Introduction
Epidemiological and preclinical studies have
reported that green tea consumption reduces the risk of cancer
development (1,2). The anticarcinogenic effects of green
tea have been attributed to the biological activities of its
polyphenol components. Green tea extract contains
(-)-epigallocatechin gallate (EGCG), (-)-epigallocatechin (EGC),
(-)-epicatechin gallate (ECG) and (-)-epicatechin (EC) (3). EGCG, the most abundant polyphenol in
green tea, inhibits cell proliferation and induces apoptosis in
tumor cells (4). In addition to
its cancer chemo-preventive activity, EGCG inhibits tumor invasion,
which is a crucial step in the metastasis of all solid tumors. In a
previous study, we demonstrated that treating mice with EGCG
resulted in marked inhibition of vascularity and proliferation of
human colon cancer xenografts in nude mice (5); however, the mechanisms involved have
yet to be fully elucidated.
Although the incidence rates of gastric cancer have
declined in several industrialized countries over the past few
decades, gastric cancer remains the most common cancer of the
digestive tract, with a poor prognosis and high mortality rate
(6). Due to local tissue invasion
and metastasis, radiation therapy and chemotherapy do not
significantly affect the survival or the quality of life of
patients with advanced gastric cancer. Thus, developing effective
therapeutic strategies against gastric cancer could help improve
treatment strategies.
Recepteur d’origine nantais (RON), a member of the
c-MET family of scatter factor receptors, plays an important role
in the occurrence, progression and metastasis of gastric cancer
(7). RON is activated through
ligand-dependent or -independent mechanisms, which lead to
responses associated with tumor development and metastasis
(8). Macrophage-stimulating
protein is the only RON ligand that has been identified thus far
(9). Upon ligand binding, RON
dimerizes, autophosphorylates and transduces a variety of signals
that regulate different downstream pathways, including
Ras/mitogen-activated protein kinase (MAPK), phosphatidylinositol
3-kinase, c-Jun N-terminal kinase, β-catenin and nuclear factor-κB
(10,11). Several human tumor tissues exhibit
aberrant expression and activation of RON, including tumors of the
breast, colon and prostate gland (12). It has been suggested that multiple
regulatory elements are required for full RON promoter activity and
gene expression (13). Since RON
plays a central role in multiple processes involved in cancer
progression and metastasis, it is an attractive target for
molecular-based cancer therapy.
In this study, we discovered that EGCG suppressed
RON expression in cancer cells and inhibited tumor growth in
vivo. We verified the EGCG-response elements in the RON
promoter in order to investigate the mechanism behind EGCG-mediated
regulation of RON.
Materials and methods
Cell culture and culture conditions
AGS human gastric cancer cells were obtained from
the American Type Culture Collection (Manassas, VA, USA) and MKN28
cells were obtained from the Korean Cell Line Bank (Seoul, Korea).
The TMK-1 human gastric cancer cell line was provided by Dr Eiichi
Tahara (Hiroshima University, Hiroshima, Japan). The cells were
cultured in RPMI-1640 supplemented with 10% fetal bovine serum
(FBS) and 1% penicillin-streptomycin at 37°C in an atmosphere
containing 5% CO2. AGS cells pretreated with 30
μM EGCG for 1 h were exposed to 200 nM phorbol 12-myristate
13-acetate (PMA) for 8 h and the levels of RON were analyzed by
western blot analysis to determine the effect of EGCG on the tumor
promoter.
Western blot analysis
Cells were suspended in ice-cold RIPA-M buffer with
1% NP-40 and cell lysates were prepared as previously described
(7). Cell lysate proteins (100
μg) were resolved on 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred to
nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). The blots
were blocked for at least 1 h at room temperature in blocking
buffer (5% non-fat dry milk in Tris-buffered saline containing
0.05% Tween-20; TBST). Anti-RONβ (Santa Cruz Biotechnology, Santa
Cruz, CA, USA) was diluted in blocking buffer and incubated with
the blots overnight at 4°C. The bound antibodies were detected with
a 1:3,000 dilution of horseradish peroxidase-conjugated secondary
antibody, according to the instructions of the enhanced
chemiluminescence kit (Amersham, Franklin Lakes, NJ, USA).
Construction of the RON promoter-reporter
construct
A construct of the RON promoter fragment, ∼3 kb in
length, was synthesized from human genomic DNA (Promega, Madison,
WI, USA) by polymerase chain reaction (PCR) using the primers
5′-GGTACCTAGCTGACC-3′ (forward) and 5′-GGGCCAAATTTAAGC-3′
(reverse). The amplified PCR products were ligated into the T&A
Vector (RBC Bioscience, Saskatoon, SK, Canada), then digested with
KpnI and BglII. The products were ligated into the
KpnI and BglII sites of the pGL3-Basic Vector
(Promega). A series of deletion constructs of the human RON
promoter fragments was synthesized by PCR using the pRON-Luc
plasmid as the template. The forward primer sequences were:
5′-CCAAGGGCCGGAAGA-3′ (−128/+173, pGL3-RON-301),
5′-TCGGCTGAGCGCTAA-3′ (−20/+173, pGL3-RON-193) and
5′-TCGTGCGTCCGCAGG-3′ (+50/+173, pGL3-RON-123). One reverse primer,
5′-GGGCCA AATTTAAGC-3′, was used to generate all three deletion
constructs. The amplified PCR products were ligated into the
T&A Vector and then digested with KpnI and BglII.
Subsequently, the products were ligated into the KpnI and
BglII sites of the pGL3-Basic Vector. Site-directed
mutagenesis was utilized to mutate potential transcriptional Egr-1
elements in the promoter region. Mutant promoter constructs were
generated using the pGL3-RON-301 construct as a template. The
primers used for mutagenesis (mutations underlined) were:
TCCGCCCGCC to
TCCATATGCC
(pGL3-Mt1) and CCCGCCCCCA to CCCAATTCCA (pGL3-Mt2). The mutated
nucleotide sequences of all mutant constructs were confirmed by DNA
sequencing.
RON promoter-reporter assay
Transcriptional regulation of RON was examined by
transient transfection of a RON promoter-luciferase reporter
construct (pGL3-RON). Gastric cancer cells (5×105) were
seeded and grown until they reached 60–70% confluence and pGL3-RON
wild-type and deletion mutants were transfected into the cells
using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA),
according to the manufacturer’s protocol. The significance of the
Egr-1 binding site as an EGCG-response element in the regulation of
RON was examined by co-transfecting the cells with pGL3-RON and
Egr-1 expression plasmids containing full-length complementary DNA
(cDNA) coding for human Egr-1 (a gift from Dr Young Han Lee, Konkuk
University, Seoul, Korea). The pRL-null plasmid encoding
Renilla luciferase was included in all the samples to
monitor transfection efficiency. At 24 h post-transfection, the
levels of firefly and Renilla luciferase activity were
measured sequentially from a single sample using the Dual-Glo
Luciferase Assay system (Promega). Firefly luciferase activity was
normalized to Renilla activity and the relative amount of
luciferase activity in the untreated cells was designated as 1.
Reverse transcription-PCR
Total RNA was extracted from AGS cells using TRIzol
reagent (Invitrogen). One microgram of total RNA was used for
first-strand cDNA synthesis using random primers and superscript
reverse transcriptase (Invitrogen). The cDNA was subjected to PCR
amplification with the Egr-1 and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) primer sets. The specific primers sequences
were: Egr-1, sense, 5′-CAGTGGCCTAGTGAGCATGA-3′ and
antisense, 5′-CCGCAAGTGGATCTTGGTAT-3′ (786 bp); GAPDH, sense,
5′-TTGTTGCCATCAATGACCCC-3′ and antisense,
5′-TGACAAAGTGGTCGTTGAGG-3′ (836 bp). The PCR conditions were:
denaturation at 94°C for 20 sec, annealing at 53°C for 20 sec and
extension at 72°C for 50 sec.
Chromatin immunoprecipitation assay
AGS cells (2×106), grown in 6-well
plates, were cross-linked with 0.5% (v/v) formaldehyde at 37°C for
5 min. The cells were sonicated for 3×20 sec prior to
centrifugation at 16,000 x g for 15 min at 4°C. The specific
DNA-bound transcription factor complexes were precipitated with 20
μl anti-Egr-1 at 4°C overnight prior to the addition of
Protein A agarose beads. The proteins were removed from the DNA by
digestion with 10 μg/ml Proteinase K at 65°C for 30 min. The
DNA was recovered from the solution using the QIAquick PCR
Purification kit (Qiagen Inc., Valencia, CA, USA) and eluted in 50
μl sterile water. Eluted DNA (20 μl) was subjected to
PCR with forward, −5′-AGGAGCCAGGCCTCCAAGGGC-3′ and reverse,
−5′-TCCCGACAGCCCCAAGATAGC-3′ primers, which flank the Egr-1 binding
sites.
Small interfering RNA transfection
Gene silencing was performed using human Egr-1
(sc-29303; Santa Cruz Biotechnology) and human RON
sequence-specific duplex small interfering RNA (siRNA) (sc-36434).
Briefly, 20 nM of siRNA oligonucleotides and 2 μl of
Lipofectamine RNAiMAX (Invitrogen) were mixed with 100 μl of
Opti-MEM serum-free medium (Hyclone, Logan, UT, USA) for each
transfection reaction in two separate tubes and incubated for 5 min
at room temperature. Subsequently, the contents of the two tubes
were combined and allowed to form siRNA-Lipofectamine complexes for
30 min at room temperature. A 900 μl volume of AGS cells
cultured in serum-free medium was combined with the
siRNA-Lipofectamine mix, plated in the wells of a 6-well tissue
culture dish and placed in a 37°C, 5% CO2 incubator for
5 h. The medium was replaced with normal growth medium.
Matrigel invasion assay
The cell invasion assay was carried out using
BioCoat Matrigel Invasion Chambers (Becton-Dickinson, Bedford, MA,
USA) with 10% FBS as the chemoattractant in the lower chamber. AGS
cells (105) in 300 μl were allowed to invade the
Matrigel for 24 h. The non-invading cells on the upper surface of
each membrane were removed from the chamber and the invading cells
on the lower surface of each membrane were stained with the
Diff-Quick Stain kit (Becton-Dickinson). Following two washes with
water, the chambers were allowed to air-dry. The number of invading
cells was counted using a phase-contrast microscope.
Animal model
Eight-week-old male athymic nude mice
(BALB/cnu/nu; Charles River, Sulzfeld, Germany) were
used for the experiments, as approved by the Institutional Animal
Care and Use Committee of the University of Regensburg and the
regional authorities. Experiments were conducted according to the
Guidelines for the Welfare of Animals in Experimental Neoplasia by
the United Kingdom Coordinating Committee on Cancer Research. The
effects of EGCG inhibition on the growth of TMK-1 human gastric
cancer cells were investigated in a subcutaneous xenograft tumor
model. TMK-1 cells (1×106) were injected subcutaneously
into the right flank of nude mice. Mice were randomized (n=8 per
group) and assigned to treatment groups. Intraperitoneal injections
of EGCG (1 /mg/mouse, twice/week) were initiated on Day 1. The
control group (n=8) was treated with the same dosage of EC. Tumor
diameters were measured every other day and tumor volumes were
calculated (width2 × length × 0.5). The experiment was
terminated on Day 27 following tumor cell injection, and the tumors
were then excised, weighed and prepared for western blot
analyses.
Results
Effect of EGCG on PMA-induced RON
expression in gastric cancer cells
To examine whether green tea catechins inhibit
PMA-induced RON expression in gastric cancer cells, AGS human
gastric carcinoma cells were pretreated with 30 μM catechins
for 1 h prior to an 8-h incubation with 200 nM of PMA, and the RON
protein levels were measured by western blot analysis. EGCG at 30
μM inhibited PMA-induced RON protein expression. However,
other tea catechins such as EC, ECG and EGC inhibited RON
expression only slightly at the same concentrations (Fig. 1A). We then examined the effect of
EGCG on transcriptional regulation of the RON gene induced by PMA.
AGS cells were transiently transfected with the promoter-reporter
construct (pGL3-RON) of the human RON gene fused to the luciferase
gene. AGS cells transfected with pGL3-RON exhibited an ∼3.5-fold
increase in promoter activity following PMA treatment (Fig. 1B). When the transfected cells were
pretreated with 30 μM catechins prior to PMA treatment, only
EGCG significantly inhibited PMA-induced RON promoter activity
(Fig. 1B). Catechins at the same
concentrations did not affect cell viability (data not shown).
TMK-1, MKN28 and AGS gastric cells were used to explore whether
EGCG was able to inhibit RON expression in various gastric cancer
cells. As shown in Fig. 1C and D,
EGCG inhibited PMA-induced RON expression and promoter activity in
TMK-1, MKN28 and AGS cells. Collectively, these results demonstrate
that EGCG suppressed the expression of the RON gene and reduced its
promoter activity in human gastric cancer cells.
 | Figure 1Effect of EGCG on PMA-induced RON
expression in gastric cancer cells. (A) AGS cells were exposed to
200 nM of PMA for 8 h and the levels of RON were determined by
western blot analysis following pretreatment with 30 μM of
EC, ECG, EGC and EGCG for 1 h. (B) AGS cells were transiently
transfected with the pGL3-RON reporter (pRON-Luc, −128/+173)
construct. Following pretreatment with 30 μM of EC, ECG, EGC
and EGCG for 1 h, the transfected cells were exposed to 200 nM of
PMA for 8 h and luciferase activity was determined using a
luminometer. (C) Following pretreatment with 30 μM of EGCG
for 1 h, TMK-1, MKN28 and AGS cells were exposed to 200 nM of PMA
for 8 h and the levels of RON were determined by western blot
analysis. (D) TMK-1, MKN28 and AGS cells were transiently
transfected with the pGL3-RON reporter (pRON-Luc, −128/+173)
construct. Following pretreatment with 30 μM of EGCG for 1
h, the transfected cells were exposed to 200 nM of PMA for 8 h and
luciferase activity was determined using a luminometer. Data are
the means ± standard deviation from triplicate measurements.
*P<0.05 vs. PMA. |
EGCG-response elements in the gastric
cancer cell RON promoter
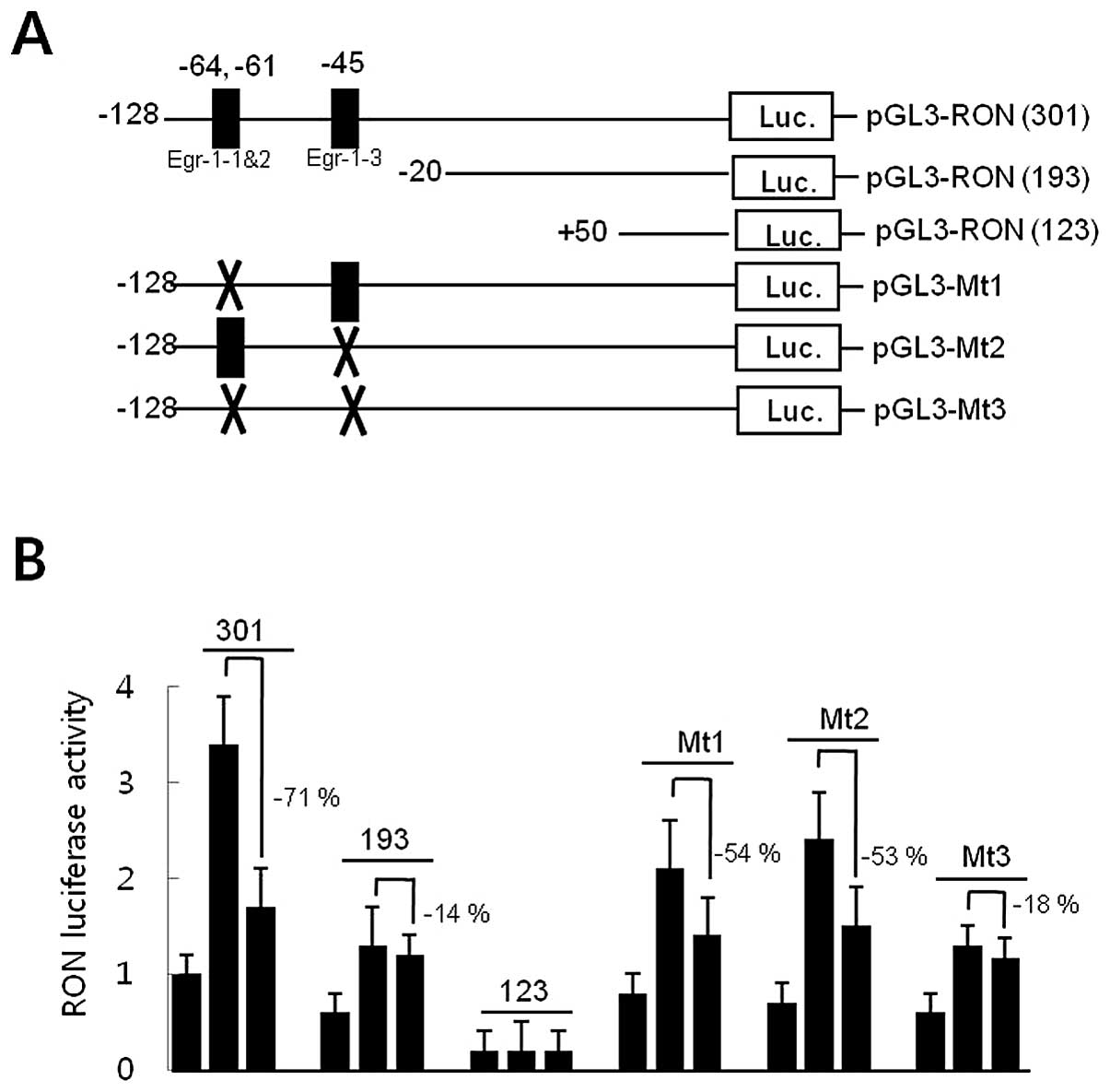
Promoter deletion analyses were performed to locate
cis-response elements in the RON promoter in response to
EGCG, in order to explore the underlying molecular mechanisms by
which EGCG reduced RON gene promoter activity in AGS gastric cancer
cells. AGS cells were transfected with pGL3-RON promoter-reporter
constructs of different lengths (Fig.
2A). AGS cells were pretreated with 30 μM of EGCG for 1
h prior to an 8-h incubation with 200 nM of PMA, and luciferase
activity was measured. As shown in Fig. 2B, EGCG significantly reduced
luciferase activity by 71% in cells transfected with pGL3-RON
(301). However, cells transfected with pGL3-RON (193) resulted in a
decrease of 14% in response to EGCG, suggesting that the −128 to
−20 DNA fragment in the RON promoter might contain the
EGCG-response element(s). A computer-aided search revealed three
consensus Egr-1 binding sites in the −128 to −20 DNA fragment of
the RON promoter. Site-directed mutagenesis of the Egr-1 binding
site was generated in the pGL3-RON plasmids (301) to examine the
role of the Egr-1 binding site in the inhibition of RON promoter
activity by EGCG (Fig. 2A). A
construct containing a mutation in the core sequence of the Egr-1-1
and -2-binding motif (CCCGCCCCCA to CCCAATTCCA, pGL3-Mt1) and the
Egr-1-3-binding motif (TCCGCCCGCC to TCCATATGCC, pGL3-Mt2) reduced PMA
responsiveness to ∼54 and 53%, respectively, of that of the
wild-type construct. However, a double-mutant promoter, containing
mutations in Egr-1-1 and -2, as well as in Egr-1-3 (pGL3-Mt3),
resulted in an 18% decrease in the response to EGCG, indicating
that the presence of the wild-type Egr-1 binding sites was required
for EGCG to reduce RON promoter activity (Fig. 2B). Collectively, these results
suggest that the Egr-1 binding sites in the RON promoter might be
the EGCG-response element acting as a cis-element to
regulate promoter activity.
Role of Egr-1 in the inhibition of RON by
EGCG in gastric cancer cells
Egr-1 mRNA level was determined by RT-PCR to verify
the inhibitory mechanisms of EGCG on Egr-1 activity in AGS gastric
cancer cells. As shown in Fig. 3A,
EGCG dose-dependently reduced PMA-induced Egr-1 expression at the
transcriptional level. A chromatin immunoprecipitation assay was
performed to further determine whether EGCG inhibits Egr-1 binding
to the putative Egr-1-binding sequence in the RON promoter. AGS
cell chromatin was immunoprecipitated with rabbit anti-Egr-1
antibody and the resulting immunoprecipitates were analyzed by PCR
using primers flanking the Egr-1-binding sequences (−407 to −112)
of the RON promoter. An evident increase in DNA band intensity was
observed in cells treated with PMA and anti-Egr-1 antibody, but not
when normal rabbit IgG was used (Fig.
3B). When the cells were pretreated with 0–30 μM of EGCG
prior to PMA treatment, the induction of Egr-1-DNA binding by PMA
was inhibited in a dose-dependent manner (Fig. 3B). To explore the role of Egr-1 in
regulating the RON promoter activity, AGS cells were co-transfected
with the RON promoter-luciferase reporter and the Egr-1 cDNA
expression plasmid (pEgr-1cDNA) at the indicated concentrations. As
shown in Fig. 3C, forced
expression of Egr-1 cDNA dose-dependently eliminated the EGCG
inhibitory effect, suggesting that the increase in the abundance of
cellular Egr-1 eradicated the inhibitory effect of EGCG on RON
promoter activity in AGS cells.
Effect of EGCG, siRON and siEgr-1 on
PMA-induced gastric cancer cell invasion
It has been suggested that RON expression is
essential for the invasive phenotype of cancer cells. The role of
PMA-induced RON in AGS cell invasion was evaluated in a modified
Boyden invasion chamber. As shown in Fig. 4, cell invasiveness increased
∼2-fold following incubation with PMA. However, cells transfected
with RON siRNA and Egr-1 siRNA partially lost the Matrigel
invasiveness induced by PMA. These results suggest that PMA-induced
Egr-1 and RON in AGS cells stimulated AGS cell invasion. In
addition, the effect of EGCG on AGS cell invasion stimulated by PMA
was examined. Cells pretreated with 30 μM of EGCG also
partially abrogated PMA-induced cell invasion. Our results suggest
that treating AGS cells with EGCG reduced RON by regulating Egr-1
and that these events contributed to a reduction in AGS gastric
cancer cell invasion.
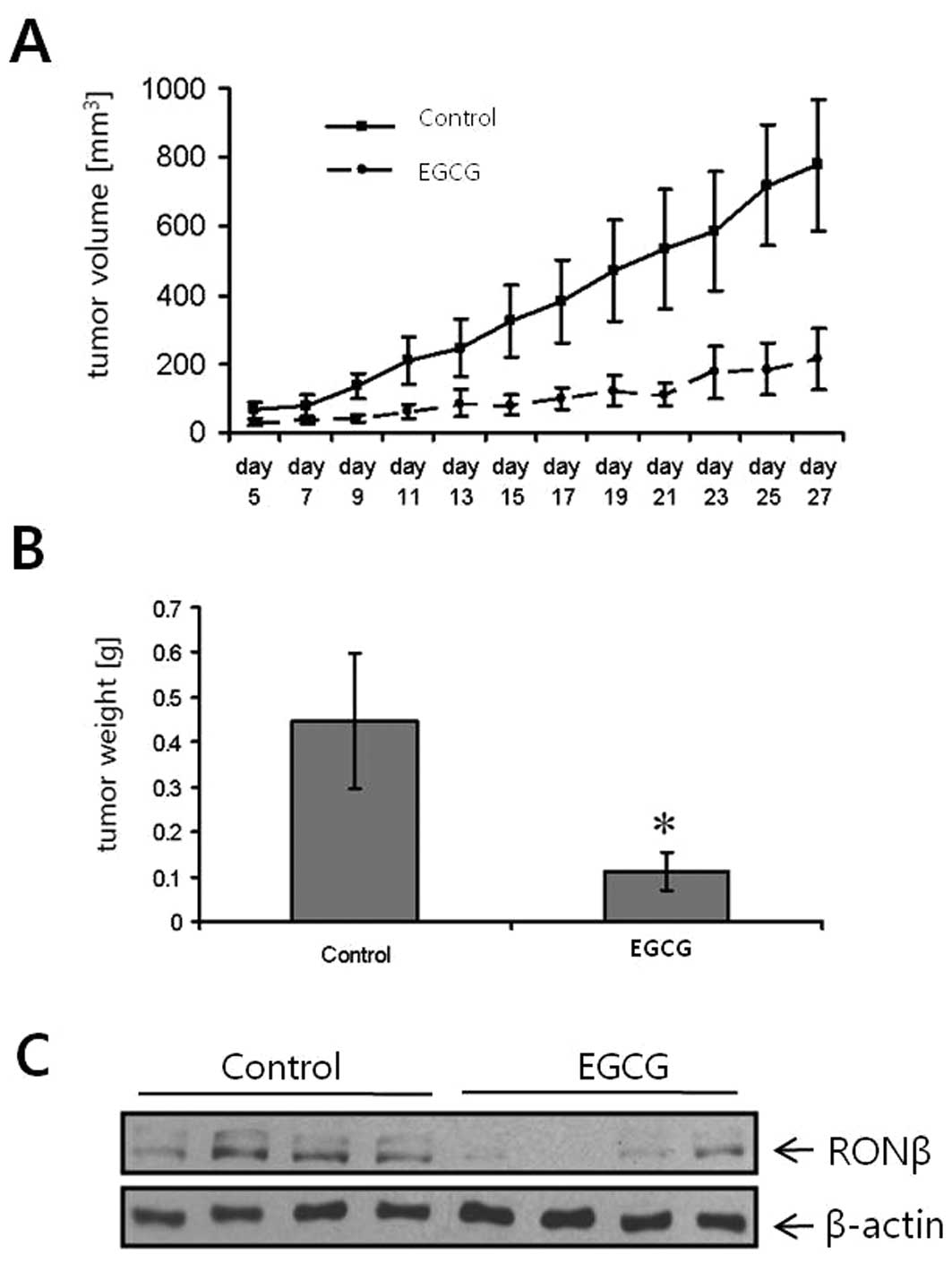
Effects of EGCG on tumor growth and RON
expression in vivo
The effects of EGCG on cancer growth in vivo
were determined in a subcutaneous gastric (TMK-1) cancer xenograft
model. Treatment with EGCG (1 mg in 0.1–0.2 ml PBS/day/mouse)
inhibited gastric tumor growth (Fig.
5A). The control group was treated with the same dosage of EC.
The potent growth-inhibitory effect was also mirrored by the
weights of the excised tumors, which were significantly decreased
in the EGCG-treated mice (Fig.
5B). Notably, in vivo RON expression levels were also
substantially decreased in mice treated with EGCG, compared to
controls (Fig. 5C). Mouse body
weight did not differ among treatment groups (data not shown). We
concluded that EGCG sufficiently inhibited gastric cancer cell
growth in vivo. Our data provide the first evidence that the
inhibition of tumor growth by EGCG may, in part, be mediated by
impairing the RON system.
Discussion
We previously delineated the role of RON in the
acquisition of the gastric cancer cell invasive phenotype and we
also identified the critical regulatory elements that are necessary
for oncogenic RON tyrosine kinase promoter activity and gene
expression (14). Tyrosine kinase
receptors regulate multiple processes involved in tumor progression
and metastasis, making them attractive molecular therapy targets.
RON is mainly transcribed at relatively low levels in normal
epithelial cells. However, the levels of RON expression in
malignant epithelial cells increase severalfold compared with those
in benign epithelium. This aberrant expression and activation of
RON has been observed in human cancer and is responsible for
various malignant behaviors in breast, colon and ovary cancer
(12,15). Increased RON expression is strongly
correlated with phosphorylation and tumor invasiveness, suggesting
that increased RON expression plays a role in the progression of
carcinomas to an invasive-metastatic phenotype (16).
In this study, we demonstrated that EGCG suppressed
RON expression by regulating Egr-1 in gastric cancer cells and that
these events may contribute to the reduction of tumor growth in
vivo. Interest in green tea as an anticancer agent in humans
has increased for several reasons: i) several epidemiological
studies have reported that green tea lowers the risk of cancer,
when consumed in large amounts (17); ii) green tea inhibits the
development and progression of skin, lung, mammary gland and
gastrointestinal tract cancer in animal models (18); iii) green tea extracts, including
purified EGCG, stimulate apoptosis in various cancer cell lines,
such as stomach, prostate, lymphoma and lung in
vitro(5); and iv) green tea
consumption is associated with few adverse events and it can be
easily obtained at a low cost (19). The anticancer effects of green tea
have been attributed to the biological activities of its polyphenol
components. EGCG is the most abundant of the green tea polyphenols,
accounting for >40% of the total polyphenolics (3). Several molecular mechanisms have been
suggested for the observed anticancer effect of EGCG, including
suppression of ligand binding to the epidermal growth factor
receptor (EGFR) (20), inhibition
of protein kinase C (21),
lipoxygenase and cyclooxygenase activities (3), induction of apoptotic cell death and
arrest of the tumor cell cycle (5).
The suppression of RON expression by EGCG occurred
at the transcriptional level, as shown by the transient
transfection study using the RON promoter-reporter construct
(Fig. 1). It has previously been
suggested that multiple regulatory elements are required for full
RON promoter activity and a portion of the 5′-flanking region of
the RON gene has been cloned (13). Similar to numerous tyrosine kinase
receptor gene promoters, the RON promoter also lacks distinct TATA
box and CCAAT sequences. However, it contains several GC boxes,
seven Sp1-binding sites, four retinoblastoma control elements,
three IL-6 response elements and two AP-2 elements (15). Putative Egr-1-binding motifs in the
RON gene promoter region of gastric cancer cells have also been
reported (14). The role of the
Egr-1 signaling pathway in the inhibition of RON gene expression by
EGCG was evaluated in the present study. Site-directed mutagenesis
of Egr-1 binding sites resulted in a decrease in the effects of
EGCG on RON expression (Fig. 2).
In addition, forced Egr-1 expression eliminated the inhibitory
effects of EGCG in a dose-dependent manner (Fig. 3). These results indicate that
interruption of Egr-1 by EGCG played a critical role in suppressing
RON gene promoter activity. A similar finding was reported by Fu
and Chen (22), who observed that
EGCG suppresses EGFR gene expression in rat hepatic stellate cells
by reducing Egr-1 activity. They suggested that inhibition of Egr-1
transactivation activity and the EGFR gene promoter activity by
EGCG occurs through an interruption in the ERK signaling pathway.
By contrast, regarding the effect of EGCG on Egr-1 in human
pulmonary epithelial cells, Moon et al reported that EGCG
induced Egr-1 expression and mediated Egr-1 nuclear translocation
via the ERK signaling pathway in pulmonary epithelial cells
(23). Comparing these findings
with the downregulation of Egr-1 by EGCG demonstrated by our
results, it can be hypothesized that Egr-1 and ERK responses are
differentially regulated by EGCG, depending on the tissue
environment and external stimuli.
The Egr-1 transcription factor is an immediate-early
response gene that is rapidly induced by various growth factors,
cytokines and DNA-damaging agents, and modulates cell
proliferation, differentiation, apoptosis and inflammation in a
variety of cells (24,25). The role of Egr-1 in tumor
development might be largely dependent on tissue type, since Egr-1
is highly expressed and plays an essential role in tumor growth and
survival in various types of cancer (26,27).
The exact mechanisms by which EGCG inhibits the activation of Egr-1
are unknown. One possible explanation is that EGCG inhibits the
kinases that are involved in the activation Egr-1. Numerous studies
have suggested a causal relationship between ERK activation and
induction of Egr-1 gene expression (28,29).
Activation of ERK induces Egr-1 gene expression mediated by the
transcription factor Elk-1, an ERK substrate (29). Elk-1 binds to multiple serum
response elements and their adjacent Ets motifs located in the
Egr-1 gene promoter and stimulates promoter activity, leading to
Egr-1 transcription (29).
Previous studies suggested that MAPK is one of the target molecules
in the signaling cascades regulated by EGCG. It was previously
discovered that EGCG treatment suppresses ERK phosphorylation,
resulting in the downregulation of vascular endothelial growth
factor in HT29 human colon adenocarcinoma cells (5). Katiyar et al(30) demonstrated that pretreatment of
human epidermal keratinocytes with EGCG inhibits ultraviolet
B-induced hydrogen peroxide production and hydrogen
peroxide-mediated phosphorylation of the MAPK signaling pathway.
EGCG modulates multiple signaling pathways, and the tyrosine kinase
receptor (30), which is a strong
metal ion chelator (31). Since
certain kinases depend on divalent cations for their activity, EGCG
may inhibit the activity of receptor kinases by chelating divalent
cations. On the other hand, the EGCG inhibitory effects could be
considered ‘non-specific’. It has been demonstrated that EGCG
non-specifically binds proteins and modulates enzyme activity,
leading to inhibition of cell cycle-related kinases, MAPK and the
activity of receptor tyrosine kinases (32). Our results do not exclude the role
of other signaling pathways in EGCG-induced suppression of RON gene
expression.
EGCG administration inhibits carcinogenesis in
several animal models. Our results suggest, for the first time,
that EGCG may exert its anticancer effects by inhibiting RON,
supporting a role for green tea in cancer chemoprevention. The
suitability of RON as a therapeutic target has been demonstrated in
an experimental study that used a novel function-blocking antibody
(33). The growth-inhibitory
effects of the RON antibody have been validated in preclinical
tumor models and a comprehensive analysis was performed on RON
expression in various human cancer entities. The authors concluded
that inhibiting RON is a potentially useful target for human cancer
therapy. However, another report revealed that the
growth-inhibitory effects of RON inhibitors may be transient
(34). In that study,
Logan-Collins et al demonstrated that silencing RON reduces
tumor growth and renders cancer cells susceptible to
chemotherapeutic agents; however, downregulation of RON is lost
over time. This transient effect was linked to an increase in c-MET
and EGFR expression, suggesting that these two oncogenic receptor
systems provide an escape mechanism from RON silencing in cancer
cells. The EGFR and c-MET findings are particularly relevant to our
study, since both are possibly inhibited by EGCG. Thus, further
studies are required to elucidate the detailed mechanism by which
EGCG inhibits RON activity and to examine whether EGCG exerts the
same effects in human cancer.
Acknowledgements
This study was supported by a research
grant (0720570) from the National Cancer Center, by a Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
(2010-0009910) and by a Medical Research Center grant
(2012-000-9442) from the Korean Science and Engineering
Foundation.
References
|
1
|
Sasazuki S, Tamakoshi A, Matsuo K, Ito H,
Wakai K, Nagata C, Mizoue T, Tanaka K, Tsuji I, Inoue M and Tsugane
S; Research Group for the Development and Evaluation of Cancer
Prevention Strategies in Japan: Green tea consumption and gastric
cancer risk: an evaluation based on a systematic review of
epidemiologic evidence among the Japanese population. Jpn J Clin
Oncol. 42:335–346. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yuan JM, Sun C and Butler LM: Tea and
cancer prevention: epidemiological studies. Pharmacol Res.
64:123–135. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stoner GD and Mukhtar H: Polyphenols as
cancer chemopreventive agents. J Cell Biochem (Suppl). 22:169–180.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yoo HG, Shin BA, Park JC, Kim HS, Kim WJ,
Chay KO, Ahn BW, Park RK, Ellis LM and Jung YD: Induction of
apoptosis by the green tea flavonol (-)-epigallocatechin-3-gallate
in human endothelial ECV 304 cells. Anticancer Res. 22:3373–3378.
2002.PubMed/NCBI
|
|
5
|
Jung YD, Kim MS, Shin BA, Chay KO, Ahn BW,
Liu W, Bucana CD, Gallick GE and Ellis LM: EGCG, a major component
of green tea, inhibits tumour growth by inhibiting VEGF induction
in human colon carcinoma cells. Br J Cancer. 84:844–850. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nagini S: Carcinoma of the stomach: A
review of epidemiology, pathogenesis, molecular genetics and
chemoprevention. World J Gastrointest Oncol. 4:156–169. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park JS, Park JH, Khoi PN, Joo YE and Jung
YD: MSP-induced RON activation upregulates uPAR expression and cell
invasiveness via MAPK, AP-1 and NF-κB signals in gastric cancer
cells. Carcinogenesis. 32:175–181. 2011.PubMed/NCBI
|
|
8
|
Feres KJ, Ischenko I and Hayman MJ: The
RON receptor tyrosine kinase promotes MSP-independent cell
spreading and survival in breast epithelial cells. Oncogene.
28:279–288. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang MH, Ronsin C, Gesnel MC, Coupey L,
Skeel A, Leonard EJ and Breathnach R: Identification of the ron
gene product as the receptor for the human macrophage stimulating
protein. Science. 266:117–119. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen YQ, Zhou YQ, Angeloni D, Kurtz AL,
Qiang XZ and Wang MH: Overexpression and activation of the RON
receptor tyrosine kinase in a panel of human colorectal carcinoma
cell lines. Exp Cell Res. 261:229–238. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang J, Rajput A, Kan JL, Rose R, Liu XQ,
Kuropatwinski K, Hauser J, Beko A, Dominquez I, Sharratt EA,
Brattain L, Levea C, Sun FL, Keane DM, Gibson NW and Brattain MG:
Knockdown of Ron kinase inhibits mutant phosphatidylinositol
3-kinase and reduces metastasis in human colon carcinoma. J Biol
Chem. 284:10912–10922. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Leonis MA, Thobe MN and Waltz SE:
Ron-receptor tyrosine kinase in tumorigenesis and metastasis.
Future Oncol. 3:441–448. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thangasamy A, Rogge J and Ammanamanchi S:
Recepteur d’origine nantais tyrosine kinase is a direct target of
hypoxiainducible factor-1alpha-mediated invasion of breast
carcinoma cells. J Biol Chem. 284:14001–14010. 2009.
|
|
14
|
Lee KE, Park JS, Khoi PN, Joo YE, Lee YH
and Jung YD: Upregulation of recepteur d’origine nantais tyrosine
kinase and cell invasiveness via early growth response-1 in gastric
cancer cells. J Cell Biochem. 113:1217–1223. 2012.
|
|
15
|
Thangasamy A, Rogge J and Ammanamanchi S:
Regulation of RON tyrosine kinase-mediated invasion of breast
cancer cells. J Biol Chem. 283:5335–5343. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou D, Pan G, Zheng C, Zheng J, Yian L
and Teng X: Expression of the RON receptor tyrosine kinase and its
association with gastric carcinoma versus normal gastric tissues.
BMC Cancer. 8:3532008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kono S, Ikeda M, Tokudome S and Kuratsune
M: A case-control study of gastric cancer and diet in northern
Kyushu, Japan. Jpn J Cancer Res. 79:1067–1074. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rogers AE, Hafer LJ, Iskander YS and Yang
S: Black tea and mammary gland carcinogenesis by
7,12-dimethylbenz(a)anthracene in rats fed control or high fat
diets. Carcinogenesis. 19:1269–1273. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fujiki H, Suganuma M, Okabe S, Sueoka E,
Suga K, Imai K, Nakachi K and Kimura S: Mechanistic findings of
green tea as cancer preventive for humans. Proc Soc Exp Biol Med.
220:225–228. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liang YC, Lin-shiau SY, Chen CF and Lin
JK: Suppression of extracellular signals and cell proliferation
through EGF receptor binding by (-)-epigallocatechin gallate in
human A431 epdeidermoid carcinoma cells. J Cell Biochem. 67:55–65.
1997. View Article : Google Scholar
|
|
21
|
Kitano K, Nam KY, Kimura S, Fujiki H and
Imanishi Y: Sealing effects of (-)-epigallocatechin gallate on
protein kinase C and protein phosphatase 2A. Biophys Chem.
65:157–164. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fu Y and Chen A: The phyto-chemical
(-)-epigallocatechin gallate suppresses gene expression of
epidermal growth factor receptor in rat hepatic stellate cells in
vitro by reducing the activity of Egr-1. Biochem Pharmacol.
72:227–238. 2006. View Article : Google Scholar
|
|
23
|
Moon Y, Lee M and Yang H: Involvement of
early growth response gene 1 in the modulation of microsomal
prostaglandin E synthase 1 by epigallocatechin gallate in A549
human pulmonary epithelial cells. Biochem Pharmacol. 73:125–135.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thiel G and Cibelli G: Regulation of life
and death by the zinc finger transcription factor Egr-1. J Cell
Physiol. 193:287–292. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shin SY, Kim JH, Baker A, Lim Y and Lee
YH: Transcription factor Egr-1 is essential for maximal matrix
metalloproteinase-9 transcription by tumor necrosis factor alpha.
Mol Cancer Res. 8:507–519. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Keates S, Keates AC, Nath S, Peek RM Jr
and Kelly CP: Trans-activation of the epidermal growth factor
receptor by cag+ Helicobacter pylori induces
upregulation of the early growth response gene Egr-1 in gastric
epithelial cells. Gut. 54:1363–1369. 2005.PubMed/NCBI
|
|
27
|
Ma J, Ren Z, Ma Y, Xu L, Zhao Y, Zheng C,
Fang Y, Xue T, Sun B and Xiao W: Targeted knockdown of EGR-1
inhibits IL-8 production and IL-8-mediated invasion of prostate
cancer cells through suppressing EGR-1/NF-kappaB synergy. J Biol
Chem. 284:34600–34606. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kaufmann K and Thiel G: Epidermal growth
factor and platelet-derived growth factor induce expression of
Egr-1, a zinc finger transcription factor, in human malignant
glioma cells. J Neurol Sci. 189:83–91. 2001. View Article : Google Scholar
|
|
29
|
Cohen DM, Gullans SR and Chin WW: Urea
inducibility of egr-1 in murine inner medullary collecting duct
cells is mediated by the serum response element and adjacent Ets
motifs. J Biol Chem. 271:12903–12908. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Katiyar SK, Afaq F, Azizuddin K and
Mukhtar H: Inhibition of UVB-induced oxidative stress-mediated
phosphorylation of mitogen-activated protein kinase signaling
pathways in cultured human epidermal keratinocytes by green tea
polyphenol (-)-epigallocatechin-3-gallate. Toxicol Appl Pharmacol.
176:110–117. 2001. View Article : Google Scholar
|
|
31
|
Khan HY, Zubair H, Ullah MF, Ahmad A and
Hadi SM: Oral administration of copper to rats leads to increased
lymphocyte cellular DNA degradation by dietary polyphenols:
implications for a cancer preventive mechanism. Biometals.
24:1169–1178. 2011. View Article : Google Scholar
|
|
32
|
Lin JK: Cancer chemoprevention by tea
polyphenols through modulating signal transduction pathways. Arch
Pharm Res. 25:561–571. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
O’Toole JM, Rabenau KE, Burns K, Lu D,
Mangalampalli V, Balderes P, Covino N, Bassi R, Prewett M,
Gottfredsen KJ, Thobe MN, Cheng Y, Li Y, Hicklin DJ, Zhu Z, Waltz
SE, Hayman MJ, Ludwig DL and Pereira DS: Therapeutic implications
of a human neutralizing antibody to the macrophage-stimulating
protein receptor tyrosine kinase (RON), a c-MET family member.
Cancer Res. 66:9162–9170. 2006.
|
|
34
|
Logan-Collins J, Thomas RM, Yu P, Jaquish
D, Mose E, French R, Stuart W, McClaine R, Aronow B, Hoffman RM,
Waltz SE and Lowy AM: Silencing of RON receptor signaling promotes
apoptosis and gemcitabine sensitivity in pancreatic cancers. Cancer
Res. 70:1130–1140. 2010. View Article : Google Scholar : PubMed/NCBI
|