Introduction
Human colorectal cancer (CRC) is one of the most
frequently diagnosed cancers in the Western world and a leading
cause of mortality in the USA. Genetic events (mutations,
deletions, genome amplifications and chromosome translocations) are
involved in the initiation and progression of CRC and their
stepwise accumulation is a driving force of malignancies (1). Epigenetic regulation also plays a
critical role in the pathogenesis of CRC. DNA methylation is a
component of the epigenetic gene-silencing complex (2), whereas histone (H3 and H4)
post-translational modifications comprise a ubiquitous component of
rapid epigenetic changes (3).
Epigenetic changes are associated with altered transcription
(4). Metastasis correlates with
the loss of epithelial differentiation, induction of epithelial
mesenchymal transition and the acquisition of a migratory
phenotype, which are controlled by epigenetic alterations caused by
the dysregulation of the transcriptome in CRC (4).
The basic nucleosome unit has four core histone
proteins (H2A, H2B, H3 and H4). Histones H3 and H4 are generally
associated with active gene transcription. Their acetylation levels
are crucial with respect to the chromatin status and regulation of
gene expression (5). Using the
H3K4 demethylase, jumonji, AT rich interactive domain 1B
(JARID1B), as a biomarker, a small subpopulation of
tumor-initiating cells was isolated from a melanoma sample
(6). JARID1B depletion has
been shown to eliminate melanoma cell growth (6). Therefore, in this study, we
investigated the effect of JARID1B depletion by lentiviral
transfer of small hairpin RNA (shRNA) molecules on CRC cells. We
identified a novel phenotype and cellular senescence in CRC induced
by continuous JARID1B depletion, as well as tumor
elimination and regression, which suggests a potential role for
JARID1B in CRC diagnosis and therapy.
Materials and methods
Immunohistochemistry
Immunohistochemical staining was performed on
4-μm sections of formalin-fixed, paraffin-embedded surgical
tumor samples. The sections were mounted, deparaffinized in xylene
and rehydrated in descending concentrations of ethanol. Antigen
retrieval was performed using citrate buffer (10 mM, pH 6.0) heated
in a pressure cooker for 5 min. The blocking of endogenous
peroxidases was accomplished by incubating the sections in 3%
hydrogen peroxide (H2O2; Wako Pure Chemical
Industries, Ltd.) for 5 min. The sections were incubated with
rabbit anti-JARID1B antibody (1:100; Novus Biologicals) overnight
at 4°C. Immunostaining for JARID1B was performed using the Envision
+ Dual Link System and Vectastain ABC kit (Vector Laboratories)
according to the manufacturer’s instructions. The sections were
counterstained with hematoxylin and eosin.
Cell culture
Three CRC cell lines (Colo201, DLD1 and HCT116) were
used. Colo201 and DLD1 cells were cultured in RPMI-1640
supplemented with 10% fetal bovine serum (FBS). HCT116 and human
embryonic kidney (HEK)-293T cells were cultured in Dulbecco’s
modified Eagle’s medium (DMEM) supplemented with 10% FBS.
Transfection was performed using FuGENE-6 (Roche) transfection
reagent according to the manufacturer’s instructions, followed by
lentiviral production in HEK-293T cells and viral infection
(Roche).
Proliferation and MTT assays
Quantification of cell proliferation was based on
measurements of bromodeoxyuridine (BrdU) incorporation during DNA
synthesis in replicating (cycling) cells using the BrdU Cell
Proliferation ELISA kit (colorimetric) (Roche). The cells
(1.0×105) were incubated with
0–1×10−2μM 5-fluorouracil (5-FU; Kyowa Hakko
Kirin Co., Ltd.) for 48 h and analyzed using the Cell Proliferation
kit I (MTT; Roche).
Flow cytometry and cell sorting
Allophycocyanin (APC)-conjugated anti-human CD44 (BD
Biosciences) and fluorescein isothiocyanate-conjugated anti-human
aldehyde dehydrogenase (ALDH; the Aldefluor kit, Aldagen) were used
to characterize cancer cells. Labeled cells (1×106) were
analyzed using the BD FACSAria II cell sorter system
(Becton-Dickinson), followed by data analysis using the Diva
program (Becton-Dickinson). The fluorescent ubiquitination-based
cell cycle indicator (Fucci)-G1 DsRed2 contains a fragment of human
Cdt1 (amino acids 30–120), which is ubiquitinated by the ubiquitin
ligase complex SCFskp2 during the S and G2 phases and
degraded by proteasomes, thereby denoting the G1 phase (7). Fucci-S/G2/M Green contains a fragment
of human geminin (amino acids 1–110) linked to enhanced green
fluorescent protein (EGFP), which is ubiquitinated by the E3 ligase
complex APCcdh1 and degraded by proteasomes during the M
and G1 phases, denoting the S, G2 and M phases (7). DsRed2 and mKO2 or EGFP and mAG were
excited by 488-nm laser lines and their emission was detected with
530/30BP and 585/42BP filters, respectively.
Reactive oxygen species (ROS) assay and
senescence-associated (SA) β-galactosidase (SA-β-gal) analysis
The ROS assay was performed as described previously
(8). To evaluate the effects of
ROS, 10 μM N-acetyl cysteine (NAC; Wako Pure Chemical
Industries, Ltd.), a general antioxidant and ROS inhibitor, was
added. The cells (2×105) were treated with 20–100
μM H2O2 (Wako Pure Chemical
Industries, Ltd.) for 1 h to induce oxidative stress. Intracellular
ROS and SA-β-gal (the Senescence Detection kit) were analyzed using
NAC (9). Intracellular ROS levels
were determined by incubating the cells for 30 min at 37°C with 5
μM CellROX™ Deep Red reagent (Invitrogen Life Technologies)
in complete medium, followed by cytometry.
Protein analysis
Western blot analysis and immunoprecipitation were
performed. Total cell lysates were prepared using lysis buffer [50
mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH
7.5), 150 mM NaCl, 1% TritonX-100] containing
ethylenediaminetetraacetic acid (EDTA)-free protease inhibitors.
Cell lysates containing 20 μg of protein were
electrophoresed on TGX™ gels (Bio-Rad). Subsequently, proteins were
transferred onto a PVDF membrane (Bio-Rad). The primary antibodies
were JARID1B (1:2,000; Novus Biologicals), c-Jun N-terminal kinase
(Jnk/Sapk; 1:1,000; Cell Signaling Technology, Danvers, MA),
phospho-Jnk/Sapk (1:1,000; Cell Signaling Technology) and β-actin
(loading control; 1:5,000; Cell Signaling Technology). Western
blotting signals were detected and quantified by image analysis
software (Multi Gauge version 3, Fujifilm). The means ± standard
deviation (SD) of three independent experiments were
determined.
Chromatin immunoprecipitation (ChIP)
analysis
ChIP analysis was performed using the ChIP-IT
Express Enzymatic kit (Active Motif, Carlsbad, CA). The antibodies
used for ChIP analysis were histone H3 (ab1791, Abcam), H3K4 me3
(ab8580, Abcam), H3K4 me2 (ab32356, Abcam) and H3K4 me1 (ab8895,
Abcam), with rabbit immunoglobulin G (IgG) (ab46540, Abcam) used as
the negative control. Immunoprecipitated DNA (100 ng) was
quantified by real-time quantitative PCR (qPCR) using the following
primers: human p16/INK4A promoter,
5′-AACCGCTGCACGCCTCTGAC-3′ (forward) and 5′-CCGCGGCTGTCGTGAAGGTT-3′
(reverse). The means ± SD of three independent experiments were
determined.
RNA interference (RNAi)
RNAi involved the transfection of small interfering
RNA (siRNA) oligos (Cosmo Bio Co., Ltd) or infection with an
shRNA-encoding lentivirus against JARID1B (NM_006618,
Sigma-Aldrich) and a control (SHC002, Sigma-Aldrich). The target
sequences were as follows: KDM5B #1 (100 μM),
GAGCCAGAGGCCAUG AAUAUT (sense) and AUAUUCAUGGCCUCUGCUC
(anti-sense), and KDM5B #2 (100 μM), GGGAACGAGUUAA
GAAAAU (sense) and AUUUUUCUUAACUAGUUCCC (antisense). siRNA oligos
were previously validated. siRNA duplexes were transfected into
subconfluent cells using Lipo-fectamine RNAiMAX (Invitrogen Life
Technologies). The shRNA target sequence was as follows:
JARID1B, 5′-CCGGC CCACCAATTTGGAAGGCATTCTCGAGAATGCCTTCC
AAATTGGTGGGTTTTT-3′.
Real-time reverse transcription PCR
(qRT-PCR)
Real-time qRT-PCR was performed using a Light Cycler
(Roche). Amplified signals were confirmed on the basis of the
dissociation curves and normalized against
glyceraldehyde-3-phosphate dehydrogenase (GAPDH). PCR primer
sequences were as follows: human GAPDH,
5′-ATGTTCGTCATGGGTGTG AA-3′ (forward) and
5′-TGAGTCCTTCCACGATACCA-3′ (reverse); human JARID1B,
5′-CGACAAAGCCAAGAGTC TCC-3′ (forward) and
5′-GGATAGATCGGCCTCGTGTA-3′ (reverse); and human
p16/INK4A, 5′-GTGTGCATGACGTGC GGG-3′ (forward) and
5′-GCAGTTCGAATCTGCACCG TAG-3′ (reverse). The means ± SD of three
independent experiments were determined.
Animal experiments
Six-week-old female NOD/SCID mice were maintained in
a pathogen-free environment. All procedures for animal studies were
approved by the Institutional Ethical Committee of the Faculty of
Medicine, Osaka University. 1×106 tumor cells (viability
>90%) per 50 μl Matrigel (BD Biosciences) were injected
subcutaneously. Tumor volume was measured by callipering the
largest diameter (A) and its perpendicular (B), and calculated
according to the NCI protocol [TV = (A × B2)/2].
Statistical analysis
Categorical variables were compared by the
Chi-square test. Continuous variables (medians/interquartile
ranges) were compared using the Wilcoxon test. Statistical analyses
were performed using JMP software (JMP version 8.01, SAS
Institute). P-values <0.05 were considered to indicate
statistically significant differences.
Results
Ubiquitous JARID1B expression in clinical
samples and various CRC cell lines
A number of studies have revealed the importance of
the correlation between different types of cancer (melanoma,
prostate and breast cancer) and epigenetic factors; however, to
date, no study has suggested the existence of such a correlation
for gastrointestinal cancer (6,10,11).
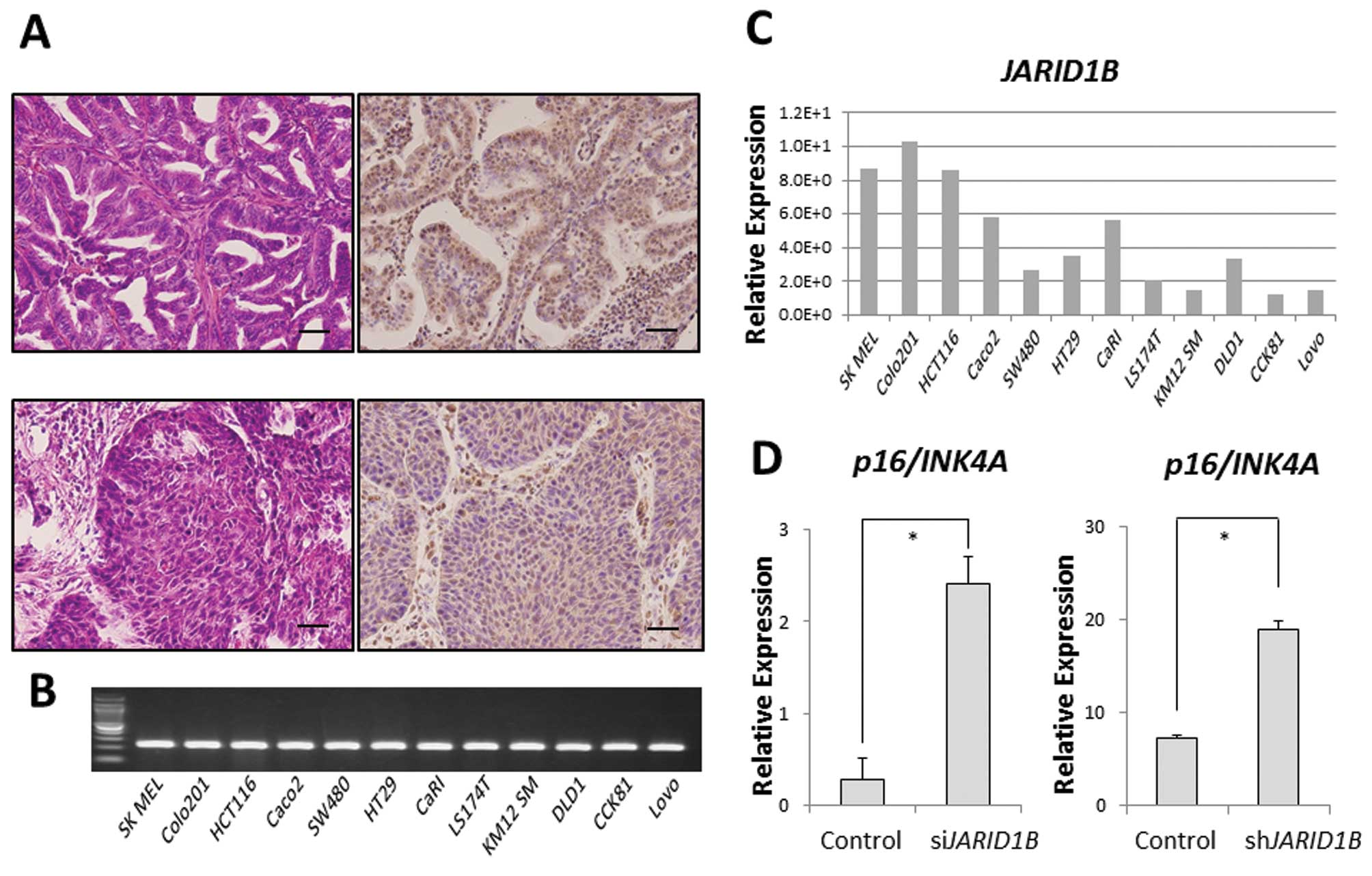
JARID1B was ubiquitously expressed in some clinical samples of
gastrointestinal cancer (modified differentiated adenocarcinoma,
Fig. 1A). In addition, we also
confirmed ubiquitous JARID1B expression in 11 CRC cell
lines. Relative JARID1B expression in 11 colon
adenocarcinoma cell lines and one melanoma cell line (SK MEL) as a
positive control was assessed using qPCR (Fig. 1B) (6). Colo201 and HCT116 cells expressed
higher JARID1B levels than DLD1 cells in vitro
(Fig. 1C). In the Colo201 and
HCT116 cells, the expression of the tumor suppressor,
p16/INK4A, was increased in JARID1B-depleted
cells compared with that in the control cells (Fig. 1D), suggesting that JARID1B
and p16/INK4A expression is inversely correlated.
JARID1B controls p16/INK4A
expression
JARID1B plays a role in the compaction of
active chromatin, which impedes the access of transcription factors
to genes and induces gene silencing (5). Thus, JARID1B expression may be
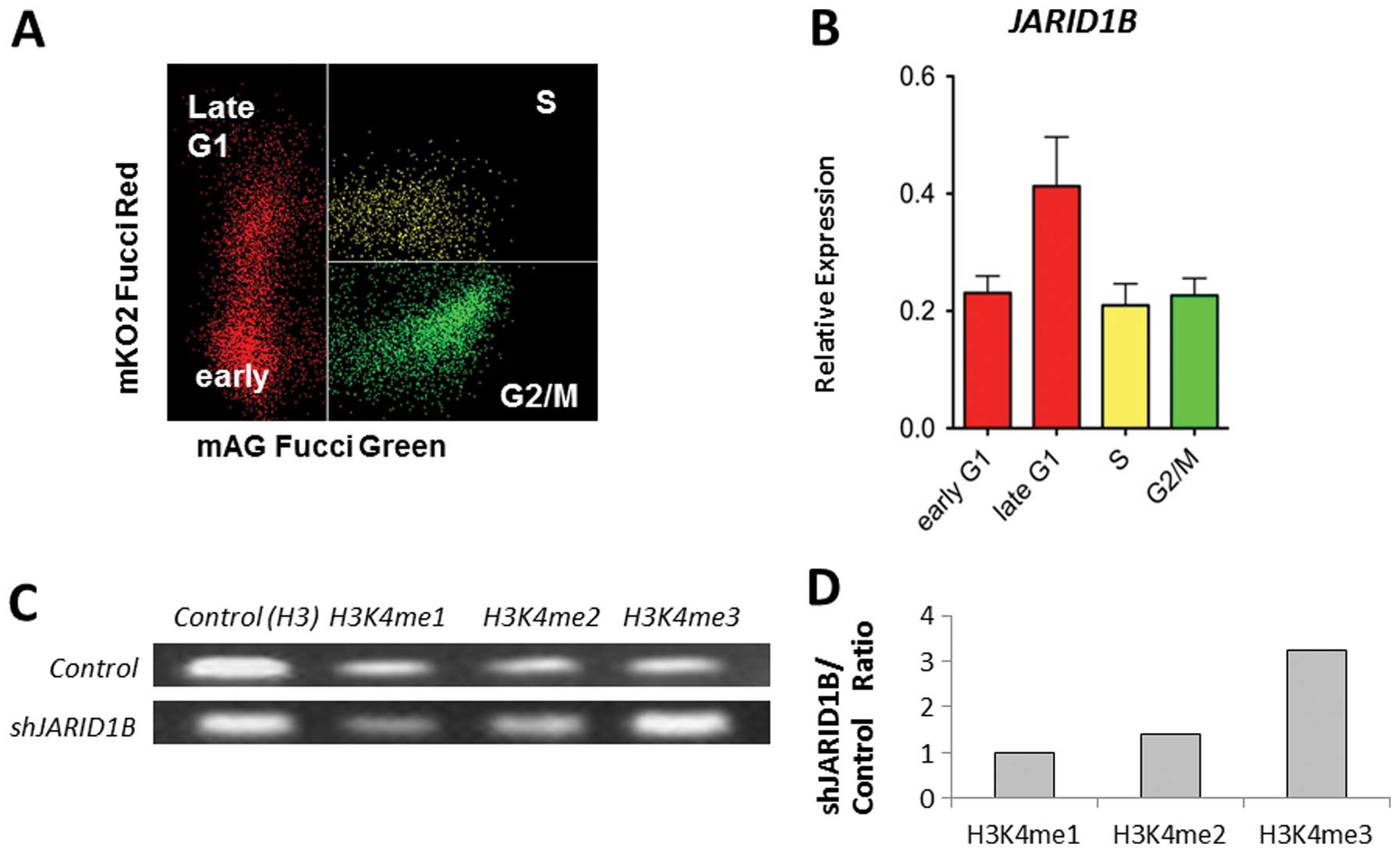
associated with cell cycle progression. Fucci transfectants of CRC
cells revealed that endogenous JARID1B expression increased
in the late G1 phase (Fig. 2A and
B), suggesting the association of JARID1B with
p16/INK4A, which plays a critical role in the G1-S
transition checkpoint (12,13).
ChIP analysis indicated that compared with the control cells,
multimethylated forms of H3K4 were preferentially associated with
the promoter sequence of the p16/INK4A genes in
JARID1B-depleted cells (Fig.
2C). JARID1B depletion led to the trimethylation of the
p16/INK4A promoter (Fig.
2D). Thus, JARID1B may play a role in active chromatin
compaction of the p16/INK4A promoter, which
contributes to gene silencing. Therefore, JARID1B depletion
may lead to p16/INK4A activation.
JARID1B depletion induces cellular
senescence in CRC cells
p16/INK4A is associated with the SA
phenotype, which occurs by the retinoblastoma-inhibiting action of
cyclin-dependant kinases, leading to G1 cell cycle arrest (13–15),
with the involvement of ROS (16).
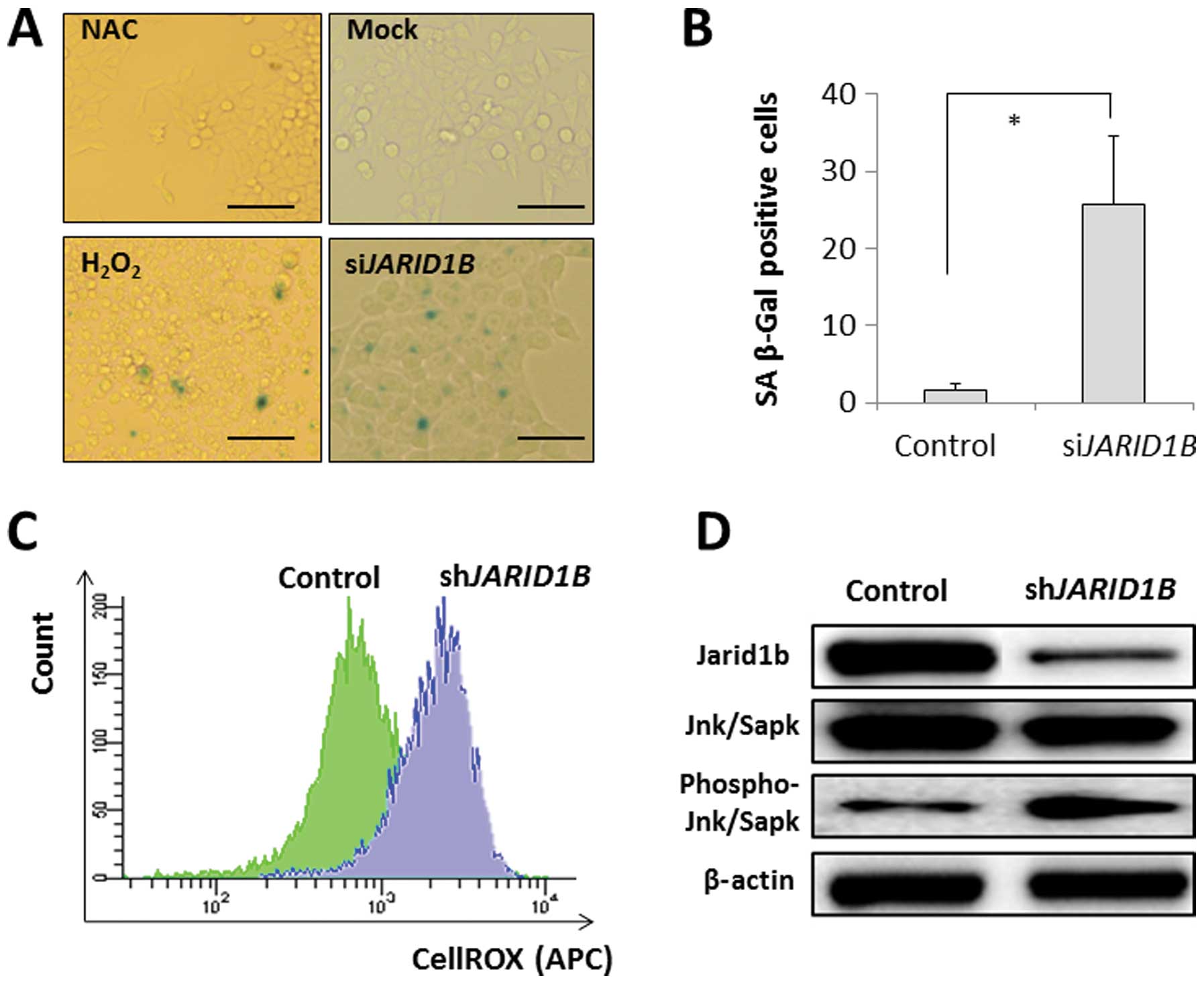
SA-β-gal activity was detected in the JARID1B-depleted CRC
cells but not in the mock-transfected control cells (Fig. 3A and B). The effect of
JARID1B depletion on SA-β-gal activity was similar to that
of H2O2 exposure with an ROS inducer in the
medium but not similar to that of the negative experimental control
with NAC in the medium (Fig. 3A and
B). This suggests that intracellular ROS may be involved in
cellular senescence induction. The results from the present study
illustrated that intracellular ROS levels were higher in
JARID1B-depleted CRC cells than in mock-transfected control
cells (Fig. 3C), which supports
the involvement of ROS in senescence induction in
JARID1B-depleted cells. Immunoblot analysis of SA phenotypes
revealed the increased phosphorylation of Jnk/Sapk, an inducer of
cellular senescence, in JARID1B-depleted cells;
JARID1B expression was decreased by RNAi (Fig. 3D).
JARID1B depletion suppresses CRC
growth
JARID1B depletion suppressed CRC cell growth
in vitro by intracellular ROS accumulation and cellular
senescence activation. Therefore, we investigated its effects on
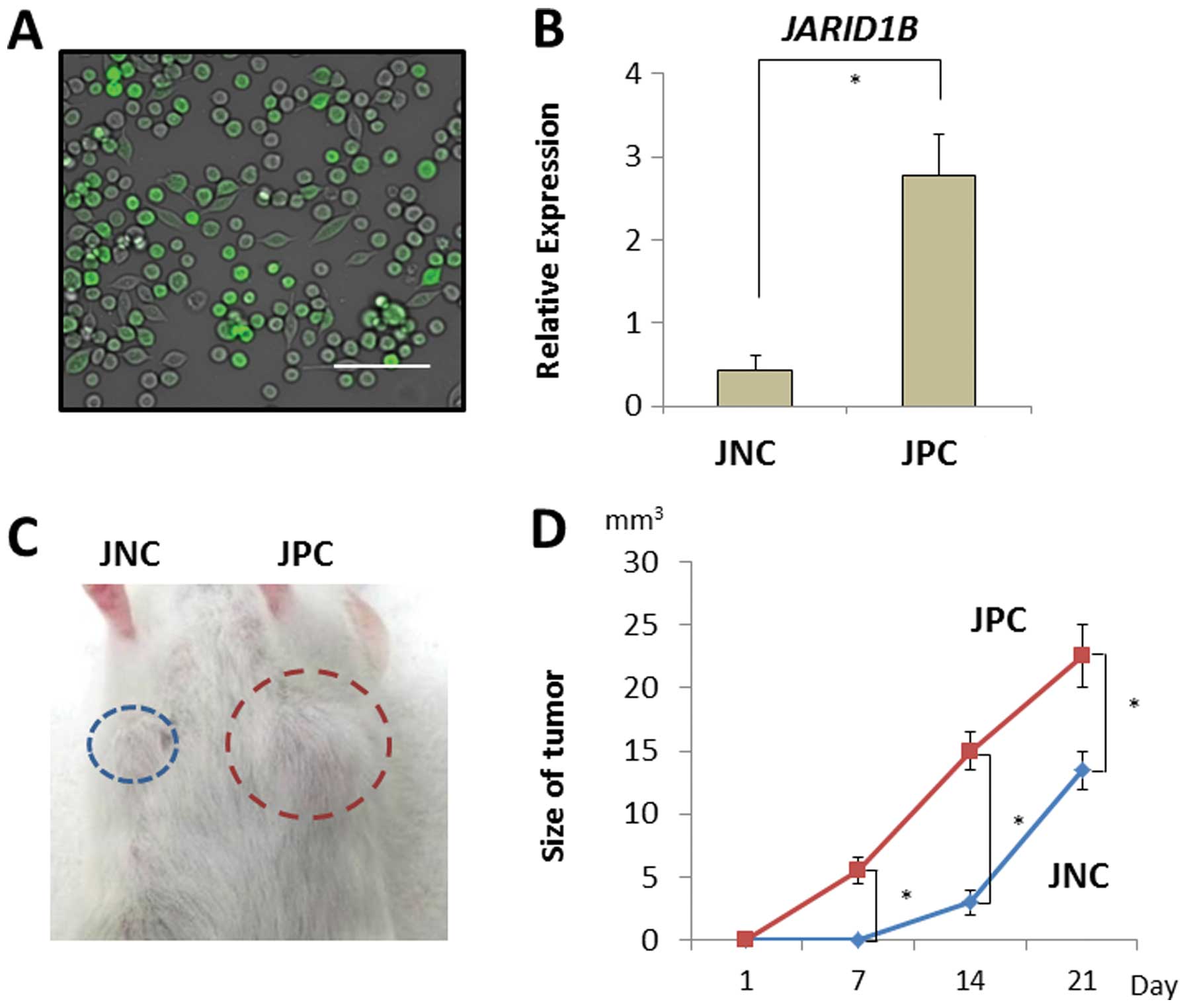
tumor growth in vivo. JARID1B-positive and -negative CRC
cells were sorted using a fluorescent tracer vector controlled by
the JARID1B promoter (Fig.
4A). CRC cells were separated on the basis of d2-Venus
expression and the fluorescent intensity depended on the endogenous
expression of the JARID1B promoter (Fig. 4B). JARID1B-positive and
-negative CRC cells were subcutaneously inoculated into
immunodeficient NOD/SCID mice to assess their tumorigenicity
(Fig. 4C). Endogenous
JARID1B-positive CRC cells produced substantial tumor growth
compared with the JARID1B-negative cells (Fig. 4D).
JARID1B depletion suppresses
therapy-resistant CRC cell growth
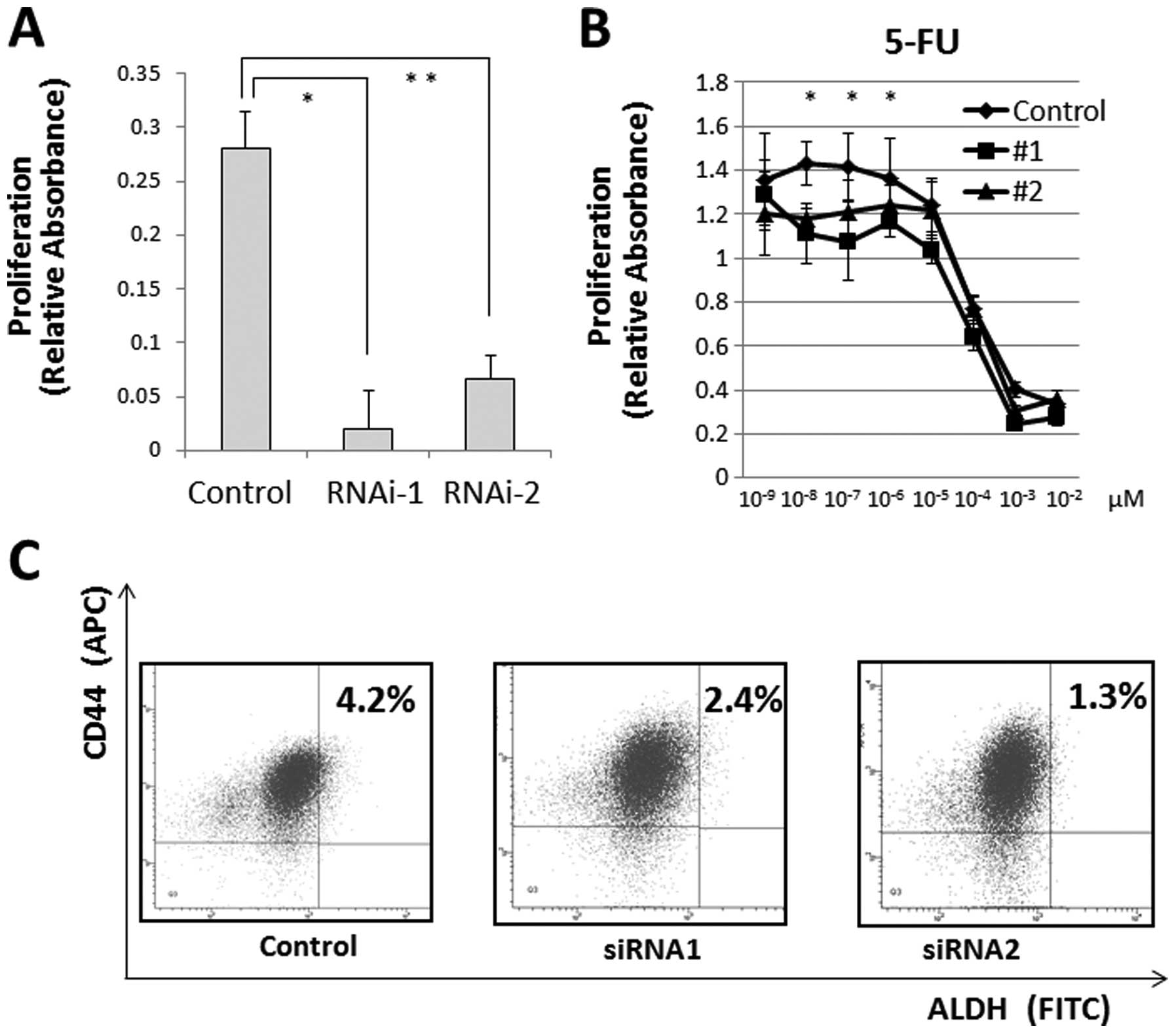
We measured cell proliferation following
JARID1B depletion and observed that JARID1B depletion
significantly suppressed cell proliferation (Fig. 5A) and cell invasion (data not
shown). Hence, we determined the resistance of CRC cells to
chemotherapy, a feature of JARID1B-depleted cells (17,18).
The MTT assay illustrated that compared with the controls,
continuous JARID1B depletion induced resistance to 5-FU in
culture (Fig. 5B), suggesting that
JARID1B depletion contributes to cancer stem cell (CSC)
suppression. The depletion of endogenous JARID1B has been
shown to suppress tumorigenicity and eliminate CSCs in melanomas
(6). Thus, we inhibited endogenous
JARID1B using shRNA and confirmed the CSC fraction.
Depletion of endogenous JARID1B reduced the
CD44+/ALDH+ CSC fraction, indicating that
continuous endogenous JARID1B inhibition contributed to the
eradication of CRC stem cells (Fig.
5C).
Discussion
Histone methylation/demethylation generally
deactivates and activates genes by controlling the access of
transcription factors to DNA. Histone dysregulation caused by
genetic and epigenetic alterations is a hallmark of cancer
(9,19). JARID1A/B-mediated
histone H3K4 demethylation contributes to the silencing of
retinoblastoma target genes in senescent cells, presumably by
compacting chromatin and silencing certain genes (20). Distinct SA changes in
histone-modification patterns are consistent with a repressive
chromatin environment in the retinoblastoma tumor suppressor
pathway (20). We found that
JARID1B depletion, i.e., the inhibition of H3K4
demethylation, stimulated p16/INK4A transcription and
suppressed tumor cell growth in vitro and in vivo,
suggesting that it plays a role in cell growth regulation in human
CRC (6).
The present findings are consistent with the notions
that JARID1B depletion induces Jnk/Sapk-related senescence
in CRC cells (21) and that
endogenous JARID1B plays a role in controlling the cellular
growth of CRCs. In cellular senescence, normal diploid cells lose
the ability to divide. This anti-proliferative stress-response
program acts as a potent tumor-suppressing mechanism (14,22).
Growth-promoting and tumor suppressor genes are important factors
controlling cancer cell proliferation. Cellular senescence can be
triggered by a number of factors, including aging, DNA damage,
oncogene activation and oxidative stress. Senescent cells have
distinctive features, including stable cell cycle arrest and
SA-β-gal activity. The tumor suppressor p16/INK4A
plays a key role in regulating senescence induction, as may the
tumor suppressor, p53. p16 acts through the retinoblastoma pathway
to inhibit cyclin-dependant kinases, leading to G1 cell cycle
arrest and senescence (14,23).
Our results demonstrate that JARID1B plays a key role in CRC
maintenance and that its continuous inhibition induces cellular
senescence.
In the present study, we present a novel hypothesis
that JARID1B suppression is an essential factor in cancer
eradication. Although CSCs play a critical role in the survival,
relapse and metastasis of malignant cancer cells (17), our data confirm the correlation
between JARID1B suppression and CSCs. ALDH and CD44, a
hyaluronic acid receptor, are considered useful markers of CRC stem
cells (18). ALDH1A1 is
responsible for CSC ALDH activity (24). It is known that
CD44+/ALDH+ double-positive cells are CSC
enrichment markers for reconstituted tumors in immunodeficient mice
(18,24). In our study, endogenous
JARID1B expression was lower in
CD44+/ALDH+ cells than in
CD44+/ALDH− or CD44−/ALDH− cells,
suggesting that slowly proliferating JARID1B-expressing
cells had a relatively undifferentiated phenotype that was
compatible with that of CSCs (17,18).
Our results demonstrate that JARID1B is involved in cell
cycle regulation and that JARID1B facilitates cellular
amplification and CSC maintenance. Future reports should further
clarify the correlation between epigenetic factors and CSCs.
Therapeutic approaches to CRC include conventional
therapies (surgical removal and chemoradiotherapy) and gene
delivery strategies. For example, continuous JARID1B
depletion could be achieved with antisense oligonucleotides or
low-molecular-weight pharmacological therapeutics (25). A combination of
p16/INK4A gene therapy and anti-JARID1B
treatment may lead to the efficient induction of a SA phenotype in
CRC cells.
Acknowledgements
We thank Dr Atsushi Miyawaki for
providing us with the Fucci System plasmids. This study was partly
supported by a Grant-in-Aid for Scientific Research from the
Ministry of Education, Culture, Sports, Science and Technology
(H.I. and M.M.); a Grant-in-Aid from the 3rd Comprehensive 10-year
Strategy for Cancer Control, Ministry of Health, Labour and Welfare
(H.I. and M.M.); a grant from the Kobayashi Cancer Research
Foundation (H.I.); and a grant from the Princess Takamatsu Cancer
Research Fund, Japan (H.I.). M.K., T.K., D.S., T.S. and H.I. were
partially supported by Chugai Co., Ltd. and Yakult Honsha Co., Ltd.
via institutional endowments.
References
|
1
|
Markowitz SD and Bertagnolli MM: Molecular
origins of cancer: Molecular basis of colorectal cancer. N Engl J
Med. 361:2449–2460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Patai AV, Molnár B, Kalmár A, Schöller A,
Tóth K and Tulassay Z: Role of DNA methylation in colorectal
carcinogenesis. Dig Dis. 30:310–315. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gargalionis AN, Piperi C, Adamopoulos C
and Papavassiliou AG: Histone modifications as a pathogenic
mechanism of colorectal tumorigenesis. Int J Biochem Cell Biol.
44:1276–1289. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brabletz T, Jung A, Spaderna S, Hlubek F
and Kirchner T: Opinion: migrating cancer stem cells-an integrated
concept of malignant tumour progression. Nat Rev Cancer. 5:744–749.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Roesch A, Fukunaga-Kalabis M, Schmidt EC,
et al: A temporarily distinct subpopulation of slow-cycling
melanoma cells is required for continuous tumor growth. Cell.
141:583–594. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sakaue-Sawano A, Kurokawa H, Morimura T,
et al: Visualizing spatiotemporal dynamics of multicellular
cell-cycle progression. Cell. 132:487–498. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haraguchi N, Ishii H, Mimori K, et al:
CD13 is a therapeutic target in human liver cancer stem cells. J
Clin Invest. 120:3326–3339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takahashi A, Imai Y, Yamakoshi K, et al:
DNA damage signaling triggers degradation of histone
methyltransferases through APC/C(Cdh1) in senescent cells. Mol
Cell. 45:123–131. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yamane K, Tateishi K, Klose RJ, et al:
PLU-1 is an H3K4 demethylase involved in transcriptional repression
and breast cancer cell proliferation. Mol Cell. 25:801–812. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xiang Y, Zhu Z, Han G, et al: JARID1B is a
histone H3 lysine 4 demethylase up-regulated in prostate cancer.
Proc Natl Acad Sci USA. 104:19226–19231. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ohtani N, Zebedee Z, Huot TJ, et al:
Opposing effects of Ets and Id proteins on p16INK4a expression
during cellular senescence. Nature. 409:1067–1070. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rayess H, Wang MB and Srivatsan ES:
Cellular senescence and tumor suppressor gene p16. Int J Cancer.
130:1715–1725. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang X, Wu X, Tang W and Luo Y: Loss of
p16(Ink4a) function rescues cellular senescence induced by telomere
dysfunction. Int J Mol Sci. 13:5866–5877. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vurusaner B, Poli G and Basaga H: Tumor
suppressor genes and ROS: complex networks of interactions. Free
Radic Biol Med. 52:7–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer and cancer stem cells. Nature.
414:105–111. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dewi DL, Ishii H, Kano Y, et al: Cancer
stem cell theory in gastrointestinal malignancies: recent progress
and upcoming challenges. J Gastroenterol. 46:1145–1157. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chicas A, Kapoor A, Wang X, et al: H3K4
demethylation by Jarid1a and Jarid1b contributes to
retinoblastoma-mediated gene silencing during cellular senescence.
Proc Natl Acad Sci USA. 109:8971–8976. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maruyama J, Naguro I, Takeda K and Ichijo
H: Stress-activated MAP kinase cascades in cellular senescence.
Curr Med Chem. 16:1229–1235. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rodier F and Campisi J: Four faces of
cellular senescence. J Cell Biol. 192:547–556. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ohtani N, Mann DJ and Hara E: Cellular
senescence: its role in tumor suppression and aging. Cancer Sci.
100:792–797. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marcato P, Dean CA, Giacomantonio CA and
Lee PW: Aldehyde dehydrogenase: its role as a cancer stem cell
marker comes down to the specific isoform. Cell Cycle.
10:1378–1384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yamamoto T, Nakatani M, Narukawa K and
Obika S: Antisense drug discovery and development. Future Med Chem.
3:339–365. 2011. View Article : Google Scholar
|