Introduction
Peritoneal dissemination is the most common mode of
metastasis in gastric cancer. Various approaches have been assessed
for the treatment of peritoneal dissemination, including systemic
chemotherapy, intraperitoneal chemotherapy (1), extensive intraoperative peritoneal
lavage (2) and aggressive surgery
(3). The clinical outcome in
gastric cancer patients with peritoneal dissemination has improved,
but sufficiently good outcomes have not yet been obtained (4,5). In
particular, acquired drug resistance and invasive scirrhous cell
dissemination, characterized by rich fibrous components, are
typical manifestations of clinical gastrointestinal disorders
(e.g., ileus, obstructive jaundice and hydronephrosis). In cancer
peritoneal dissemination, human peritoneal mesothelial cells
(HPMCs), which are classified as epithelium in the broadest sense
of the term, serve as a protective anatomical barrier and play an
important role in the immunological response to infection and wound
healing; they also contribute to cancer cell growth and fibrosis
via epithelial-mesenchymal transition (EMT) (6,7).
EMT is an essential mechanism that guides proper
development during several phases of embryogenesis (8). It is also related to pathological
changes such as organ fibrosis (9)
and tumour metastasis (10). EMT
is characterised by loss of cell-cell adhesion and apical-basal
polarity, followed by a shift in cytoskeletal dynamics toward front
end-back end polarity and cell migration (11).
Transforming growth factor-β (TGF-β), the prototype
member of a mammalian superfamily of growth factors that includes
activins, bone morphogenetic proteins (BMPs), inhibins and nodal,
is a common initiator of EMT. TGF-β and related factors evoke
pleiotropic cellular responses through binding to transmembrane
serine-threonine kinase receptor type I (TβR-I) and TβR type II
(TβR-II) (12). The activated
TGF-β receptors stimulate the phosphorylation of receptor-regulated
Smad2 and Smad3 proteins (R-Smad), which in turn form complexes
with Smad4 (co-Smad). This complex translocates from the cytoplasm
into the nucleus, where the Smads regulate the transcription of
target genes. The activity of inhibitory Smad7 (I-Smad) is the
opposite of that exhibited by R-Smads; I-Smad downregulates TGF-β
signalling (13). Some studies
have previously shown that endogenous Smad2, Smad3 and Smad4 bind
to microtubules in several cell lines and that this binding
provides a negative regulatory mechanism to modulate TGF-β activity
(14).
Paclitaxel (PTX), derived from the bark of the
Pacific yew, Taxus brevifolia, is an antineoplastic agent
that stabilizes polymerized microtubules and enhances microtubule
assembly and thus arrests the cell cycle in the G0/G1 (low dose)
and G2/M (high dose) phases, leading to cell death (15,16).
PTX has been used in the treatment of peritoneal dissemination of
ovarian and gastric cancers (17,18).
Clinically, PTX has also been reported to improve intestinal
stenosis due to fibrosis associated with the progression of
peritoneal dissemination (19). At
low concentrations, PTX has antiproliferative and antimigratory
effects in vascular smooth muscle and endothelial cells (20,21)
and has been widely applied clinically, e.g., in drug-eluting
stents (22,23). Importantly, PTX has been shown to
modulate TGF-β signalling, interrupting fibrosis in a murine
systemic sclerosis model and in gallbladder myofibroblasts
(24,25).
The aim of the present study was to evaluate the
inhibitory effects of TGF-β-induced EMT in HPMCs at the cytostatic
concentration of the antineoplastic agents PTX, 5-fluorouracil
(5-FU) and cisplatin (CDDP).
Materials and methods
Antineoplastic agents
5-FU and CDDP were purchased from Sigma-Aldrich Inc.
(USA). PTX was kindly provided by the Bristol-Myers Squibb Co.
(Japan) and reconstituted in distilled water at appropriate
concentrations and stored at −20°C until use.
Cell lines and cell culture
HPMCs were isolated from surgical specimens of the
human omentum, as previously described (26). Omental specimens were obtained,
with informed consent, from patients undergoing elective abdominal
surgery. Tissue samples were collected in ice-cold phosphate
buffered saline (PBS) to minimize cell degeneration. Contaminating
red blood cells were removed by extensive PBS washes and samples
were incubated in pre-warmed PBS containing 0.125% trypsin/EDTA
(Gibco/Invitrogen, USA) for 30 min at 37°C. The suspension was
passed through a 100-μm-pore nylon mesh (Becton-Dickinson,
Japan) to remove undigested fragments and was then centrifuged at
1,500 rpm for 5 min. Dissociated cells were cultured in RPMI-1640
medium (Gibco/Invitrogen) supplemented with 10% heat-inactivated
fetal bovine serum (Nichirei Bioscience Inc., Japan), 100 IU/ml
penicillin, 100 mg/ml streptomycin (Gibco/Invitrogen) and 2 mM
glutamine (Nissui Pharmaceutical Co. Ltd., Japan). The cells were
seeded in gelatin-coated 75-cm2 flasks (BD BioCoat, USA)
and cultured in 10 ml of medium at 37°C in a humidified atmosphere
of 5% CO2 in air.
Human gastric cancer cell lines (MKN45) were
obtained from the American Type Culture Collection (USA). MKN45
cells were cultured in the media indicated above for HPMC. The
cells were grown to confluence and were harvested by trypsinisation
with 0.25% trypsin/EDTA. Confluent HPMCs were trypsinised with
0.125% trypsin/EDTA before use. HPMCs were then transferred to
serum-free medium for 24 h, after which they were continuously
exposed to 5 ng/ml of recombinant human TGF-β1 (Sigma-Aldrich,
Inc., USA) for 48 h. Finally, they were transferred to RPMI-1640
containing 10% FBS, which caused a shift in the morphology of the
cells, resulting in activated HPMCs (a-HPMCs). HPMCs were used from
passage 1 to 3 in all experiments.
Cell growth assay
The viability of HPMC and MKN45 cells treated with
antineoplastic agents was determined by a standard
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT)
assay. MKN45 cells were seeded at 5×103 per well in
96-well plates and were incubated overnight at 37°C. HPMC cells
were seeded at 5×103 per well on gelatin-coated 96-well
microplates (BD BioCoat). After incubation, the supernatant was
discarded and replaced with fresh serum-free medium. Antineoplastic
agents were dissolved in PBS and added to the cell culture medium
at various concentrations (5-FU, 0-10 μM; CDDP, 0–10
μM; PTX, 0–100 nM). At 48 h after exposure to antineoplastic
agents, the supernatant was discarded and MTT solution was added to
each well (final concentration, 500 μg/ml) and incubated at
37°C for 3 h. Then, the supernatant was removed and 150 μl
of dimetylsulphoxide (DMSO; Wako, Japan) was added. The absorbance
of the solution was read at 540 nM with a microplate reader
(Bio-Rad 550; Bio-Rad, Japan). The percentage inhibition was
determined by comparing the cell density of the drug-treated cells
with that of untreated controls. All the experiments were repeated
a minimum of 3 times.
Histology and immunofluorescence
Subconfluent HPMCs were transferred to serum-free
medium for 24 h, after which they were continuously exposed to one
of the following treatments: 0.5 or 1 μM of 5-FU, 5 or 10
μM of CDDP and 1 or 5 nM of PTX for 1 h. Subsequently, 5
ng/ml of recombinant human TGF-β1 was added. After 48-h exposure to
TGF-β1, morphological changes in HPMCs were observed by
phase-contrast microscopy. Cells were then harvested for
immunostaining.
For visualizing E-cadherin and α-SMA in HPMCs, the
cells were grown on 4-well collagen type I-coated culture slides
(BD BioCoat) and then fixed in a mixture of methanol and acetone
(1:1) for 15 min. Immunostaining was performed as reported earlier
(27). Briefly, the slides were
immersed in methanol containing 0.3% H2O2 for
30 min, blocked with 3.3% normal goat serum in PBS and incubated
with the anti-E-cadherin antibody (H-108, rabbit polyclonal IgG,
diluted 1:100; Santa Cruz Biotechnology, Inc. USA) and anti-α-SMA
(1A4, mouse monoclonal IgG, diluted 1:100; DakoCytomation, Denmark)
at 4°C overnight. Following the PBS washes, immunoreactivity was
visualized by incubating the sections with anti-mouse IgG antibody
conjugated with Alexa Fluor® 488 and anti-rabbit IgG
antibody conjugated with Alexa Fluor® 546 (Molecular
Probes/Invitrogen, USA) (1:400) for 1 h at room temperature. The
slides were observed with an immunofluorescence microscope
(BX50/BX-FLA; Olympus, Japan).
Western blotting
Immunoblot analysis was performed as described
previously (28). The cells were
lysed in RIPA buffer [50 mmol/l Tris-HCl (pH 8.0), 150 mmol/l
sodium chloride, 0.5 w/v% sodium deoxycholate, 0.1 w/v% sodium
dodecyl sulphate, 1.0 w/v% NP-40 substitute (Wako, Japan)]
containing 1% protease inhibitor cocktail (Sigma-Aldrich, Inc.).
The protein concentration of each sample was measured using a BCA
protein assay kit (Pierce Biotechnology, USA). Whole-cell lysates
were prepared in denaturing SDS sample buffer and subjected to
SDS-PAGE (Atto Co. Ltd., Japan). Proteins were transferred to PVDF
membranes (Bio-Rad, USA) and were then blocked with commercial
gradient buffer (EzBlock, Atto Corp.) at room temperature for 30
min. The immunoblots were visualized using an ECL Plus kit (GE
Healthcare Japan Ltd., Japan). The antibody-antigen complex was
detected using an ECL western blotting detection kit (GE Healthcare
Japan Ltd.) and the Light-Capture system (Atto). We used the
following primary antibodies: anti-E-cadherin (H-108, rabbit
polyclonal IgG, diluted 1:1,000; Santa Cruz Biotechnology, Inc.),
anti-α-SMA (1A4, mouse monoclonal IgG, diluted 1:5,000;
DakoCytomation), anti-Smad2/3 (E-20, goat polyclonal IgG, diluted
1:1,000; Santa Cruz Biotechnology, Inc.) and anti-β-actin (AC-15,
mouse monoclonal IgG, diluted 1:10,000; Sigma).
Relative quantification by real-time
quantitative PCR
Total RNA was extracted from HPMCs with an RNeasy
mini kit (Qiagen, USA) and treated with an RNase-free DNase set
(Qiagen), following the manufacturer’s recommendations. An Agilent
2100 Bioanalyzer microfluidic assay (Agilent Technologies, USA) was
used to assess RNA integrity. Spectrophotometric and fluorometric
methods were combined to quantitate RNA. cDNA was generated from
RNA using a reverse transcription kit (Applied Biosystems, USA).
Total RNA (1 μg) was reverse transcribed in a total volume
of 20 μl by using 100 U of reverse transcriptase, 2.0
μl 10X RT buffer, 2.0 μl 10X random primers and 1.0
μl of 20 U/μl RNase inhibitor. The mixture was
incubated for 10 min at 25°C, 120 min at 37°C and 5 min at 85°C; it
was then rapidly cooled on ice. The cDNA samples were stored at
−20°C.
Real-time qPCR was performed on an M×3005P Multiplex
Quantificative PCR system with the MxPro QPCR software (Stratagene,
USA). TaqMan® Universal Master Mix (Applied Biosystems)
was used for PCR. In a final volume of 20 μl, 1 μl of
cDNA was amplified using the following TaqMan® assays
(Applied Biosystems): Smad2 (Hs00998181_gH), collagen type I
(Hs01076775_g1), GAPDH control reagents and β-actin control
reagents. The PCR cycling conditions were as follows: 50°C for 2
min; 95°C for 10 min; and 40 cycles of 95°C for 15 sec and 60°C for
1 min. All qPCR reactions were performed in triplicate. The
threshold cycle (Ct) method was used for quantification. For
relative quantification, Smad2 and collagen type I mRNA levels,
normalized to endogenous house-keeping controls (GAPDH and
β-actin), were divided by normal control sample values (normal HPMC
samples) to generate the relative quantification to calibrator
(rel. quant. to cal.). All the experiments were repeated a minimum
of 3 times.
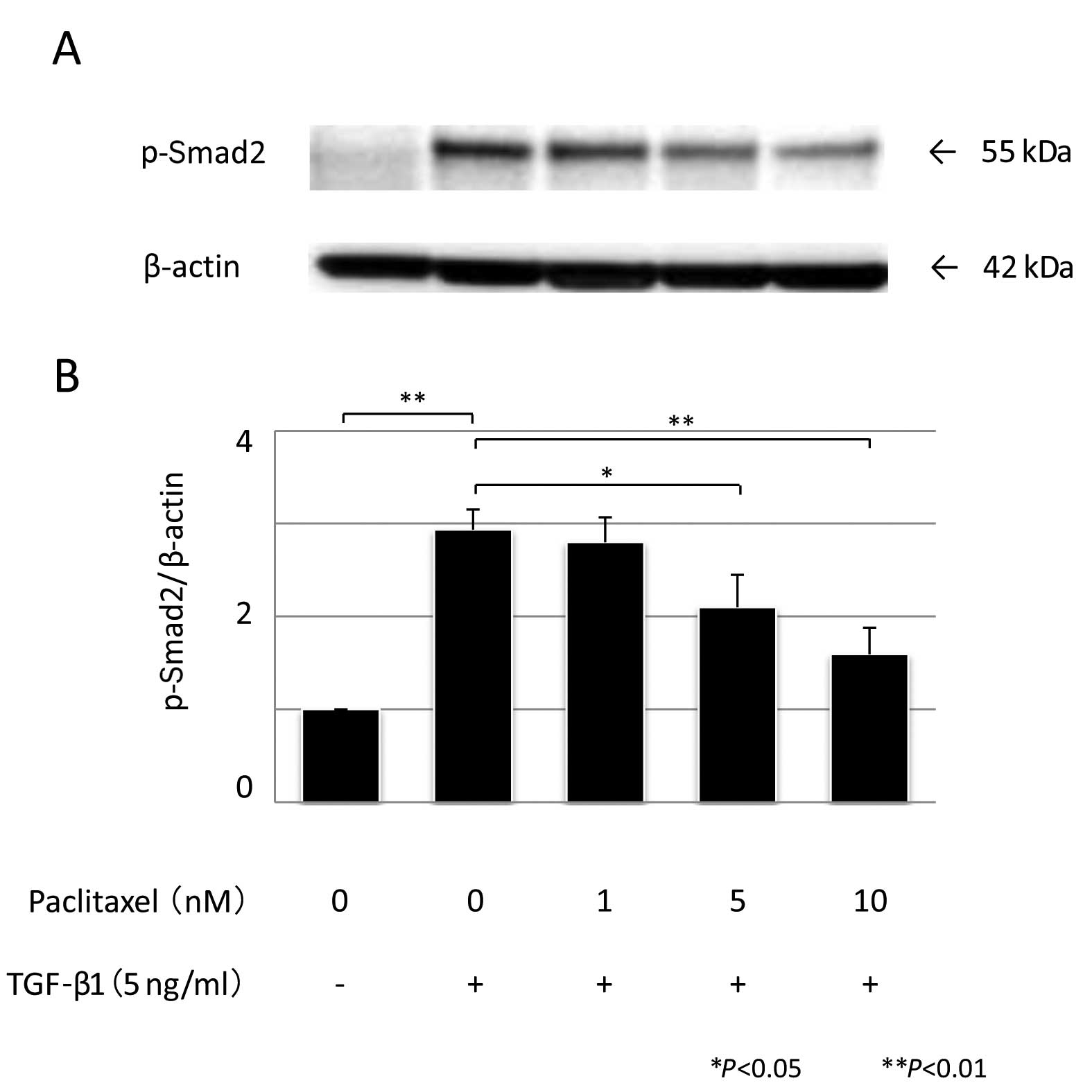
Quantitation of phosphorylated Smad2
To determine whether the modulation of TGF-β1
transcriptional activity by PTX correlated with a change in the
phosphorylation state of Smad2, we performed western blot analysis.
Subconfluent HPMCs were prepared as mentioned earlier; 1 or 5 nM of
PTX was added for 1 h, followed by exposure to 5 ng/ml of
recombinant human TGF-β1 and western blotting was performed using
an anti-phospho-Smad2 antibody (Ser 465/467, A5S, rabbit polyclonal
IgG, diluted 1:1,000; Millipore, USA). The antibody-antigen complex
was detected using a Light-Capture system (Atto) and was then
quantified using the CS analyzer program (Atto). All the
experiments were repeated 3 times.
Statistical analysis
Values are expressed as means ± standard error (SE).
One-way analysis of variance (ANOVA) was performed using SPSS 10.0
(SPSS Inc., USA). Significance was defined as P<0.05.
Results
Determination of minimum cytostatic
concentration of anti-neoplastic agents
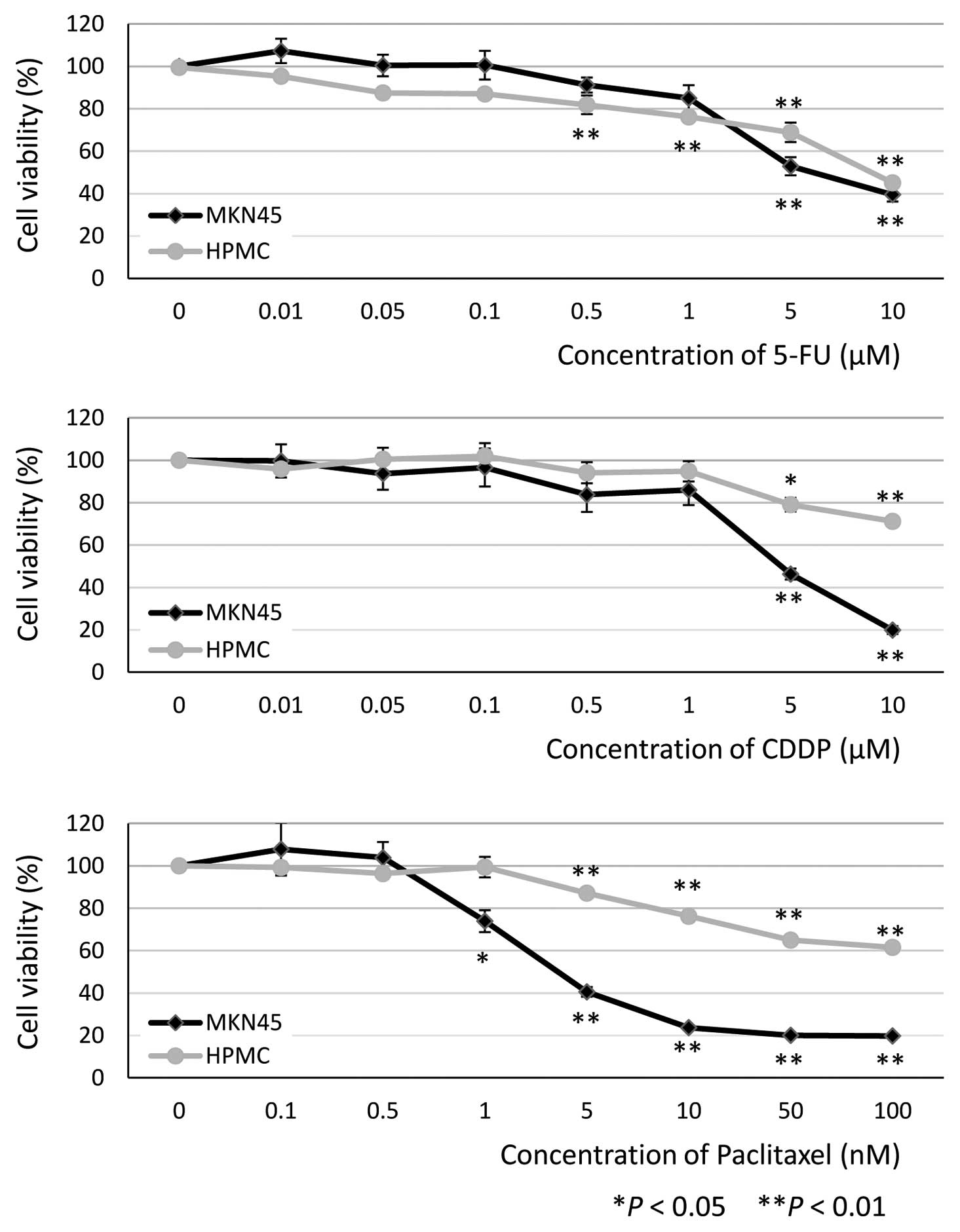
MTT assays were performed in HPMC and MKN45 cells
(as a comparator) to determine the minimum cytostatic
concentrations of the antineoplastic agents. The concentrations
required for significant inhibition of HPMC viability at 48 h were
0.5 μM 5-FU (81.9±4.4%, P<0.01, n=9), 5 μM CDDP
(79.0±3.2%, P=0.032, n=9) and 5 nM PTX (87.1±1.5%, P<0.01, n=9)
(Fig. 1). On the basis of these
results, we decided to use the following concentrations for
subsequent experiments: 0.5 and 1 μM 5-FU, 5 and 10
μM CDDP and 1, 5 and 10 nM PTX.
Effect of PTX on morphological changes,
and E-cadherin and α-SMA expression in HPMCs
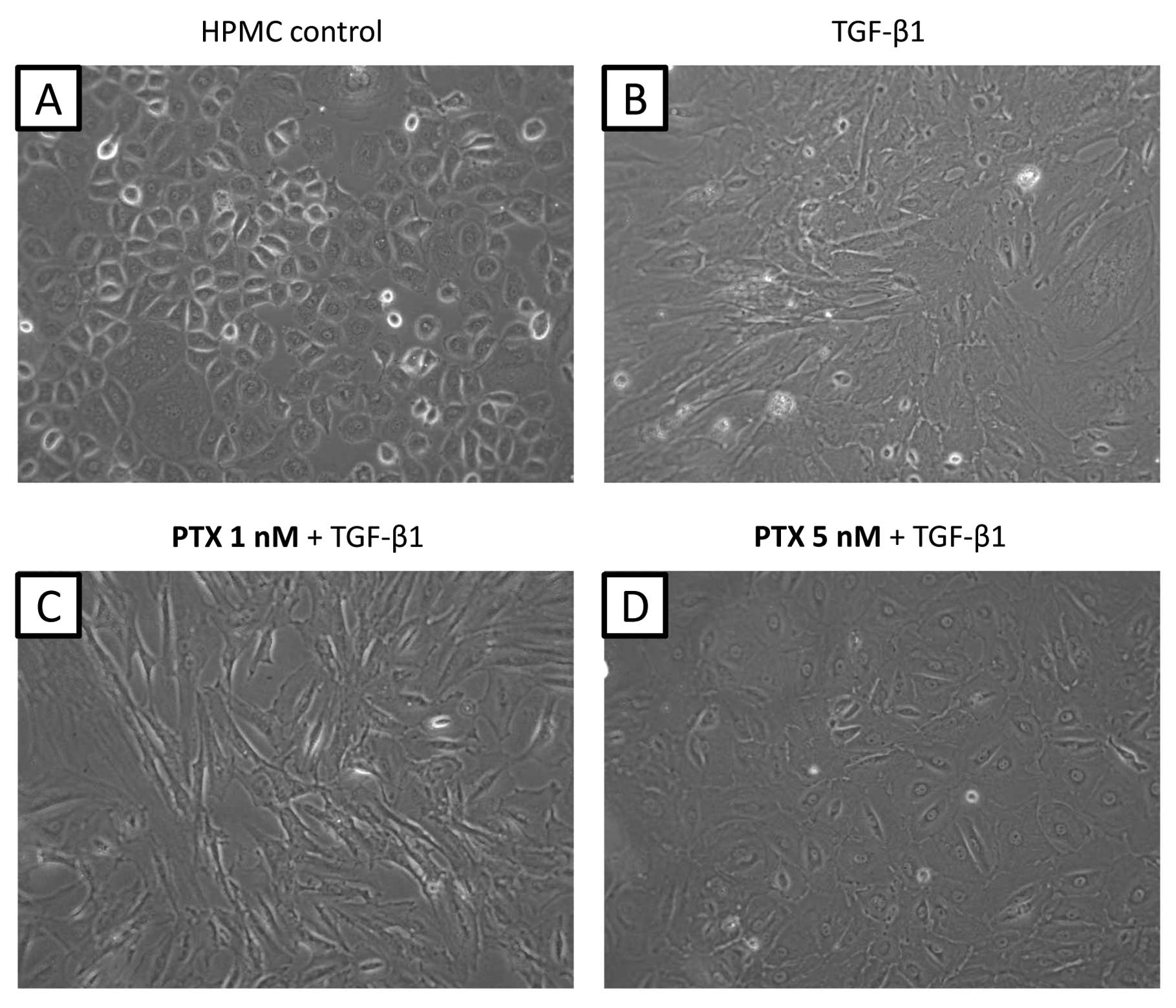
Morphological changes were observed in cultured
HPMCs after adding TGF-β1 (5 ng/ml) for 48 h. Control HPMCs without
TGF-β1 treatment showed an epithelial morphology with a cobblestone
appearance (Fig. 2A); however,
treated HPMCs converted to a morphology with a spindle fibroblastic
pattern (Fig. 2B). Pre-treatment
with 1 nM PTX did not suppress the morphological changes induced by
TGF-β1 (Fig. 2C). However, cells
pre-treated with 5 nM PTX appeared rounded in shape suggesting that
at this dose, PTX had an inhibitory effect on the action of TGF-β1
(Fig. 2D). This inhibition of
morphological changes was not observed with other antineoplastic
agents (data not shown).
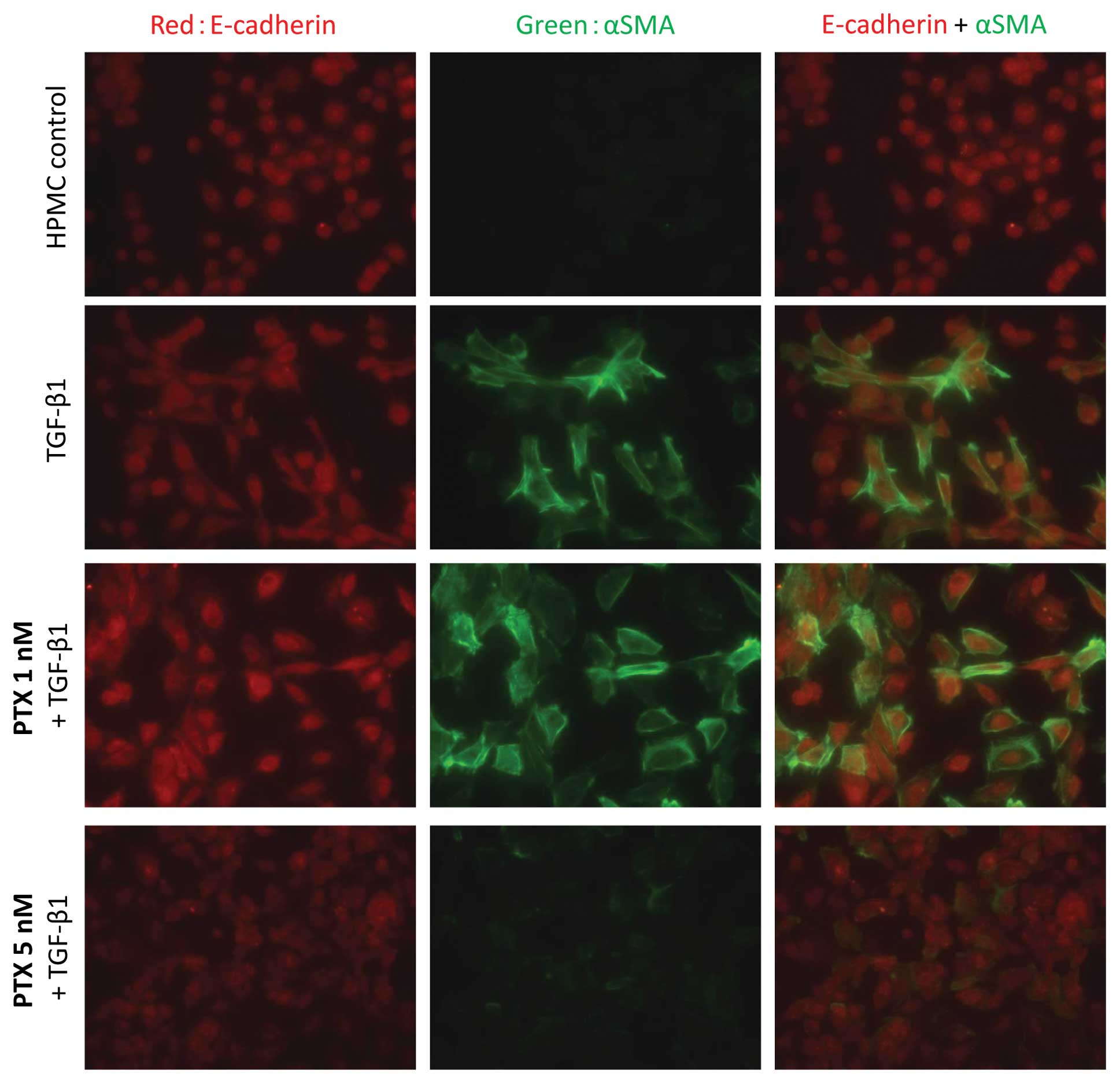
Immunofluorescence analysis of the morphological
changes showed that α-SMA expression increased on TGF-β1 treatment
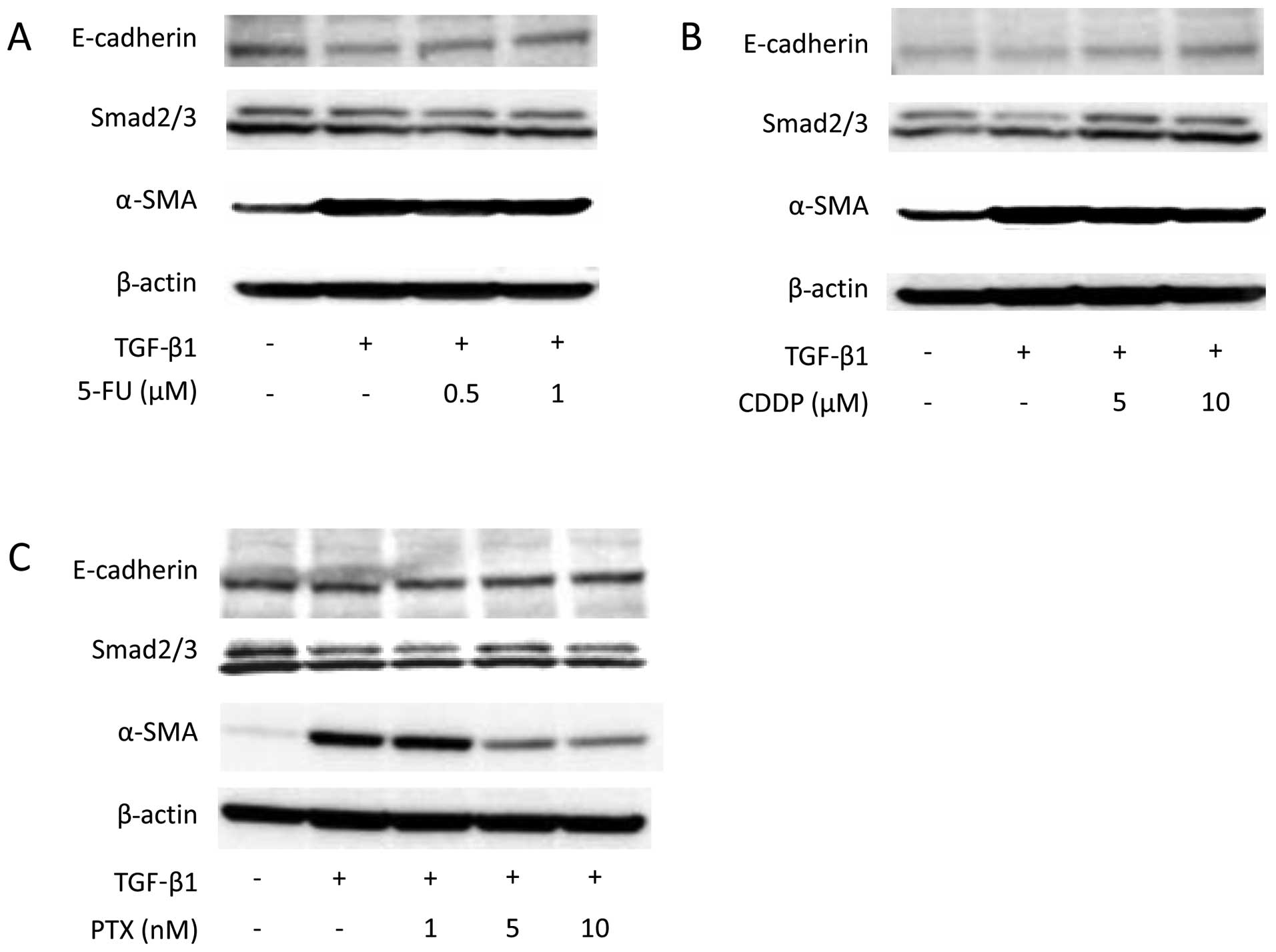
and was suppressed by pre-treatment with 5 nM PTX (Fig. 3). Western blot analysis also showed
that α-SMA expression increased on TGF-β1 stimulation. Suppression
of α-SMA expression was not detected with either 5-FU or CDDP
treatment (Fig. 4); however,
pre-treatment with 5 and 10 nM PTX did suppress TGF-β1-induced
α-SMA expression.
Effect of antineoplastic agents on
collagen production and TGF/Smad signalling in HPMC
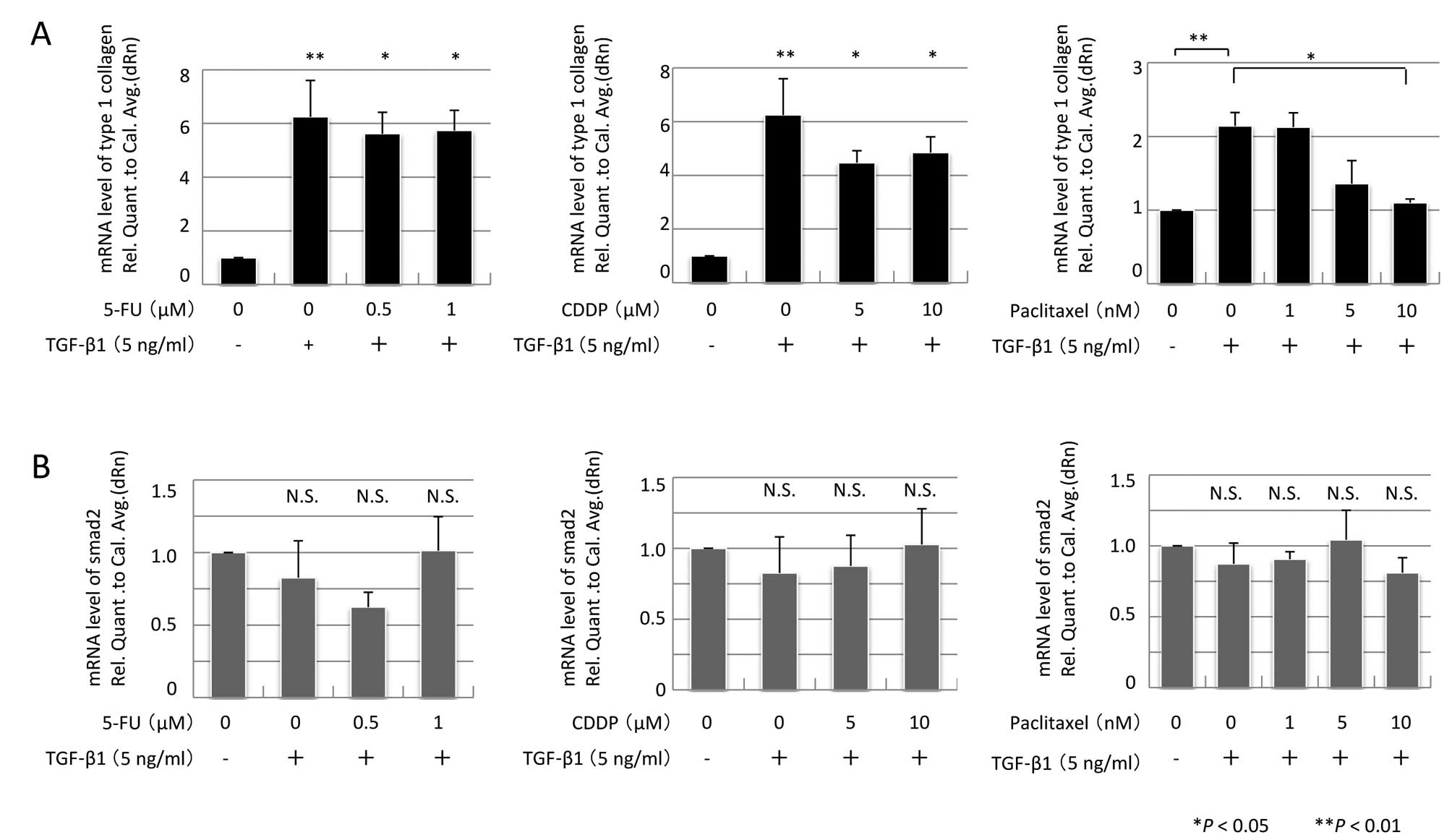
Collagen I mRNA expression was assessed by real-time
qPCR. TGF-β1 stimulation induced parallel increases in both α-SMA
and collagen I mRNA expression (Fig.
5A). Pre-treatment with 5-FU or CDDP did not suppress collagen
I mRNA expression; however, pre-treatment with 10 nM PTX promoted
attenuation of collagen I mRNA expression (P=0.031, n=3).
To investigate the TGF/Smad pathway, Smad2/3 and
Smad2 mRNA expression were assessed, but there were no alterations
in their expression (Figs. 4 and
5B). In contrast, TGF-β1 induced a
fourfold increase in Smad2 phosphorylation (P<0.01, n=3).
Pre-treatment with either 5 or 10 nM PTX significantly suppressed
TGF-β1-induced phosphorylation of Smad2 (P=0.042 and P <0.01,
respectively, n=3) (Fig. 6).
Discussion
In the present study, we investigated the minimum
cytostatic concentration of PTX that would not cause cytotoxicity
in HPMCs. We had previously reported that TGF-β1-mediated
activation of HPMCs is one of the origins of cancer-associated
fibroblasts and can promote peritoneal fibrosis (6). Low-dose PTX might have the potential
to reduce TGF-β1-mediated activation of HPMCs in the peritoneal
microenvironment.
The diffusely infiltrating human scirrhous gastric
carcinoma is characterized by cancer cell infiltration and
proliferation accompanied with extensive stromal fibrosis (29). At the peritoneal dissemination
site, cancer cells usually generate a supportive microenvironment
by producing stroma-modulating growth factors such as the
fibroblast growth factor (FGF) family, platelet-derived growth
factor (PDGF), epidermal growth factor (EGF) ligands, vascular
endothelial growth factor (VEGF) family, interleukins and TGF-β
(30,31). In particular, TGF-β1 expression is
correlated with the malignant potential of scirrhous gastric cancer
(32) and contributes to adhesion,
migration and invasion in the peritoneal dissemination of scirrhous
gastric cancer (33). In addition,
TGF-β produced by orthotopic fibroblasts has been shown to
contribute to cell growth and extensive stromal fibrosis at the
primary cancer site (34) and
stimulates both the invasion and adhesion of scirrhous gastric
cancer cells to the peritoneum (35,36).
In Japanese populations, 3-h infusions of PTX at the
clinical dosages of 105, 135, 180, 210, 240 and 270
mg/m2 have been shown to result in peak plasma
concentrations of 2600–14,000 nM. Peak plasma levels ranging from
40 to 120 nM have been obtained on administering PTX over 24 h
(37). However, inhibition of
TGF-β/Smad signalling can be achieved with very low doses of PTX.
In addition, inhibition of migration and proliferation potential by
different concentrations of PTX has been observed in tumour cell
lines (38), epithelial cells
(39), fibroblasts (40,41)
and vascular smooth muscle cells (21). However, no studies have
investigated the effect of PTX on HPMCs and no guidelines are
available regarding the appropriate concentration for inhibiting
fibrosis.
We found that the minimum cytostatic concentration
of PTX was 5 nM. Interestingly, in the gastric cancer cell line,
MKN45, exposure to 5 nM PTX resulted in lesser cell viability than
the 50% inhibitory concentration (IC50), suggesting that
the cytostatic concentration of PTX for HPMCs might be cytotoxic to
gastric cancer cells. We have also verified that TGF-β1 induces a
morphological change in HPMC and an associated elevation in α-SMA
expression. These fibroblastic changes in HPMCs contribute to
fibrosis by inducing collagen synthesis. Low-dose PTX (5 nM)
inhibited a series of changes associated with EMT.
The Smad pathway plays a major role in the EMT
process. R-Smads (Smad2 and Smad3) are composed of 3 regions: the
N-terminal Mad-homology (MH) 1 domain that has DNA-binding
activity, the C-terminal MH2 domain that has protein-binding
properties and a middle linker region (42). The C-terminal phosphorylation of
R-Smads is mediated by the activated TβR-I receptor, whereas middle
linker region phosphorylation is mediated by mitogen-activated
protein kinase (MAPK) (13). Other
recent studies have shown that TGF-β can also activate non-Smad
signalling cascades, including the MAPK pathway, leading to
activation of MAPK-Erk, Jun N-terminal kinase (JNK) and p38MAPK
(43,44). In rat peritoneal mesothelial cells,
the JNK-Smad3 pathway contributes to peritoneal fibrosis (45), but the phosphorylation of R-Smads
at the middle linker regions has been suggested to be cell specific
(46). Therefore, we focused on
the Smad pathway, which is common to many cells and on Smad2
phosphorylation, instead of Smad3 in HPMCs.
In the present study, we showed that TGF-β1
stimulation resulted in the phosphorylation of Smad2 in HPMCs and
that this phosphorylation was inhibited by pre-treatment with 5 nM.
In contrast to our findings, Wendling et al reported that
5-FU blocked TGF-β actions in human fibroblasts although this was
in a different cell line and with a different concentration of 5-FU
(47). We also determined that
CDDP did not alter the action of TGF-β in HPMCs.
In conclusion, we have shown that low-dose PTX can
significantly suppress TGF-β/Smad signalling by inhibiting Smad2
phosphorylation and decrease stromal fibrosis in human peritoneum
cells. While our study does not prove that this happens in
vivo, low-dose PTX has been shown to attenuate fibrosis in a
rat model of unilateral ureteral obstruction (48). Furthermore, low-dose PTX (5 nM)
prevented peritoneal fibrosis and was cytotoxic to gastric cancer
cells. Therefore, combination therapies with low-dose cytostatic
PTX and other cytotoxic antineoplastic agents could potentially
become the expected regimen for peritoneal dissemination of gastric
cancer. We hope that the results of the present study will provide
an impetus for future investigations of novel treatment strategies
for fibrotic peritoneal dissemination of gastric cancer.
Acknowledgements
We are grateful to the members of the
Department of Gastroenterologic Surgery of Kanazawa University for
their helpful suggestions. We thank Dr Tomohiko Wakayama and
Professor Shoichi Iseki for providing technical support for
fluorescence microscopy.
References
|
1
|
Fushida S, Kinoshita J, Yagi Y, Funaki H,
Kinami S, Ninomiya I, Fujimura T, Nishimura G, Kayahara M and Ohta
T: Dual anti-cancer effects of weekly intraperitoneal docetaxel in
treatment of advanced gastric cancer patients with peritoneal
carcinomatosis: a feasibility and pharmacokinetic study. Oncol Rep.
19:1305–1310. 2008.
|
|
2
|
Shimada S, Tanaka E, Marutsuka T, Honmyo
U, Tokunaga H, Yagi Y, Aoki N and Ogawa M: Extensive intraoperative
peritoneal lavage and chemotherapy for gastric cancer patients with
peritoneal free cancer cells. Gastric Cancer. 5:168–172. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yonemura Y, Kawamura T, Nojima N, Bandou
E, Keizou T, Fujita H, Michiwa Y, Fujimura T, Fushida S, Ajisaka H
and Miwa K: Postoperative results of left upper abdominal
evisceration for advanced gastric cancer. Hepatogastroenterology.
47:571–574. 2000.PubMed/NCBI
|
|
4
|
Koizumi W, Narahara H, Hara T, Takagane A,
Akiya T, Takagi M, Miyashita K, Nishizaki T, Kobayashi O, Takiyama
W, Toh Y, Nagaie T, Takagi S, Yamamura Y, Yanaoka K, Orita H and
Takeuchi M: S-1 plus cisplatin versus S-1 alone for first-line
treatment of advanced gastric cancer (SPIRITS trial): a phase III
trial. Lancet Oncol. 9:215–221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shirao K, Boku N, Yamada Y, Yamaguchi K,
Doi T, Takiuchi H, Nasu J, Nakamura K, Fukuda H and Ohtsu A:
Randomized phase III study of 5-fluorouracil continuous infusion
(5FUci) versus methotrexate and 5-FU sequential therapy (MF) in
gastric cancer with peritoneal metastasis (JCOG0106). Proc ASCO.
27(Suppl 15): abs. 4545. 2009.
|
|
6
|
Tsukada T, Fushida S, Harada S, Yagi Y,
Kinoshita J, Oyama K, Tajima H, Fujita H, Ninomiya I, Fujimura T
and Ohta T: The role of human peritoneal mesothelial cells in the
fibrosis and progression of gastric cancer. Int J Oncol.
41:476–482. 2012.PubMed/NCBI
|
|
7
|
Lv ZD, Na D, Ma XY, Zhao C, Zhao WJ and Xu
HM: Human peritoneal mesothelial cell transformation into
myofibroblasts in response to TGF-β1 in vitro. Int J Mol Med.
27:187–193. 2011.PubMed/NCBI
|
|
8
|
Hay ED: An overview of
epithelio-mesenchymal transformation. Acta Anat. 154:8–20. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kalluri R and Neilson EG:
Epithelial-mesenchymal transition and its implications for
fibrosis. J Clin Invest. 112:1776–1784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nawshad A, Lagamba D, Polad A and Hay ED:
Transforming growth factor-beta signaling during
epithelial-mesenchymal transformation: implications for
embryogenesis and tumor metastasis. Cells Tissues Organs.
179:11–23. 2005. View Article : Google Scholar
|
|
12
|
Miyazono K, Suzuki H and Imamura T:
Regulation of TGF-beta signaling and its roles in progression of
tumors. Cancer Sci. 94:230–234. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dong C, Li Z, Alvarez R Jr, Feng XH and
Goldschmidt-Clermont PJ: Microtubule binding to Smads may regulate
TGF beta activity. Mol Cell. 5:27–34. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Donaldson KL, Goolsby GL, Kiener PA and
Wahl AF: Activation of p34cdc2 coincident with taxol-induced
apoptosis. Cell Growth Differ. 5:1041–1050. 1994.PubMed/NCBI
|
|
16
|
Gelmon K: The taxoids: paclitaxel and
docetaxel. Lancet. 344:1267–1272. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ishigami H, Kitayama J, Kaisaki S,
Hidemura A, Kato M, Otani K, Kamei T, Soma D, Miyato H, Yamashita H
and Nagawa H: Phase II study of weekly intravenous and
intraperitoneal paclitaxel combined with S-1 for advanced gastric
cancer with peritoneal metastasis. Ann Oncol. 21:67–70. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Markman M, Bundy BN, Alberts DS, Fowler
JM, Clark-Pearson DL, Carson LF, Wadler S and Sickel J: Phase III
trial of standard-dose intravenous cisplatin plus paclitaxel versus
moderately high-dose carboplatin followed by intravenous paclitaxel
and intraperitoneal cisplatin in small-volume stage III ovarian
carcinoma: an intergroup study of the Gynecologic Oncology Group,
Southwestern Oncology Group and Eastern Cooperative Oncology Group.
J Clin Oncol. 19:1001–1007. 2001.
|
|
19
|
Sakurai Y, Yoshida I, Tonomura S, Sakai W,
Nakamura Y, Imazu H, Matsubara T and Ochiai M: Weekly
administration of paclitaxel attenuated rectal stenosis caused by
multiple peritoneal recurrence 8 years after the resection of
gastric carcinoma. Gastric Cancer. 6:243–249. 2003.
|
|
20
|
Jordan MA, Toso RJ, Thrower D and Wilson
L: Mechanism of mitotic block and inhibition of cell proliferation
by taxol at low concentrations. Proc Natl Acad Sci USA.
90:9552–9556. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Axel DI, Kunert W, Göggelmann C, Oberhoff
M, Herdeg C, Küttner A, Wild DH, Brehm BR, Riessen R, Köveker G and
Karsch KR: Paclitaxel inhibits arterial smooth muscle cell
proliferation and migration in vitro and in vivo using local drug
delivery. Circulation. 96:636–645. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Colombo A, Drzewiecki J, Banning A, Grube
E, Hauptmann K, Silber S, Dudek D, Fort S, Schiele F, Zmudka K and
Guagliumi G: Randomized study to assess the effectiveness of slow-
and moderate-release polymer-based paclitaxel-eluting stents for
coronary artery lesions. Circulation. 108:788–794. 2003. View Article : Google Scholar
|
|
23
|
Stone GW, Ellis SG, Cox DA, Hermiller J,
O’Shaughnessy C, Mann JT, Turco M, Caputo R, Bergin P, Greenberg J,
Popma JJ and Russell ME; TAXUS-IV Investigators: A polymer-based,
paclitaxel-eluting stent in patients with coronary artery disease.
N Engl J Med. 350:221–231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu X, Zhu S, Wang T, Hummers L, Wigley
FM, Goldschmidt-Clermont PJ and Dong C: Paclitaxel modulates
TGFbeta signaling in scleroderma skin grafts in immunodeficient
mice. PLoS Med. 2:1334–1442. 2005.PubMed/NCBI
|
|
25
|
Choi HS, Savard CE, Choi JW, Kuver R and
Lee SP: Paclitaxel interrupts TGF-beta1 signaling between
gallbladder epithelial cells and myofibroblasts. J Surg Res.
141:183–191. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yung S, Li FK and Chan TM: Peritoneal
mesothelial cell culture and biology. Perit Dial Int. 26:162–173.
2006.PubMed/NCBI
|
|
27
|
Wakayama T, Sai Y, Ito A, Kato Y, Kurobo
M, Murakami Y, Nakashima E, Tsuji A, Kitamura Y and Iseki S:
Heterophilic binding of the adhesion molecules poliovirus receptor
and immunoglobulin superfamily 4A in the interaction between mouse
spermatogenic and Sertoli cells. Biol Reprod. 76:1081–1090. 2007.
View Article : Google Scholar
|
|
28
|
Yagi Y, Fushida S, Harada S, Tsukada T,
Kinoshita J, Oyama K, Fujita H, Ninomiya I, Fujimura T, Kayahara M,
Kinuya S, Yashiro M, Hirakawa K and Ohta T: Biodistribution of
humanized anti-VEGF monoclonal antibody/bevacizumab on peritoneal
metastatic models with subcutaneous xenograft of gastric cancer in
mice. Cancer Chemother Pharmacol. 66:745–753. 2010. View Article : Google Scholar
|
|
29
|
Yashiro M, Chung YS, Nishimura S, Inoue T
and Sowa M: Fibrosis in the peritoneum induced by scirrhous gastric
cancer cells may act as ‘soil’ for peritoneal dissemination.
Cancer. 77:1668–1675. 1996.PubMed/NCBI
|
|
30
|
Nakazawa K, Yashiro M and Hirakawa K:
Keratinocyte growth factor produced by gastric fibroblasts
specifically stimulates proliferation of cancer cells from
scirrhous gastric carcinoma. Cancer Res. 63:8848–8852. 2003.
|
|
31
|
Elenbaas B and Weinberg RA: Heterotypic
signaling between epithelial tumor cells and fibroblasts in
carcinoma formation. Exp Cell Res. 264:169–184. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kinugasa S, Abe S, Tachibana M, Hishikawa
Y, Yoshimura H, Monden N, Dhar DK, Nagasue N and Nagaoka S: Over
expression of transforming growth factor-beta1 in scirrhous
carcinoma of the stomach correlates with decreased survival.
Oncology. 55:582–587. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shinto O, Yashiro M, Kawajiri H, Shimizu
K, Shimizu T, Miwa A and Hirakawa K: Inhibitory effect of a TGFbeta
receptor type-I inhibitor, Ki26894, on invasiveness of scirrhous
gastric cancer cells. Br J Cancer. 102:844–851. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yashiro M, Chung YS and Sowa M: Role of
orthotopic fibroblasts in the development of scirrhous gastric
carcinoma. Jpn J Cancer Res. 85:883–886. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Inoue T, Chung YS, Yashiro M, Nishimura S,
Hasuma T, Otani S and Sowa M: Transforming growth factor-beta and
hepatocyte growth factor produced by gastric fibroblasts stimulate
the invasiveness of scirrhous gastric cancer cells. Jpn J Cancer
Res. 88:152–159. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Koyama T, Yashiro M, Inoue T, Nishimura S,
Hirakawa YS and Chung K: TGF-beta1 secreted by gastric fibroblasts
up-regulates CD44 H expression and stimulates the peritoneal
metastatic ability of scirrhous gastric cancer cells. Int J Oncol.
16:355–362. 2000.PubMed/NCBI
|
|
37
|
Tamura T, Sasaki Y, Nishiwaki Y and Saijo
N: Phase I study of paclitaxel by three-hour infusion: hypotension
just after infusion is one of the major dose-limiting toxicities.
Jpn J Cancer Res. 86:1203–1209. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Matsuoka H, Furusawa M, Tomoda H and Seo
Y: Difference in cytotoxicity of paclitaxel against neoplastic and
normal cells. Anticancer Res. 14:163–167. 1994.PubMed/NCBI
|
|
39
|
Gloushankova NA, Lyubimova AV, Tint IS,
Feder HH, Vasiliev JM and Gelfand IM: Role of the microtubular
system in morphological organization of normal and
oncogene-transfected epithelial cells. Proc Natl Acad Sci USA.
91:8597–8601. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schiff PB and Horwitz SB: Taxol stabilizes
microtubules in mouse fibroblast cells. Proc Natl Acad Sci USA.
77:1561–1565. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhu S, Goldschmidt-Clermont PJ and Dong C:
Transforming growth factor-β-induced inhibition of myogenesis is
mediated through Smad pathway and is modulated by microtubule
dynamic stability. Circ Res. 94:617–625. 2004.
|
|
42
|
Shi Y, Hata A, Lo RS, Massagué J and
Pavletich NP: A structural basis for mutational inactivation of the
tumour suppressor Smad4. Nature. 388:87–93. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shi Y and Massague J: Mechanisms of TGF-
signaling from cell membrane to the nucleus. Cell. 113:685–700.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ten Dijke P, Goumans MJ, Itoh F and Itoh
S: Regulation of cell proliferation by Smad proteins. J Cell
Physiol. 191:1–16. 2002.PubMed/NCBI
|
|
45
|
Liu Q, Mao H, Nie J, Chen W, Yang Q, Dong
X and Yu X: Transforming growth factor β1 induces
epithelial-mesenchymal transition by activating the JNK-Smad3
pathway in rat peritoneal mesothelial cells. Perit Dial Int.
28:S88–S95. 2008.
|
|
46
|
Saika S, Yamanaka O, Ikeda K,
Kim-Mitsuyama S, Flanders KC, Yoo J, Roberts AB, Nishikawa-Ishida
I, Ohnishi Y, Muragaki Y and Ooshima A: Inhibition of p38MAP kinase
suppresses fibrotic reaction of retinal pigment epithelial cells.
Lab Invest. 85:838–850. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wendling J, Marchand A, Mauviel A and
Verrecchia F: 5-fluorouracil blocks transforming growth
factor-beta-induced alpha 2 type I collagen gene (COL1A2)
expression in human fibroblasts via c-Jun NH2-terminal
kinase/activator protein-1 activation. Mol Pharmacol. 64:707–713.
2003. View Article : Google Scholar
|
|
48
|
Zhang D, Sun L, Xian W, Liu F, Ling G,
Xiao L, Liu Y, Peng Y, Haruna Y and Kanwar YS: Low-dose paclitaxel
ameliorates renal fibrosis in rat UUO model by inhibition of
TGF-beta/Smad activity. Lab Invest. 90:436–447. 2010. View Article : Google Scholar : PubMed/NCBI
|