
an ELISA reader (VERSA man, Molecular Devices, Sunnyvale, CA, USA)
and used to calculate the number of viable cells as previously
described (37,38). Viability of cells treated with the
different EDCs was calculated relative to the control
(DMSO-treated) cells.
To demonstrate the connection between E2 or BPA
action and ER signaling, BG-1 cells were co-treated with E2 or BPA
along with ICI 182,780 (a typical ER antagonist), PPT (an ERα
agonist), or DPN (an ERβ agonist). The concentrations of ICI
182,780, PPT and DPN were 10−7, 10−8 and
10−8 M, respectively. To evaluate the effect of GEN on
BG-1 cell proliferation, the cells were also co-treated with a
combination of GEN and E2 or BPA. GEN was added at concentrations
of 1.0, 2.5, 5.0, 7.5 and 10×10−5 M in the presence of
10−9 M of E2 or 10−5 M of BPA. After treating
these reagents, identical experimental procedures were performed
using MTT as in the treatment of E2 or BPA. All experiments were
done at least three times.
Total RNA extraction
BG-1 cells were seeded at a density of
3.0×105 cells per well in a 6-well plate, and then
treated with DMSO, E2, BPA, or a combination of GEN and E2 or BPA.
The concentrations of E2, BPA, and GEN were 10−9,
10−5 and 10−4 M, respectively. Total RNA was
extracted at various time-points (0, 6, and 24 h) using TRIzol
reagent (Invitrogen Life Technologies) according to the
manufacturer’s instructions. The concentration of total RNA was
measured with a spectrophotometer (Optizen, Mecasys, Deajeon,
Republic of Korea) at 260/280 nm. Total RNA (1 μg) was then
dissolved in dietyl pyrocarbonated-deionzed water (DEPC-DW) for
cDNA synthesis.
Semi-quantitative reverse transcription
(RT) PCR
cDNA was synthesized from total RNA by RT-PCR. The
reaction mixture contained murine leukemia virus reverse
transcriptase (M-MLV RT; iNtRON Biotechnology, Sungnam, Republic of
Korea), 200 pM nonamer random primer (iNtRON Biotechnology), dNTPs
(iNtRON Biotechnology), RNase inhibitor (iNtRON Biotechnology), and
RT buffer (iNtRON Biotechnology). cDNA synthesis was performed at
37°C for 1 h and 95°C for 5 min. p21, cyclin D1, and GAPDH cDNAs
were amplified by PCR with specific forward and reverse primers,
Taq polymerase, PCR buffer, and dNTP mixture, and each cDNA
template as previously described. Sequences of the forward and
reverse primers along with the predicted sizes of each gene product
are shown in Table I. The RT-PCR
products were separated on a 1.5% agarose gel and the size of each
gene band was estimated by comparison with 100-bp size ladders
(iNtRON Biotechnology). The gels were scanned and the band
densities were quantified using Gel Doc 2000 (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). All experiments were done at least three
times.
 | Table IPrimer sequences and predicted
product sizes for the semi-quantitative RT-PCR. |
Table I
Primer sequences and predicted
product sizes for the semi-quantitative RT-PCR.
| Target gene | Sequences | Product size
(bp) |
|---|
| p21 | Sense:
5′-AGGCACCGAGGCACTCAGAG-3′
Antisense: 5′-TGACAGGTCCACATGGTCTTCC-3′ | 370 |
| cyclin D1 | Sense:
5′-TCTAAGATGAAGGAGACCATC-3′book
Antisense: 5′-GCGGTAGTAGGACAGGAAGTTGTT-3′ | 354 |
| GAPDH | Sense:
5′-ATGTTCGTCATGGGTGTGAACCA-3′
Antisense: 5′-TGGCAGGTTTTTCTAGACGGCAG-3′ | 351 |
Western blot analysis
Western blotting was performed to assess the protein
expression of cyclin D1 and p21 in BG-1 cells. The cells were
cultured to a density of 1.0×106 cells per of 100-mm
dish and then treated with DMSO, E2, BPA, or combinations of GEN
and E2 or BPA for 24 and 48 h. The concentrations of E2, BPA, and
GEN were 10−9, 10−5 and 10−4 M,
respectively. After treatment, the cells were suspended in 100
μl of 1X RIPA buffer (50 mM Tris-HCl, pH 8.0.; 150 mM NaCl,
1% NP-40, 0.5% deoxycholic acid, and 0.1% SDS). Total protein
concentrations were determined using bicinchoninic acid (BCA;
Sigma-Aldrich Corp.) and 50 μg of total protein were then
separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The
proteins were transferred to a polyvinylidene difluoride (PVDF)
membrane (Bio-Rad Laboratories, Inc.), and the membranes were
blocked with 5% bovine serum albumin (BSA; Sigma-Aldrich Corp.) for
2 h at room temperature. The membranes were then incubated with
mouse monoclonal anti-p21 (1:4,000; Cell Signaling Technology,
Inc., Danvers, MA, USA), mouse monoclonal anti-cyclin D1 (1:2,000;
Abcam, Hanam-city, Republic of Korea), or mouse monoclonal
anti-GAPDH (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA)
antibodies for 2 h at room temperature. The membranes were
subsequently probed with anti-mouse IgG HRP-conjugated secondary
antibody (1:3,000; Santa Cruz Biotechnology) for 2 h at room
temperature. Target proteins were detected with a West-Q
Chemiluminescent Substrate Plus kit (GenDEPOT, Barker, TX, USA).
All experiments were done at least three times.
Statistical analysis
All data were analyzed with GraphPad Prism software
(San Diego, CA, USA). The in vitro data are presented as the
mean ± SD. Statistical analyses were performed using a one-way
ANOVA followed by Dunnett’s multiple comparison test and Student’s
t-test. P-values <0.05 were considered to be statistically
significant.
Results
Cell proliferation effect by E2 or BPA on
BG-1 cells
To evaluate the effect of E2 or BPA on cell
proliferation, BG-1 cells were cultured with vehicle (0.1% DMSO,
control), E2 (10−10–10−6 M), or BPA
(10−10–10−5 M) for 5 days. E2 effectively
increased the viability of BG-1 cells at concentrations of
10−10–10−7 M in a dose-dependent manner
(Fig. 1A). At concentrations of
10−7 M and above, BPA also promoted cell proliferation
(Fig. 1B). Although higher
concentrations of BPA were needed to induce significant cell
proliferation compared to E2, BPA was shown to exert an estrogenic
effect on the BG-1 cells by mimicking E2 action.
Effects of E2 or BPA on the proliferation
of cells co-treated with ER modulators
To determine whether increased cell proliferation
promoted by E2 or BPA was mediated by ER signaling, BG-1 cells were
co-treated with various ER modulators along with E2 or BPA and cell
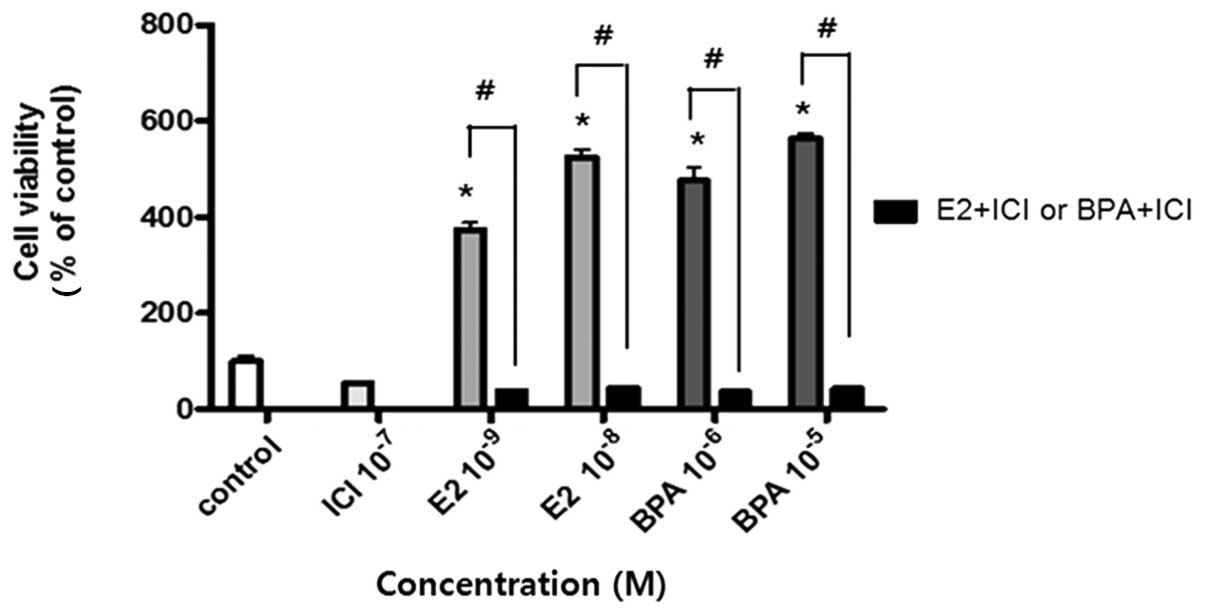
viability was measured. When the cells were co-treated with ICI
182,780 (a well-known ER antagonist) and E2 (10−9 and
10−8 M) or BPA (10−6 and 10−5 M),
cell proliferation increased by treatment with E2 or BPA alone was
dramatically reduced (Fig. 2). ICI
182,780, also called Fulvestrant, is an intact ER antagonist that
does not exert any agonist effects, working both by downregulating
and degrading the ER (39,40). Based on the result showing that E2
or BPA could not induce cell proliferation when the ER was
inactivated by ICI 182,780, it was hypothesized that the
proliferation of BG-1 cells was mediated by ER signaling via E2 or
BPA.
We next determined which ER isoform, ERα or ERβ, was
associated with the positive effect of E2 or BPA on cell
proliferation. For this, the cells were co-treated with PPT or DPN
(agonists of ERα and ERβ, respectively) and E2 or BPA. As shown in
Fig. 3A and B, PPT in combination
with E2 or BPA promoted BG-1 cell growth compared to a single
treatment of E2 or BPA (for 10−11 and 10−10 M
of E2, and for 10−9, 10−8 and 10−7
M of BPA). On the other hand, DPN in combination with E2 or BPA had
no effect on cell proliferation (Fig.
3C and D). These data showed that BG-1 cell proliferation was
mainly mediated by ERα and thus E2 or BPA induced cell growth via
ERα signaling.
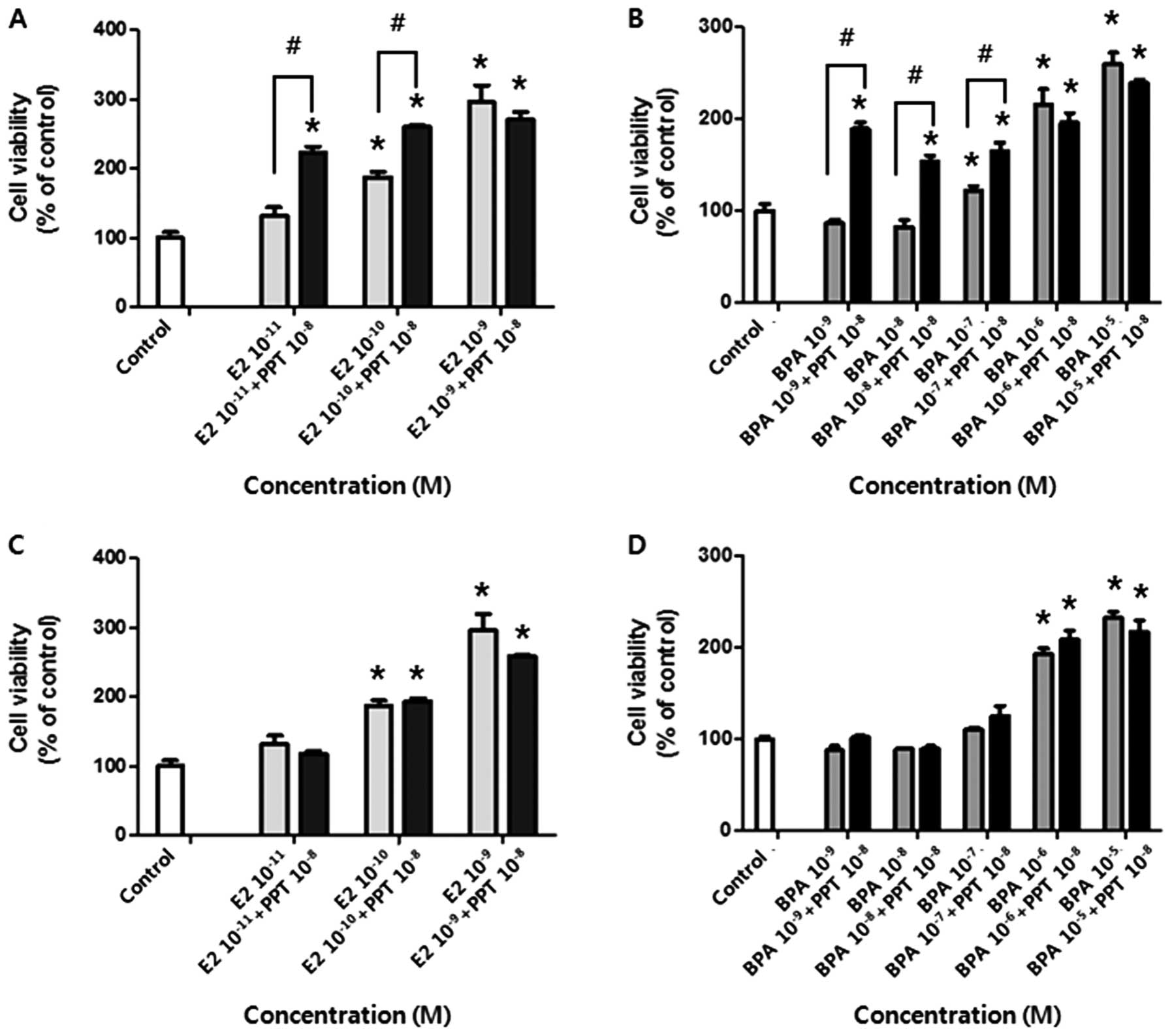 | Figure 3Viability of BG-1 human ovarian
cancer cells following co-treatment with E2 or BPA and PPT, an ERα
agonist, or DPN, an ERβ agonist. Cells were treated with DMSO (0.1
or 0.2%) as a control, E2 (10−11, 10−10 and
10−9 M), or BPA (10−9, 10−8,
10−7, 10−6 and 10−5 M) in the
presence or absence of PPT (10−8 M) or DPN
(10−8 M) for 5 days. Cell viability was measured using
an MTT assay. (A) The effect of E2 on cell proliferation in the
presence or absence of PPT. (B) The effect of BPA on cell
proliferation in the presence or absence of PPT. (C) The effect of
E2 on cell proliferation in the presence or absence of DPN. (D) The
effect of BPA on cell proliferation in the presence or absence of
DPN. Data represent the mean ± SD of triplicate experiments.
*Significant elevation in cell viability following
treatments with E2, BPA, and a respective combination of PPT or DPN
compared to the control (p<0.05 based on Dunnett’s multiple
comparison test). #Significant elevation or reduction in
cell viability by co-treatment compared to treatment with E2 or BPA
alone (p<0.05 according to Student’s t-test). |
Anti-proliferative effect by GEN on E2 or
BPA-induced cell proliferation
To evaluate the effect of GEN on BG-1 cell
proliferation promoted by E2 or BPA, BG-1 cancer cells were treated
with a combination of E2 or BPA and GEN. GEN (5.0, 7.5, and
10×10−5 M with E2 or 2.5, 5.0, 7.5 and
10×10−5 M with BPA) strongly suppressed the cell growth
induced by E2 (10−9 M) or BPA (10−5 M) as
shown in Fig. 4. These findings
demonstrate that GEN has an anti-proliferative effect and reduces
cancer cell growth promoted by E2 or BPA.
Effects of E2 and BPA alone or in
combination with GEN on mRNA expression of cell cycle-related
genes
We next examined the mechanism underlying the
effects of E2 and BPA (alone or in combination with GEN) on BG-1
cell proliferation through changes in the mRNA expression of cell
cycle-related genes. For this, we performed semi-quantitative
RT-PCR on total RNA samples isolated from the cells treated with
these agents. First, mRNA levels of cyclin D1 (a factor responsible
for cell cycle progression) were significantly increased by
treatment with E2 for 6 h or BPA for 6 and 24 h compared to the
control. In contrast, cyclin D1 mRNA expression was considerably
reduced by co-treatment with E2 or BPA and GEN for both 6 and 24 h
compared to administration of E2 or BPA alone (Fig. 5A and C). The mRNA levels of p21 (a
factor that causes cell cycle arrest) were significantly decreased
by treatment with E2 or BPA for 6 h compared to the control. On the
other hand, these mRNA levels were significantly increased by
co-treatment with BPA or E2 and GEN for 6 and 24 h compared to
exposure to E2 or BPA alone (Fig. 5B
and D). Alterations in the expression of cell cycle-related
genes such as cyclin D1 and p21 may explain the effect of E2 or BPA
on cell proliferation and the anti-proliferative activity of
GEN.
Effects of E2 and BPA alone or in
combination with GEN on the protein expression of cell
cycle-related genes
To confirm that E2 and BPA altered the expression of
genes involved in cell cycle regulation, we conducted a western
blot analysis using antibodies specific for cyclin D1 and p21. As
shown in Fig. 6, the protein
levels of cyclin D1 were increased by E2 or BPA after 24 h of
treatment compared to the control. These levels were decreased by
co-treatment with GEN for 24 and 48 h compared to treatment with E2
or BPA alone (Fig. 6A and B). On
the other hand, the protein expression of p21 was reduced by E2 or
BPA after 24 h compared to the control. This effect was reversed by
a co-treatment with GEN compared to treatment with E2 or BPA alone
(Fig. 6A and C). These findings
coincided with the changes we observed in mRNA expression and
further validate the effect of E2 or BPA on cell proliferation and
the anti-proliferative activity of GEN.
Discussion
It was recently found that estrogens are important
factors in the initiation and progression of cancers, including
breast and ovarian carcinomas. Since then, there has been a growing
concern that EDCs, especially xenoestrogens, might potentially have
carcinogenic effects on estrogen-sensitive organs (1,2,5–7,41).
In the present study, we demonstrated that both E2 and BPA, a
typical xenoestrogen, have the capacity to stimulate ovarian cancer
cell proliferation. When added to cell culture medium devoid of
estrogenic compounds, E2 and BPA significantly promoted the
proliferation of BG-1 ovarian cancer cells. This increased cell
proliferation was reversed by co-treatment with ICI 182,780, a
well-known ER antagonist (39).
Therefore, it was determined that E2 mediated the growth of BG-1
cells via ER signaling, and BPA exerted an estrogenic effect by
mimicking E2 action.
Estrogen signaling is mainly mediated via two
subtypes of ERs, ERα and ERβ, that are differentially expressed in
various tissues and have unique functions (22,42).
There is a careful balance between the actions of these two
distinct receptor isoforms (43).
Both have been reported to affect cellular proliferation and cell
cycle events (44). However, ERβ
may have an inhibitory effect on G2 and M phases of the cell cycle
(43) whereas ERα was shown to be
linked to cell cycle progression through the stimulation of cyclin
D1 gene expression and induction of cell proliferation (44). cyclin D1 is a key regulator of the
cell cycle that acts by binding to the retinoblastoma (Rb) protein
and directing CDK4 and CDK6 to hyperphosphorylate Rb, leading to
the progression from the G1 to S phase and cell growth (45). It was reported that E2-ERα mediates
the dissociation of p21, a CKD inhibitor, from the cyclin E-CDK2
complex, the activation of cyclin-CDK complexes, and passage from
the G1 to S phase (46). E2 was
also found to enhance ERα binding to p53, a major tumor suppressor,
and inhibit p21 transcription (47). Based on these findings, it can be
said that E2 manipulates cell cycle progression and the
proliferation rate of cancer cells by modulating the activities of
cyclin-CDK complexes during G1 phase (43,46).
In agreement with this hypothesis, E2 and BPA were shown in the
present study to induce BG-1 cell proliferation by upregulating
cyclin D1 and downregulating p21 via ERα signaling (Fig. 7). Interaction between ERα and E2 or
BPA was implied based on the finding that increases in cell
proliferation by E2 or BPA were further augmented by a co-treatment
with PPT, an ERα agonist (48),
but not by DPN, an ERβ agonist (48). These data showed that BPA acts as a
distinct xenoestrogen in BG-1 ovarian cancer cells by mimicking E2
through similar mechanisms. In our previous study (49), we also examined the estrogenic
effect of BPA mediated by gene expression alterations in BG-1
ovarian cancer cells using a microarray analysis. We found that BPA
induces the transcription of E2-responsive genes such as RAB31,
cyclin D1, cdk-4, insulin-like growth factor binding protein 4, and
anti-mullerian hormone in a manner similar to E2.
In the present study, we also demonstrated the
anticancer activity of GEN, a typical phytoestrogen, against
carcinogenicity resulting from treatment with E2 or BPA. GEN is the
most abundant isoflavone in soybean products and is known to have
various biological activities (33). Among these, its anti-cancer effects
against a diverse number of cancers including breast and prostate
carcinomas have been considered to be most noteworthy (32,33).
In the present study, we performed a cell viability assay to
evaluate the effects of co-treatment with GEN and E2 or BPA. GEN
effectively suppressed BG-1 ovarian cancer cell proliferation
promoted by E2 or BPA. This anti-proliferative effect of GEN was
achieved by reversing the effects of E2 or BPA on the expression of
cell cycle-related genes. Unlike the actions of E2 or BPA, GEN
suppressed the expression of cyclin D1 and enhanced the expression
of p21 when administered with E2 or BPA, thereby leading to cell
cycle arrest in G1 phase (Fig. 7).
Further studies are required to understand the mechanisms
underlying the anti-proliferative activities of GEN. In particular,
elucidating the impact of GEN on ER signaling in
estrogen-responsive cancers will be helpful for explaining the
neutralizing or inhibitory effect of GEN on cancer progression
induced by diverse types of EDCs.
In conclusion, our findings demonstrated that GEN,
although classified as a natural xenoestrogen, acts as a
chemopreventive agent by abolishing the carcinogenic risks
associated with BPA, a potent chemosynthetic EDC, and E2.
Acknowledgements
This study was supported by a National
Research Foundation of Korea (NRF) grant (no. 2011-0015385) funded
by the Ministry of Education, Science and Technology (MEST) of the
Republic of Korea government. In addition, this study was also
supported by Priority Research Centers Program through the NRF
funded by the Ministry of Education, Science and Technology
(2011-0031403).
References
|
1
|
Pike MC, Krailo MD, Henderson BE,
Casagrande JT and Hoel DG: ‘Hormonal’ risk factors, ‘breast tissue
age’ and the age-incidence of breast cancer. Nature. 303:767–770.
1983.
|
|
2
|
Santen RJ, Boyd NF, Chlebowski RT, et al:
Critical assessment of new risk factors for breast cancer:
considerations for development of an improved risk prediction
model. Endocr Relat Cancer. 14:169–187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Missmer SA, Eliassen AH, Barbieri RL and
Hankinson SE: Endogenous estrogen, androgen, and progesterone
concentrations and breast cancer risk among postmenopausal women. J
Natl Cancer Inst. 96:1856–1865. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Russo J, Hasan Lareef M, Balogh G, Guo S
and Russo IH: Estrogen and its metabolites are carcinogenic agents
in human breast epithelial cells. J Steroid Biochem Mol Biol.
87:1–25. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rodriguez C, Patel AV, Calle EE, Jacob EJ
and Thun MJ: Estrogen replacement therapy and ovarian cancer
mortality in a large prospective study of US women. JAMA.
285:1460–1465. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baldwin WS, Curtis SW, Cauthen CA,
Risinger JI, Korach KS and Barrett JC: BG-1 ovarian cell line: an
alternative model for examining estrogen-dependent growth in vitro.
In Vitro Cell Dev Biol Anim. 34:649–654. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Giacalone PL, Daures JP, Ouafik L, Martin
PM, Laffargue F and Maudelonde T: Steroids and adrenomedullin
growth patterns in human ovarian cancer cells:
estrogenic-regulation assay. Gynecol Oncol. 91:651–656. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grady D, Gebretsadik T, Kerlikowske K,
Ernster V and Petitti D: Hormone replacement therapy and
endometrial cancer risk: a meta-analysis. Obstet Gynecol.
85:304–313. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chung SH, Franceschi S and Lambert PF:
Estrogen and ERalpha: culprits in cervical cancer? Trends
Endocrinol Metab. 21:504–511. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Soto AM and Sonnenschein C: Environmental
causes of cancer: endocrine disruptors as carcinogens. Nat Rev
Endocrinol. 6:363–370. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Crisp TM, Clegg ED, Cooper RL, et al:
Environmental endocrine disruption: an effects assessment and
analysis. Environ Health Perspect. 106(Suppl 1): 11–56. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cabaravdic M: [Xenoestrogen effects of
chemical compounds: influence on the breast cancer]. Med Arh.
60:97–100. 2006.(In Bosnian).
|
|
13
|
Lee HR, Hwang KA, Park MA, Yi BR, Jeung EB
and Choi KC: Treatment with bisphenol A and methoxychlor results in
the growth of human breast cancer cells and alteration of the
expression of cell cycle-related genes, cyclin D1 and p21, via an
estrogen receptor-dependent signaling pathway. Int J Mol Med.
29:883–890. 2012.
|
|
14
|
Park MA, Hwang KA and Choi KC: Diverse
animal models to examine potential role(s) and mechanism of
endocrine disrupting chemicals on the tumor progression and
prevention: do they have tumorigenic or anti-tumorigenic property?
Lab Anim Res. 27:265–273. 2011. View Article : Google Scholar
|
|
15
|
Park MA, Hwang KA, Lee HR, Yi BR, Jeung EB
and Choi KC: Cell growth of BG-1 ovarian cancer cells is promoted
by di-n-butyl phthalate and hexabromocyclododecane via upregulation
of the cyclin D and cyclin-dependent kinase-4 genes. Mol Med Rep.
5:761–766. 2012.PubMed/NCBI
|
|
16
|
Wolstenholme JT, Rissman EF and Connelly
JJ: The role of Bisphenol A in shaping the brain, epigenome and
behavior. Horm Behav. 59:296–305. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee HR, Kim TH and Choi KC: Treatment with
bisphenol A leads to the promotion of human breast cancer cells and
alteration of cell cycle-related gene expression, cyclin E and p27.
J Biomed Res. 12:215–233. 2011.
|
|
18
|
Brede C, Fjeldal P, Skjevrak I and
Herikstad H: Increased migration levels of bisphenol A from
polycarbonate baby bottles after dishwashing, boiling and brushing.
Food Addit Contam. 20:684–689. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Joskow R, Barr DB, Barr JR, Calafat AM,
Needham LL and Rubin C: Exposure to bisphenol A from bis-glycidyl
dimethacrylate-based dental sealants. J Am Dent Assoc. 137:353–362.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kang JH, Kito K and Kondo F: Factors
influencing the migration of bisphenol A from cans. J Food Prot.
66:1444–1447. 2003.PubMed/NCBI
|
|
21
|
Maffini MV, Rubin BS, Sonnenschein C and
Soto AM: Endocrine disruptors and reproductive health: the case of
bisphenol-A. Mol Cell Endocrinol. 254–255:179–186. 2006.PubMed/NCBI
|
|
22
|
Fernandez SV and Russo J: Estrogen and
xenoestrogens in breast cancer. Toxicol Pathol. 38:110–122. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Prins GS, Birch L, Tang WY and Ho SM:
Developmental estrogen exposures predispose to prostate
carcinogenesis with aging. Reprod Toxicol. 23:374–382. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vandenberg LN, Maffini MV, Sonnenschein C,
Rubin BS and Soto AM: Bisphenol-A and the great divide: a review of
controversies in the field of endocrine disruption. Endocr Rev.
30:75–95. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bai W, Oliveros-Saunders B, Wang Q,
Acevedo-Duncan ME and Nicosia SV: Estrogen stimulation of ovarian
surface epithelial cell proliferation. In Vitro Cell Dev Biol Anim.
36:657–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Choi KC, Kang SK, Tai CJ, Auersperg N and
Leung PC: Estradiol up-regulates antiapoptotic Bcl-2 messenger
ribonucleic acid and protein in tumorigenic ovarian surface
epithelium cells. Endocrinology. 142:2351–2360. 2001.PubMed/NCBI
|
|
27
|
Lindgren P, Backstrom T, Mahlck CG,
Ridderheim M and Cajander S: Steroid receptors and hormones in
relation to cell proliferation and apoptosis in poorly
differentiated epithelial ovarian tumors. Int J Oncol. 19:31–38.
2001.PubMed/NCBI
|
|
28
|
Ptak A and Gregoraszczuk EL: Bisphenol A
induces leptin receptor expression, creating more binding sites for
leptin, and activates the JAK/Stat, MAPK/ERK and PI3K/Akt
signalling pathways in human ovarian cancer cell. Toxicol Lett.
210:332–337. 2012. View Article : Google Scholar
|
|
29
|
Ptak A, Wrobel A and Gregoraszczuk EL:
Effect of bisphenol-A on the expression of selected genes involved
in cell cycle and apoptosis in the OVCAR-3 cell line. Toxicol Lett.
202:30–35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sirtori CR, Arnoldi A and Johnson SK:
Phytoestrogens: end of a tale? Ann Med. 37:423–438. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mense SM, Hei TK, Ganju RK and Bhat HK:
Phytoestrogens and breast cancer prevention: possible mechanisms of
action. Environ Health Perspect. 116:426–433. 2008.PubMed/NCBI
|
|
32
|
Ravindranath MH, Muthugounder S, Presser N
and Viswanathan S: Anticancer therapeutic potential of soy
isoflavone, genistein. Adv Exp Med Biol. 546:121–165. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li HQ, Luo Y and Qiao CH: The mechanisms
of anticancer agents by genistein and synthetic derivatives of
isoflavone. Mini Rev Med Chem. 12:350–362. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yi BR, Kang NH, Hwang KA, Kim SU, Jeung EB
and Choi KC: Antitumor therapeutic effects of cytosine deaminase
and interferon-beta against endometrial cancer cells using
genetically engineered stem cells in vitro. Anticancer Res.
31:2853–2861. 2011.
|
|
35
|
Yi BR, O SN, Kang NH, et al: Genetically
engineered stem cells expressing cytosine deaminase and
interferon-beta migrate to human lung cancer cells and have
potentially therapeutic anti-tumor effects. Int J Oncol.
39:833–839. 2011.PubMed/NCBI
|
|
36
|
Kim KY, Yi BR, Lee HR, et al: Stem cells
with fused gene expression of cytosine deaminase and
interferon-beta migrate to human gastric cancer cells and result in
synergistic growth inhibition for potential therapeutic use. Int J
Oncol. 40:1097–1104. 2012.
|
|
37
|
Kang NH, Yi BR, Lim SY, et al: Human
amniotic membrane-derived epithelial stem cells display anticancer
activity in BALB/c female nude mice bearing disseminated breast
cancer xenografts. Int J Oncol. 40:2022–2028. 2012.
|
|
38
|
Kang NH, Hwang KA, Yi BR, et al: Human
amniotic fluid-derived stem cells expressing cytosine deaminase and
thymidine kinase inhibits the growth of breast cancer cells in
cellular and xenograft mouse models. Cancer Gene Ther. 19:412–419.
2012. View Article : Google Scholar
|
|
39
|
Krell J, Januszewski A, Yan K and Palmieri
C: Role of fulvestrant in the management of postmenopausal breast
cancer. Expert Rev Anticancer Ther. 11:1641–1652. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kansra S, Yamagata S, Sneade L, Foster L
and Ben-Jonathan N: Differential effects of estrogen receptor
antagonists on pituitary lactotroph proliferation and prolactin
release. Mol Cell Endocrinol. 239:27–36. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pelekanou V and Leclercq G: Recent
insights into the effect of natural and environmental estrogens on
mammary development and carcinogenesis. Int J Dev Biol. 55:869–878.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Moghadam SJ, Hanks AM and Keyomarsi K:
Breaking the cycle: An insight into the role of ERalpha in
eukaryotic cell cycles. J Carcinog. 10:252011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Heldring N, Pike A, Andersson S, et al:
Estrogen receptors: how do they signal and what are their targets.
Physiol Rev. 87:905–931. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dos Santos E, Dieudonne MN, Leneveu MC, et
al: Effects of 17beta-estradiol on preadipocyte proliferation in
human adipose tissue: involvement of IGF1-R signaling. Horm Metab
Res. 42:514–520. 2010.PubMed/NCBI
|
|
45
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Foster JS and Wimalasena J: Estrogen
regulates activity of cyclin-dependent kinases and retinoblastoma
protein phosphorylation in breast cancer cells. Mol Endocrinol.
10:488–498. 1996.PubMed/NCBI
|
|
47
|
Konduri SD, Medisetty R, Liu W, et al:
Mechanisms of estrogen receptor antagonism toward p53 and its
implications in breast cancer therapeutic response and stem cell
regulation. Proc Natl Acad Sci USA. 107:15081–15086. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li J and McMurray RW: Effects of estrogen
receptor subtype-selective agonists on autoimmune disease in
lupus-prone NZB/NZW F1 mouse model. Clin Immunol. 123:219–226.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hwang KA, Park SH, Yi BR and Choi KC: Gene
alterations of ovarian cancer cells expressing estrogen receptors
by estrogen and bisphenol a using microarray analysis. Lab Anim
Res. 27:99–107. 2011. View Article : Google Scholar
|