Introduction
Among human malignancies, head and neck cancer is
the sixth most common cancer in the world (1). Head and neck cancer is an aggressive
and life-threatening disease with poor morbidity and high mortality
in advanced disease. More than 40,000 new cases of head and neck
squamous cell carcinoma (HNSCC) are diagnosed in the United States
each year, with 12,000 US deaths annually. Survival rates have not
improved significantly for patients with HNSCC in the past thirty
years despite active clinical and basic research addressing this
issue. Treatment for HNSCC includes surgical resection,
chemotherapy and radiation therapy; however, approximately 50% of
all patients have advanced disease at the time of diagnosis often
requiring use of all three treatment modalities. Therefore, it is
important to discover new biomarkers in a cancer-specific manner
and to develop new methods that provide sensitive and reliable
biomarkers of HNSCC for detection, treatment response and
prognosis.
Genetic alterations are a hallmark of human cancer,
with the activation of proto-oncogenes and inactivation of tumor
suppressor genes, either through deletion or inactivating point
mutations, being well defined (2).
In addition to these genetic alterations, changes in DNA
methylation, an epigenetic process present in mammalian cells, are
also a hallmark of human cancer (3). Silencing of tumor suppressor genes by
means of promoter hypermethylation plays an important role in head
and neck carcinogenesis (4).
Methylation of the CpG islands in the promoter regions of tumor
suppressor genes is frequently observed with resultant reduced gene
expression (5,6). To discover the new cancer-specific
hypermethylated genes, gene expression profiling via
oligonucleotide microarray-based approach is a reliable technology
for whole genome epigenetic research (7). Measuring promoter hypermethylation by
using real-time quantitative methylation-specific PCR (QMSP) allows
an objective, robust, and rapid assessment of promoter methylation
status (8–11).
Previously, we employed a pharmacologic unmasking
expression array technique using a 12K gene expression array to
identify epigenetically inactivated genes in HNSCC (12). In this study, we expanded this
approach using a whole genome 47K array platform and then performed
bisulfite DNA sequencing for 126 selected genes and QMSP for seven
selected genes after evaluating the bisulfite sequencing results,
to validate HNSCC-specific methylation in novel genes.
Materials and methods
Cell lines
We used 2 human head and neck cancer cell lines,
JHU-011 and JHU-012, which were developed from a laryngeal primary
and a neck node metastasis of different HNSCC patients, at the
Department of Otolaryngology-Head and Neck Surgery, Johns Hopkins
University. Cell lines were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum and 1%
penicillin-streptomycin. All media components were obtained from
Life Technologies Invitrogen Corp. All cell lines tested negative
for any mycoplasma contamination.
5-aza-2′-deoxycytidine treatment
Cell lines were treated with 5-aza-2′-deoxycytidine
(5-aza-dC, a demethylating agent) and trichostatin A (TSA, a
histone deacetylase inhibitor) as previously described (7,12).
Briefly, we seeded all cell lines (1×106) in their
respective culture medium and maintained them for 24 h before
treating them with 5 μM 5-aza-dC (Sigma, St. Louis, MO, USA)
for 5 days and 300 nM for the final 24 h. We renewed medium
containing 5-aza-dC every 24 h during the treatment and handled
control cells similarly, without adding 5-aza-dC. Stock solutions
of 5-aza-dC (Sigma) and TSA (Sigma) were dissolved in DMSO (Sigma)
and ethanol (100%), respectively.
Tissue samples
After obtaining institutional review board approval
and appropriate informed consent, the HNSCC patients and control
population (healthy subjects enrolled in a community screening
study) were recruited at the Johns Hopkins School of Medicine,
Department of Otolaryngology-Head and Neck Surgery. Five normal
mucosa samples from healthy individuals by
uvulopalatopharyngoplasty (UPPP) technique, 13 HNSCC tumors for
mRNA expression array experiments, 22 salivary rinses and 14
mucosal samples from a healthy population and 33 HNSCC tumor
samples were collected. Salivary rinses were obtained by brushing
oral cavity and oropharyngeal surfaces with an exfoliating brush
followed by rinse and gargle with 20 ml normal saline solution. The
brush was gently agitated to release the obtained material into
saline. Following centrifugation, the supernatant was discarded and
DNA was isolated from the pellet. Tumors were snap frozen and
microdissected on a cryostat to ≥75% purity. DNA from 22 salivary
rinse samples and 14 normal mucosa samples from healthy individuals
were analyzed as a control, to investigate the normal promoter
methylation status of seven newly identified candidate genes,
MAP2K3 (mitogen-activated protein kinase kinase 3) (n=18),
MAP3K3 (mitogen-activated protein kinase kinase kinase 3)
(n=18), GNG7 (guanine nucleotide-binding protein, γ-7)
(n=17), GALNT10 (UDP-N-acetyl-α-D-galactosamine:polypeptide
N-acetylgalactosaminyltransferase 10) (n=18), PPFIBP2 (PTPRF
interacting protein, binding protein 2) (n=17), TUSC2 (tumor
suppressor candidate 2) (n=16) and TXNIP (thioredoxin
interacting protein) (n=16). The methylation status of these genes
was analyzed in 33 fresh HNSCC tumor samples.
RNA isolation, cDNA synthesis and probe
hybridization
We prepared total RNA from 13 HNSCC tumors, 5 normal
mucosa samples and cell lines using the RNeasy Mini kit (Qiagen,
Valencia, CA, USA). Total RNA quality was checked via the Nanodrop
Spectrophotometer and Agilent Bioanalyzer total RNA series II kit.
Total RNA (1 μg) was combined with 2 μl T7 oligo(dT)
primer and 2 μl Poly-A controls and brought to a volume of
12 μl. The samples were incubated at 70°C for 10 min. A
master mix of 4 μl first strand buffer, 2 μl DTT and
1 μl 10 mM dNTPs was added to the samples, followed by a
2-min incubation at 42°C. Superscript II (Invitrogen, Carlsbad, CA,
USA) (1 μl) was added to each sample and the samples were
incubated at 42°C for 1 h for first strand cDNA synthesis. A master
mix of 91 μl water, 30 μl 5X second strand buffer, 3
μl 10 mM dNTPs, 4 μl DNA polymerase I, 1 μl
E. coli DNA ligase and 1 μl RNase H was added to each
sample and the samples were incubated for 2 h at 16°C for second
strand cDNA synthesis. T4 DNA polymerase (2 μl) was added
followed by a 5-min incubation at 16°C. EDTA (10 μl, 0.5 M)
was added to stop the reaction. cDNA cleanup was performed with the
Affymetrix GeneChip Sample Cleanup Module, according to the
manufacturer’s instructions. The cDNA was eluted in 14 μl
elution buffer. The final elution volume (∼12 μl) was
combined with 28 μl of IVT mix master mix (4 μl 10X
IVT labeling buffer, 12 μl labeling NTP mix, 4 μl
labeling enzyme mix, 8 μl RNAse-free water) for the in
vitro transcription cRNA synthesis reaction. The samples were
incubated overnight for 16 h at 37°C followed by a hold at 4°C.
Cleanup was performed as per the manufacturer’s protocol using the
Affymetrix GeneChip Sample Cleanup Module. The final elution volume
was ∼19 μl. Concentration was checked via a nanodrop
spectrophotometer. Fragmentation buffer (5X) (6 μl) was then
combined with 15 μg cRNA and incubated at 95°C for 35 min to
fragment the cRNA. The samples were ice quenched and combined with
the hybridization cocktail. After 10 min of pre-hybridizing Human
U133 Plus 2.0 Genome array at 45°C, 60 rpm, 200 μl of
cocktail was loaded onto each array and the arrays were hybridized
for 16 h at 45°C, 60 rpm. The cocktail was removed and the arrays
were stained and washed using the Affymetrix GeneChip Fluidics
Station 450 and FS450_001 fluidics script. All arrays were scanned
in the Affymetrix GeneChip Scanner 3000 and the raw analysis was
performed with Affymetrix GeneChip Operating System (GCOS) 1.4.
Following RNA isolation, the expertise, facilities and
instrumentation for Affymetrix GeneChip experimentation were
performed at the JHU microarray core facility. All reagents needed
for cDNA synthesis and probe hybridization were provided by
Affymetrix (Santa Clara, CA, USA)
Microarray data analysis
mRNA gene expression profiling was performed using
Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays containing
47K probe sets (Affymetrix). Signal intensity and statistical
significance were established for each transcript initially using
dChip version 2008 (13) and then
significance analysis of microarrays (SAM) (14) software to analyze and normalize the
array data. Default settings for dChip were used, including the
perfect match/mismatch difference model, invariant set
normalization and check single/probe/array outlier algorithm. The
mRNA gene expression profiling data has been deposited in the Gene
Expression Omnibus (GEO) database with the accession no.
GSE29330.
DNA extraction and bisulfite
treatment
DNA was isolated as previously described (15). In brief, DNA was obtained by
phenol/chloroform extraction following overnight incubation with
proteinase K (Boehringer-Mannheim, Germany) at 48°C. DNA from tumor
and control samples was subjected to bisulfite treatment using
Epitect Bisulfite Modification kit (Qiagen) according to the
manufacturer’s instructions.
Bisulfite sequencing
Bisulfite sequence analysis was performed to
determine the methylation status in the promoter regions of 126
genes obtained from gene expression profiling in normal mucosal and
HNSCC tumor samples. Bisulfite-treated DNA was amplified for the 5′
region that included at least a portion of the CpG island within
1–2 kb of the first exon of 126 genes, using primer sets (data not
shown). The promoter regions of the genes were found from the
database of the University of California, Santa Cruz, USA (UCSC)
(http://genome.ucsc.edu/). Primer sequences were
determined by the MethPrimer program (16) showing the CpG islands in the
promoter regions of 126 genes for bisulfite sequencing. The primers
for bisulfite sequencing were designed to hybridize to regions in
the promoter without CpG dinucleotides. PCR products were
gel-purified using the QIAquick Gel Extraction kit (Qiagen)
according to the manufacturer’s instructions. Each amplified DNA
sample was sequenced by the Applied Biosystems 3700 DNA analyzer
using nested, forward or reverse primers and BD terminator dye
(Applied Biosystems, Foster City, CA, USA).
Quantitative methylation-specific
PCR
To determine if the methylated genes in tumor
samples were cancer-specific, we investigated promoter methylation
in 22 normal saliva, 14 age-matched normal mucosa from healthy
individuals that were analyzed as a control, to investigate the
normal promoter methylation status of seven newly identified
candidate genes (MAP3K3, MAP2K3, GNG7, GALNT10, PPFIBP2,
TUSC2 and TXNIP) and in 33 HNSCC tumor samples by QMSP.
Primer and probe sequences were determined by the MethPrimer
program showing the CpG islands in the promoter regions of seven
genes selected after bisulfite sequencing (Table I). Lymphocytes obtained from a
healthy individual were in vitro methylated using excess
SssI methyltransferase (New England Biolabs Inc., Beverly,
MA, USA) to generate completely methylated DNA that was used as a
positive control standard. To quantitate the relative percent of
methylation, we computed the ratio between the QMSP values of the
gene of interest relative to an internal control,
ACTB(15) (β-actin)
(Table I) (gene of
interest/reference gene × 100). Fluorogenic PCR was carried out in
a reaction volume of 20 μl consisting of 600 nM of each
primer; 200 nM of probe; 0.6 U of platinum Taq polymerase
(Invitrogen); 200 μM of each dATP, dCTP, dGTP and dTTP; 1X
Rox Dye reference and 1X buffer [16.6 mM of ammonium sulfate; 67 mM
of Trizma (Sigma); 6.7 mM of magnesium chloride; 10 mM of
mercaptoethanol; and 0.1% dimethylsulfoxide]. Thirty nanograms of
bisulfite-treated DNA were used in each real-time QMSP reaction.
Amplifications were carried out in 384-well plates in a 7900
Sequence Detector system (Perkin-Elmer Applied Biosystems, Norwalk,
CT, USA) and were analyzed by SDS 2.3 (Sequence Detector System)
(Applied Biosystems). Each reaction was performed in
triplicate.
 | Table IPrimers and probe sequences of
selected genes for validation by QMSP. |
Table I
Primers and probe sequences of
selected genes for validation by QMSP.
| Gene | Probe 5′-3′
(6-FAM-5′-3′-6-TAMRA) | Forward 5′-3′
(primer) | Reverse 5′-3′
(primer) |
|---|
| ACTB |
ACCACCACCCAACACACAATAACAAACACA |
TGGTGATGGAGGAGGTTTAGTAAGT |
AACCAATAAAACCTACTCCTCCCTTAA |
| MAP3K3 |
GGGAGTCGGGCGTTGTTTCGATG |
ATGCGTAGAGGCGGGGGTTT |
ACACGATAAACCAATCCCGCC |
| MAP2K3 |
GGGCGACGTTTGTTGGCGTTAGG |
CGTGTTGTTTCGTTATCGGGTA |
AACTATCTCCCGACGCTACTC |
| GNG7 |
GCGCGGGATTCGAATTCGCGAAAT |
CGGAGTTGGTATGTAGGATTCG |
CCCCGACTACGAAAAACCGAA |
| GALNT10 |
CGTTTCGGTTCGGTATTTTGTAGCG |
TCGTAAAGTTTTAGAGGGCGG |
AATCTCTACGCTACAAACTCGA |
| PPFIBP2 |
ACGAGGTAGGTTCGAAGGGGCG |
TAATCGGAGTTGTGCGGAGGA |
CCTATTCCCGAAAAACCGACC |
| TUSC2 |
CGGAAGCGGAAGTGAGGTTTTCGT |
AGGGCGTTTATTGGTTTCGTTT |
CGCAATCCGCACTACCATAAC |
| TXNIP |
CGAGGGTAGTACGAGTTTTCGGGT |
GCGATTTTATTGATTGGTCGGG |
CGTCTCTATATAATAACCCGAACC |
Results
Clinicopathological characteristics of
control subjects and patients with HNSCC
Table II describes
the demographic parameters of the sample populations used in this
study. The mean age of normal mucosal subjects was 43.4 years
(range 24–65). Forty-two percent of controls were tobacco users.
Normal mucosal and tumor subjects had a similar male and Caucasian
predominance. Among tumor patients, smoking rate was 78% and
alcohol consumption was 69%. Tumor samples (n=33) were obtained
from patients with stage I (7.4%), stage II (22%), stage III (26%)
and stage IV (44%) lesions. These were from primary tumors of the
oral cavity (n=9), oropharynx (n=7), hypopharynx (n=2), larynx
(n=8), maxillary sinus (n=2), nasal floor (n=1), salivary gland
(n=1) and unknown primary/neck (n=3). Male and Caucasian status was
less prevalent in the normal salivary rinse subjects and 36% were
tobacco users. Individuals from whom the normal salivary rinse was
obtained were slightly younger than the population of head and neck
cancer patients, with a mean age of 52.2 years (range 19–83) and
61.4 years (range 36–88), respectively. In Table III, the clinical findings of the
mRNA expression cohort are given.
| Table IIQMSP results and demographics of the
patients with HNSCC. |
Table II
QMSP results and demographics of the
patients with HNSCC.
| Tumor sample | GNG7 | TXNIP | TUSC2 | PPFIBP2 | GALNT10 | MAP2K3 | Age | Gendera | Racea | Smoking | Alcohol | Tumor site | Stage |
|---|
| 1 | N | N | N | N | N | Y | 67 | M | C | Yes | Yes | Nasal floor | 2 |
| 2 | N | N | Y | N | N | Y | 57 | M | C | Yes | Yes | Larynx | 2 |
| 3 | N | N | N | Y | N | Y | 61 | M | C | No | No | Neck | 3 |
| 4 | Y | Y | N | N | N | Y | 60 | F | C | Yes | Yes | Larynx | 4 |
| 5 | N | N | N | N | N | Y | 55 | M | AA | Yes | Yes | Larynx | 2 |
| 6 | Y | N | N | N | N | Y | 54 | M | C | No | Yes | Oropharynx | 4 |
| 7 | Y | Y | N | N | N | Y | 64 | M | AA | Yes | Yes | Hypopharynx | 3 |
| 8 | Y | N | N | N | N | Y | 55 | F | A | Yes | No | Oral cavity | 1 |
| 9 | Y | Y | N | N | N | N | 80 | M | C | Yes | No | Oral cavity | |
| 10 | Y | N | N | N | N | Y | 54 | F | C | No | | Oral cavity | 4 |
| 11 | N | Y | Y | Y | N | Y | 62 | M | C | Yes | Yes | Oropharynx | 4 |
| 12 | N | N | N | N | N | N | 72 | M | C | Yes | Yes | Hypopharynx | 3 |
| 13 | N | N | N | N | N | Y | 42 | M | C | Yes | No | Larynx | 2 |
| 14 | Y | Y | Y | N | N | Y | 66 | M | C | Yes | No | Oropharynx | 4 |
| 15 | N | N | N | N | N | Y | 74 | M | C | Yes | | Larynx | 2 |
| 16 | Y | N | N | N | N | Y | 58 | M | A | Yes | Yes | Oropharynx | 4 |
| 17 | Y | N | N | N | Y | Y | 56 | F | C | Yes | Yes | Oral Cavity | 2 |
| 18 | Y | N | N | N | N | Y | 43 | M | C | Yes | Yes | Oropharynx | 4 |
| 19 | Y | N | N | N | N | Y | 68 | M | C | Yes | Yes | Oropharynx | 4 |
| 20 | Y | N | N | N | N | Y | 63 | M | A | Yes | | Oral cavity | |
| 21 | Y | N | N | N | N | Y | 64 | F | C | No | Yes | Oral cavity | 3 |
| 22 | N | N | N | N | N | Y | 88 | M | C | Yes | Yes | Oral cavity | |
| 23 | Y | N | N | N | N | Y | 42 | M | C | Yes | No | Oral cavity | 3 |
| 24 | N | N | N | N | N | Y | 51 | M | C | Yes | Yes | Larynx | 4 |
| 25 | N | N | Y | N | N | Y | 80 | M | C | No | Yes | Neck | |
| 26 | Y | N | Y | N | N | Y | 58 | M | C | Yes | Yes | Larynx | 3 |
| 27 | Y | N | N | N | N | Y | 71 | M | C | Yes | Yes | Neck | |
| 28 | N | N | Y | N | N | Y | 48 | M | C | Yes | Yes | Oropharynx | 4 |
| 29 | Y | N | N | N | N | Y | 61 | M | C | Yes | No | MSb | 1 |
| 30 | Y | N | N | Y | N | Y | 77 | M | C | No | No | Salivary gland | |
| 31 | Y | N | N | Y | N | Y | 67 | M | C | | | Larynx | 3 |
| 32 | Y | N | N | N | N | Y | 36 | M | C | Yes | No | Oral Cavity | 4 |
| 33 | N | Y | N | N | Y | Y | 74 | F | C | No | Yes | MSb | 4 |
| Table IIIClinical characteristics of mRNA
expression array cohort. |
Table III
Clinical characteristics of mRNA
expression array cohort.
| Case | Diagnosis | Age | Gendera | Racea | Overall stage | T | N | M | Site | Tobacco | Alcohol |
|---|
| 1 | Normal | 20 | M | C | NA | NA | NA | NA | Left tonsil | No | No |
| 2 | Normal | 28 | M | C | NA | NA | NA | NA | Right tonsil | No | No |
| 3 | Normal | 28 | M | C | NA | NA | NA | NA | Uvula | No | No |
| 4 | Normal | 28 | M | C | NA | NA | NA | NA | Left tonsil | NA | NA |
| 5 | Normal | 30 | M | C | NA | NA | NA | NA | Uvula | No | No |
| 6 | Cancer | 62 | M | C | 3 | 3 | 0 | 0 | Larynx | No | Yes |
| 7 | Cancer | 80 | F | C | 1 | 1 | 2A | 0 | Oral cavity | No | NA |
| 8 | Cancer | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 9 | Cancer | 82 | M | C | 3 | 3 | 0 | 0 | Larynx | Yes | NA |
| 10 | Cancer | 74 | M | C | NA | NA | NA | NA | NA | NA | NA |
| 11 | Cancer | 71 | M | C | 4 | 4 | 0 | 0 | Oral cavity | Yes | No |
| 12 | Cancer | 56 | M | AA | NA | NA | NA | NA | NA | NA | NA |
| 13 | Cancer | 62 | F | AA | 1 | 1 | 0 | 0 | Oral cavity | Yes | Yes |
| 14 | Cancer | 58 | M | C | 3 | 2 | 2B | 0 | Oropharynx | No | Yes |
| 15 | Cancer | 61 | M | C | NA | NA | NA | NA | NA | NA | NA |
| 16 | Cancer | 89 | M | C | NA | NA | NA | NA | NA | Yes | Yes |
| 17 | Cancer | 63 | M | C | 2 | 2 | 0 | 0 | Larynx | Yes | Yes |
| 18 | Cancer | 50 | F | C | 4 | 3 | 2B | 0 | Oropharynx | Yes | Yes |
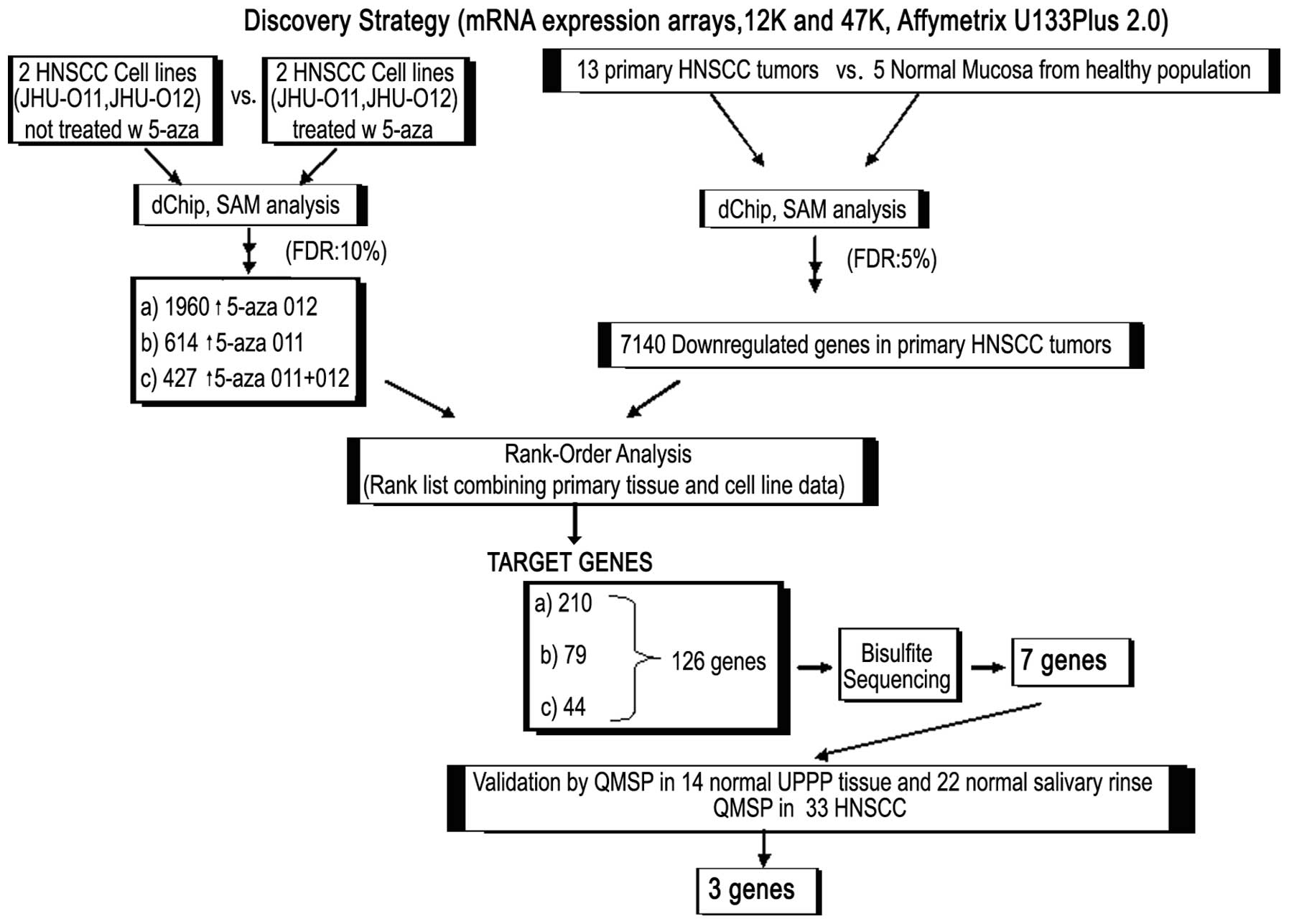
Workflow of gene methylation discovery
approach
We performed pharmacological unmasking analysis on
two HNSCC cancer cell lines JHU-O11 and JHU-O12 by treating cells
with or without 5-aza-dC (as a control group), followed by RNA
extraction and microarray analysis using Affymetrix U133 Plus 2.0.
The array data were analyzed initially by dChip and then SAM. We
performed a four-phase strategy to obtain the unmasked genes in the
cells treated with 5-aza and downregulated genes in primary tumors.
In the first phase, we compared the cell lines, either JHU-012 or
JHU-011, before treatment to the cell lines treated with 5-aza, in
order to identify genes that were reexpressed ≥2-fold. We found
1,960 genes that were upregulated by 5-aza-dC in the JHU-O12 cell
line. SAM output was obtained at a delta value of 2.05 with a false
discovery rate (FDR) of 10% and the d-score cut-off was 1.17. We
found 614 reexpressed genes in 5-aza-treated JHU-O11 (SAM output;
delta=2.089, FDR=10%, d-score cut-off=2.8); 427 genes were commonly
upregulated in both cell lines when the cell lines were normalized
and analyzed together (SAM output; delta=1.44, FDR=10%, d-score
cut-off=1.88) (Fig. 1). In the
second phase of our analysis, we further extracted RNA and
performed the 47K mRNA expression array analysis on 13 primary
HNSCC tumors and 5 normal mucosal samples from non-cancer control
patients. Following initial dChip and SAM analysis (SAM output;
delta=1.247, FDR=10%, d-score cut-off=0.24), we found 7,140
downregulated genes in primary HNSCC tumors compared with normal
mucosa. In the third phase, we investigated the three data sets
(Fig. 1) below: a) SAM output of
1,960 upregulated genes after 5-aza treatment of JHU-012 vs. SAM
output of 7,140 downregulated genes in primary HNSCC. We found that
210 genes that were upregulated by 5-aza-dC in the JHU-O12 cell
line and showed downregulation in tumor samples. b) SAM output of
614 upregulated genes after 5-aza treatment of JHU-011 vs. SAM
output of 7,140 downregulated genes in primary HNSCC. We found 79
genes that were upregulated by 5-aza-dC in the JHU-O11 cell line
and showed downregulation in tumor samples. c) SAM output from
analyzing both cell lines together in the same SAM computation, of
427 upregulated genes after 5-aza treatment of JHU-011 and JHU-012
vs. SAM output of 7,140 downregulated genes in primary HNSCC. We
found 44 genes that were upregulated by 5-aza-dC in the JHU-O11 and
JHU-012 cell lines and showed downregulation in tumor samples,
suggesting that methylation might be involved in gene
downregulation.
In the fourth phase of our strategy, we rank-ordered
the results of upregulated genes obtained from these 3 data sets
and found 126 common genes. We then examined promoter regions of
the 126 genes for CpG islands and performed bisulfite sequencing
analysis of the promoter region of these genes. We found that seven
genes showed a differential methylation pattern between normal and
neoplastic samples (Fig. 1).
Table IV shows the d-scores, fold
change and q-values of these genes by SAM analysis in human and
cell line samples. After validation of these genes in a cohort of
33 HNSCC patients and normal salivary and mucosal samples from
healthy individuals by QMSP, we found 3 genes of interest
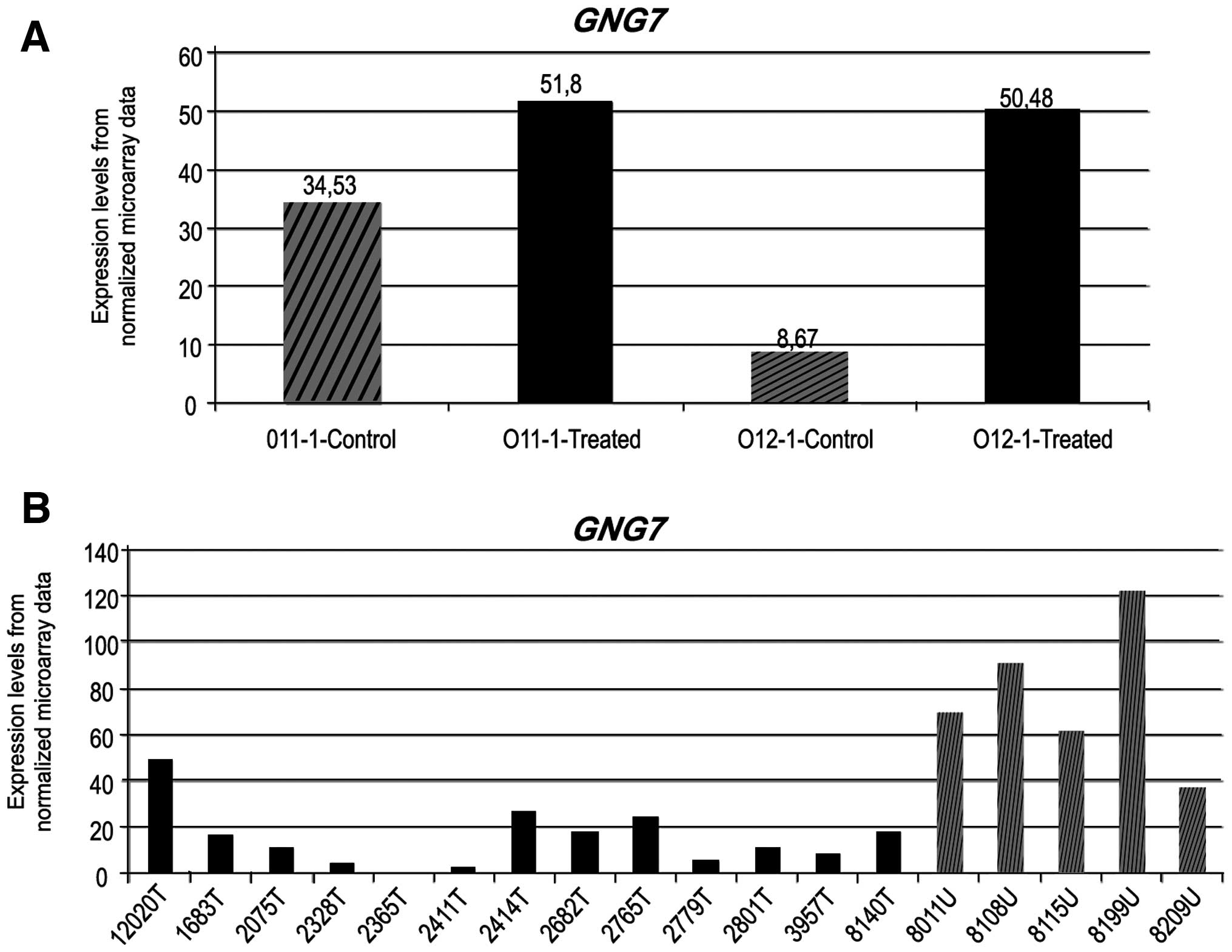
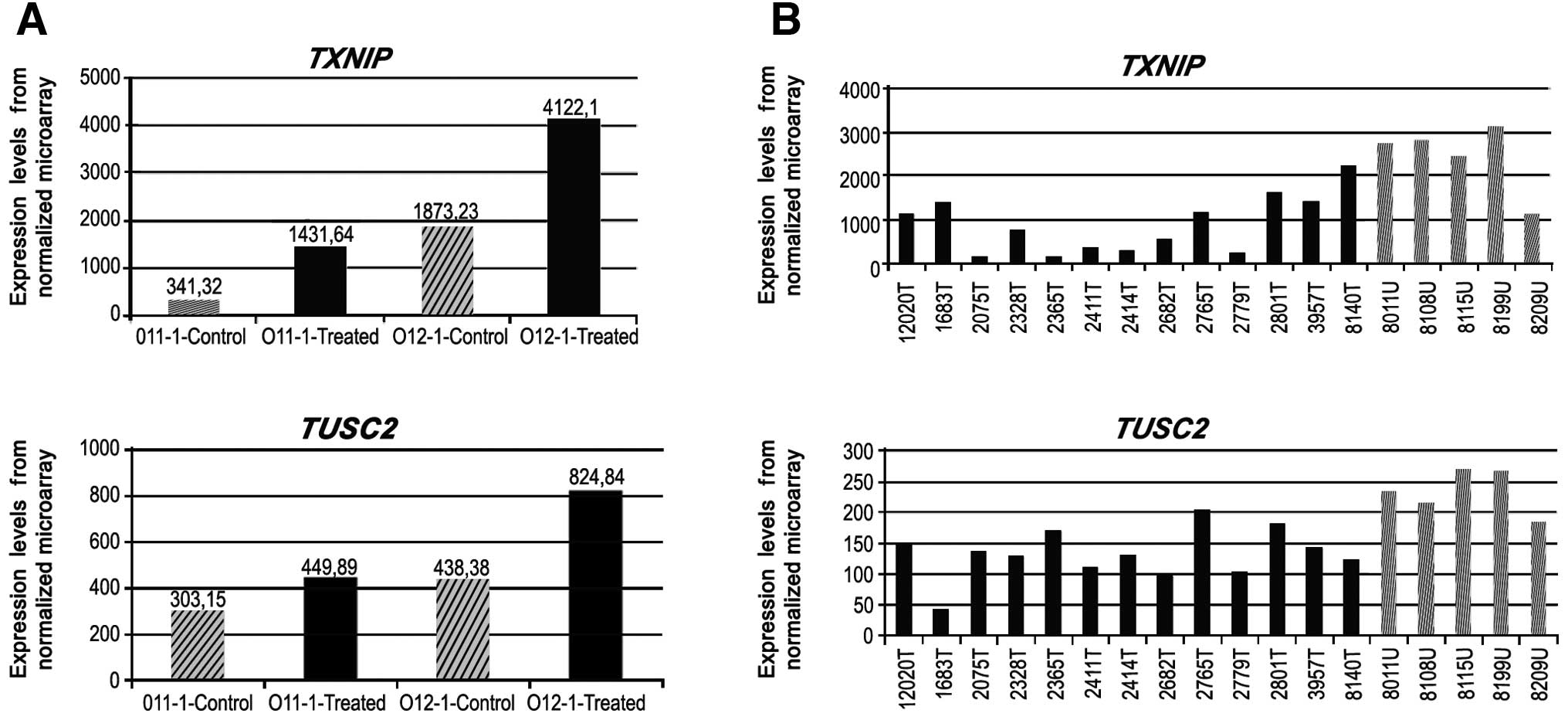
(GNG7, TXNIP and TUSC2). The upfold expression
arrays of these genes are given in Figs. 2 and 3. A 1.5-fold upregulation in GNG7
expression was observed in 5-aza-treated JHU-011, whereas the rate
of change was 5.8-fold in 5-aza-treated metastatic JHU-012 cells
(Fig. 2). The TXNIP gene
was upregulated 4.19-fold in JHU-011 cells, whereas a 2.2-fold
change was observed in the JHU-012 cells. The TUSC2 gene was
upregulated in a similar manner in both cell lines (Fig. 3).
| Table IVThe results of SAM analysis in human
and cell line specimens. |
Table IV
The results of SAM analysis in human
and cell line specimens.
Downregulated genes
from the SAM output of human tumor vs. normal mucosal tissues
| Upregulated genes
from the SAM analysis of 5-aza non-treated vs. 5-aza-treated
JHU-011 + JHU-012 cells
|
|---|
| Rank | Gene ID | Score
(d) | Fold
change | q-value
(%) | Rank | Gene ID | Score
(d) | Fold
change | q-value
(%) |
|---|
| 5506 | GALNT10 | −2.84 | 0.55 | 3.37 | 61 | GALNT10 | 8.38 | 2.28 | 2.55 |
| 5189 | MAP2K3 | −2.92 | 0.65 | 2.89 | 13 | MAP2K3 | 12.07 | 2.55 | 0.00 |
| 3406 | MAP3K3 | −3.44 | 0.73 | 1.47 | 309 | MAP3K3 | 5.15 | 2.12 | 7.68 |
| 1939 | PPFIBP2 | −3.99 | 0.58 | 0.86 | 389 | PPFIBP2 | 4.75 | 1.99 | 9.28 |
| | | | | Upregulated genes
from the SAM analysis of non-treated vs. 5-aza-treated JHU-012 cell
lines |
|
| 34 |
GNG7 | −8.32 | 0.21 | 0.00 | 313 |
GNG7 | 13.95 | 5.34 | 4.02 |
| 813 |
TUSC2 | −4.82 | 0.57 | 0.37 | 408 |
TUSC2 | 12.02 | 1.44 | 4.56 |
| | | | | Upregulated gene
from the SAM analysis of non-treated vs. 5-aza-treated JHU-011 cell
lines |
|
| 1317 |
TXNIP | −4.32 | 0.36 | 0.61 | 362 |
TXNIP | 9.55 | 5.39 | 7.40 |
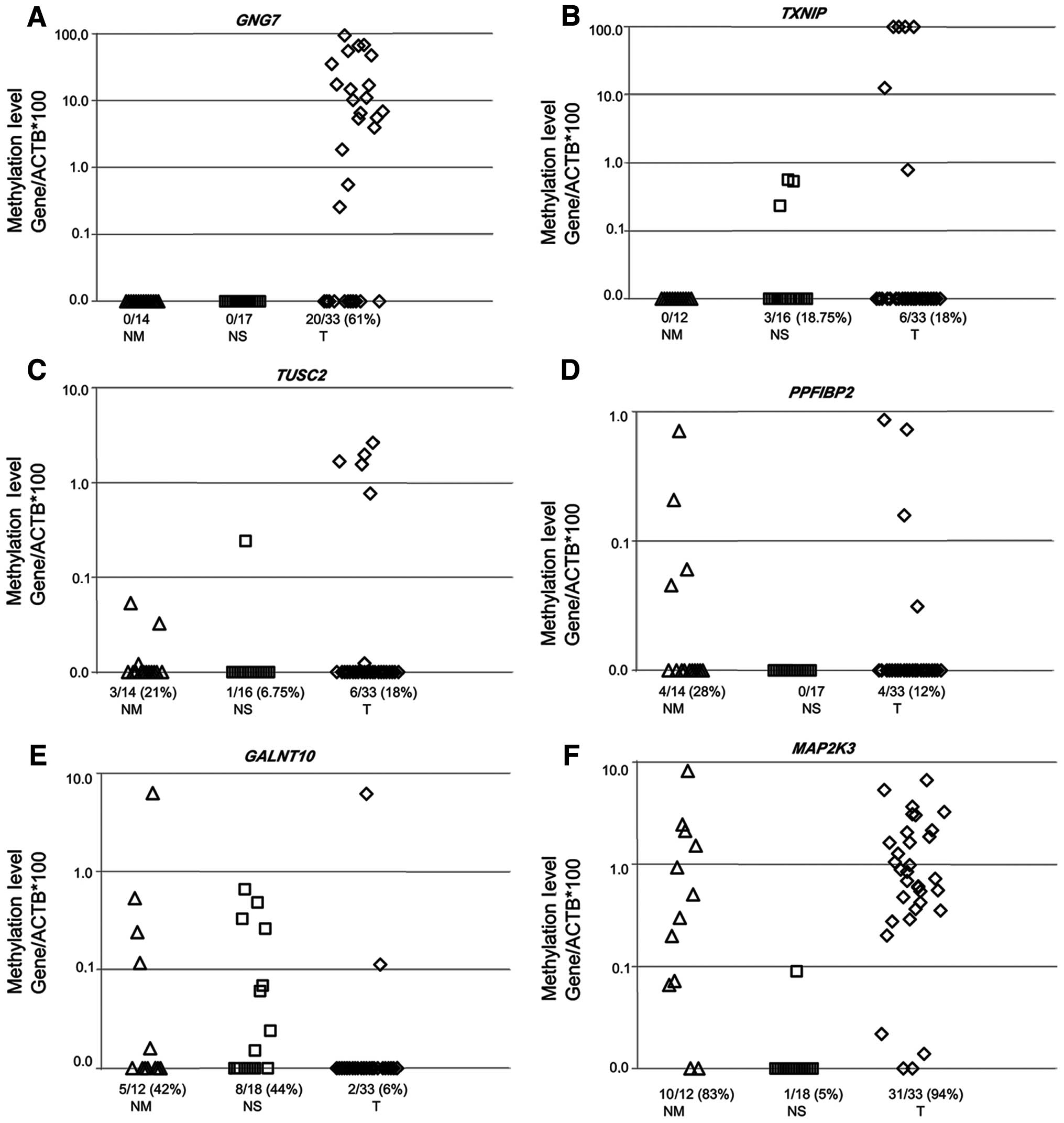
Genes specifically methylated in HNSCC
tumors
We then performed bisulfite sequencing analysis on
the promoter region of 126 genes as described above in our
discovery approach using 4 normal mucosal and 4 HNSCC samples.
Seven genes (GNG7, GALNT10, TXNIP, TUSC2, PPFIBP2, MAP2K3
and MAP3K3) were found to have no methylation in normal
mucosal samples but displayed high methylation frequency in HNSCC
samples (Table V). This high
specificity prompted us to further investigate the methylation
frequency in a larger cohort of normal mucosal and HNSCC specimens.
We then performed QMSP on 22 saliva and 14 normal mucosal samples
from healthy individuals and 33 HNSCC tumor samples for seven genes
selected. The GNG7 gene showed no methylation in normal
mucosal samples (0/14) and normal salivary rinses (0/17). The
methylation rate was 61% (20/33) on the promoter region of the
GNG7 gene in primary HNSCC tumor samples and these tumors
harbored high methylation values, mostly between 10 and 100%.
TXNIP, TUSC2, PPFIBP2, GALNT10 and MAP2K3
demonstrated varying degrees of methylation on their promoter
regions in normal mucosa, normal salivary rinses and HNSCC tumor
samples respectively (Fig. 4). We
observed no methylation in the promoter region of MAP3K3
[0/14 (0), 0/18 (0) and 0/33 (0)] gene in normal mucosa, normal
salivary rinses and HNSCC tumor samples, respectively.
| Table VMethylation analysis of candidate
genes in 4 normal mucosa and 4 tumor samples by bisulfite
sequencing. |
Table V
Methylation analysis of candidate
genes in 4 normal mucosa and 4 tumor samples by bisulfite
sequencing.
| Probe name | Gene ref ID | Gene name | Chromosome
location | Normal mucosa
tissue, n (%) | HNSCC tumor tissue,
n (%) |
|---|
| 215499_at | NM_145109 | MAP2K3 |
chr17:21,128,561–21,159,144 | 0 of 4 (0) | 4 of 4 (100) |
| 203514_at | NM_002401 | MAP3K3 |
chr17:59,053,533–59,127,402 | 0 of 4 (0) | 4 of 4 (100) |
| 220296_at | NM_198321 | GALNT10 |
chr5:153,550,488–153,780,003 | 0 of 4 (0) | 4 of 4 (100) |
| 201010_s_at | NM_006472 | TXNIP |
chr1:144,149,819–144,153,985 | 0 of 4 (0) | 4 of 4 (100) |
| 203273_s_at | NM_007275 | TUSC2 |
chr3:50,337,345–50,340,672 | 0 of 4 (0) | 4 of 4 (100) |
| 212841_s_at | NM_003621 | PPFIBP2 |
chr11:7,491,577–7,631,567 | 0 of 4 (0) | 2 of 4 (50) |
| 206896_s_at | NM_052847 | GNG7 |
chr19:2,462,218–2,653,746 | 0 of 4 (0) | 1 of 3 (33) |
Discussion
In the present study, we combined a proven
pharmacologically demethylating-unmasking strategy with an expanded
47K expression microarray platform to identify novel
cancer-specific methylated genes. Among the 47,000 transcripts of
the Affymetrix Human Genome U133 Plus 2.0 expression arrays, we
first identified seven genes of interest. We then performed QMSP on
22 saliva and 14 mucosal samples from healthy individuals and 33
HNSCC tumor samples for the seven genes selected and verified 3
genes showing hypermethylation in the promoter regions of HNSCC
tissues and minimal or absent methylation in normal salivary rinses
and mucosal samples. The GNG7 gene was the most marked,
showing no methylation in normal mucosal samples and normal
salivary rinses respectively, while 61% of primary HNSCC tumor
samples were methylated. TXNIP methylation values were less
than 1% in normal salivary rinses and the promoter was not
methylated in normal mucosal samples, whereas the methylation
levels were over 10% in tumors. Similarly, the TUSC2 gene
was found to be methylated in 0.1% of normal mucosa samples and
showed almost no methylation (under 1%) in normal salivary rinses.
Other genes were not found to be useful as potential
biomarkers.
We found that the guanine nucleotide-binding
protein, γ-7 (GNG7) promoter was specifically methylated in
HNSCC. GNG7 is located on chromosome 19 and is a member of
the guanine nucleotide-binding proteins (G proteins) which are
involved as a modulator or transducer in various transmembrane
signaling systems. The β and γ chains are required for the GTPase
activity, for replacement of GDP by GTP and for G protein-effector
interaction; they are important in the regulation of adenylyl
cyclase signaling in certain regions of the brain and have a role
in the formation or stabilization of a G protein heterotrimer
[G(olf) subunit α-β-γ-7] that is required for adenylyl cyclase
activity in the striatum. GNG7 showed homozygous deletions
in cell lines of classical Hodgkins lymphoma (17). Decreased expression of GNG7
identified by Ray et al(18) was confirmed in pancreatic
malignancies (19) and esophageal
cancer (20). Expression of G-γ-7
mRNA was downregulated in extrahepatic cholangiocarcinoma (EHCC)
tissue compared to pericancerous bile duct and normal bile duct
tissues and in poorly differentiated EHCC tissues (21).
Thioredoxin-interacting protein is encoded by the
TXNIP gene and interacts with thioredoxin and ZBTB32. This
gene functions as an oxidative stress mediator by inhibiting
thioredoxin activity or by limiting its bioavailability and acts as
a transcriptional repressor, between transcription factors and
co-repressor complexes and its overexpression induces G0/G1 cell
cycle arrest and is necessary for the maturation of natural killer
cells (22–25).
Tumor suppressor candidate 2 is encoded by the
TUSC2 gene which is a highly conserved lung cancer candidate
gene (26,27). In malignant pleural mesothelioma
(28) and nasopharyngeal carcinoma
(29), expression of the
TUSC2 gene was found to be downregulated. TUSC2 is
often deleted in lung, breast, head and neck, renal and other types
of cancer (30) and was reported
to be methylated in human lung cancer cells (31). Furthermore, large-scale analysis of
TUSC2 expression in lung cancer and in bronchial squamous
metaplastic and dysplastic lesions showed reduced expression levels
of TUSC2 compared to normal hyperplastic epithelia,
indicating it could be an early event in cancer progression
(32).
It is known that the cell culture may influence DNA
methylation and present larger stretches of methylation events and
some drawbacks when compared to primary tumors (33). Therefore, as a future step, this
issue may be investigated by use of a larger panel of HNSCC cell
lines. However, this fact does not invalidate our findings and the
data of our study showing that the GNG7 gene is a promising
candidate tumor suppressor gene and biomarker for HNSCC.
It would be helpful to test independent cohorts to
evaluate the utility of these genes as HNSCC biomarkers. Additional
studies in larger, prospective cohorts would also determine the
prognostic significance of detection of these markers in saliva,
serum or plasma samples from HNSCC patients.
Acknowledgements
This study was supported by the
National Institute of Dental and Craniofacial Research
(R37DE012588), The National Institute of Dental and Craniofacial
Research/The National Cancer Institute (SPORE-P50DE019032) and
Scientific Research Projects Coordination Unit of Istanbul
University (UDP-22027). S. Dasgupta is supported by Elsa U Pardee
Foundation. This report/analysis is based on a web database
application provided by Research Information Technology Systems
(RITS)-https://www.rits.onc.jhmi.edu/.
References
|
1
|
Jemal A, Siegel R, Ward E, Murray T, Xu J,
Smigal C and Thun MJ: Cancer statistics, 2006. CA Cancer J Clin.
56:106–130. 2006. View Article : Google Scholar
|
|
2
|
Fearon ER and Vogelstein B: A genetic
model of colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baylin SB, Herman JG, Graff JR, Vertino PM
and Issa JP: Alterations in DNA methylation: a fundamental aspect
of neoplasia. Adv Cancer Res. 72:141–196. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dulaimi E, Hillinck J, Ibanez de Caceres
I, Al-Saleem T and Cairns P: Tumor suppressor gene promoter
hypermethylation in serum of breast cancer patients. Clin Cancer
Res. 10:6189–6193. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leonhardt H and Cardoso MC: DNA
methylation, nuclear structure, gene expression and cancer. J Cell
Biochem. 35(Suppl): S78–S83. 2000. View Article : Google Scholar
|
|
6
|
Herman JG and Baylin SB: Gene silencing in
cancer in association with promoter hypermethylation. N Engl J Med.
349:2042–2054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yamashita K, Upadhyay S, Osada M, et al:
Pharmacologic unmasking of epigenetically silenced tumor suppressor
genes in esophageal squamous cell carcinoma. Cancer Cell.
2:485–495. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bernard PS and Wittwer CT: Real-time PCR
technology for cancer diagnostics. Clin Chem. 48:1178–1185.
2002.PubMed/NCBI
|
|
9
|
Eads CA, Danenberg KD, Kawakami K, Saltz
LB, Blake C and Shibata D: MethyLight: a high-throughput assay to
measure DNA methylation. Nucleic Acids Res. 28:E322000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cottrell SE and Laird PW: Sensitive
detection of DNA methylation. Ann NY Acad Sci. 983:120–130. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jerónimo C, Usadel H, Henrique R, Oliveira
J, Lopes C, Nelson WG and Sidransky D: Quantitation of
GSTP1 methylation in non-neoplastic prostatic tissue and
organ-confined prostate adenocarcinoma. J Natl Cancer Inst.
93:1747–1752. 2001.
|
|
12
|
Tokumaru Y, Yamashita K, Osada M, et al:
Inverse correlation between cyclin A1 hypermethylation and p53
mutation in head and neck cancer identified by reversal of
epigenetic silencing. Cancer Res. 64:5982–5987. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li C and Wong WH: Model-based analysis of
oligonucleotide arrays: Expression index computation and outlier
detection. Proc Natl Acad Sci USA. 98:31–36. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tusher VG, Tibshirani R and Chu G:
Significance analysis of microarrays applied to the ionizing
radiation response. Proc Natl Acad Sci USA. 98:5116–5121. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Carvalho AL, Jeronimo C, Kim MM, et al:
Evaluation of promoter hypermethylation detection in body fluids as
a screening/diagnosis tool for head and neck squamous cell
carcinoma. Clin Cancer Res. 14:97–107. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li LC and Dahiya R: MethPrimer: designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Giefing M, Arnemann J, Martin-Subero JI,
et al: Identification of candidate tumour suppressor gene loci for
Hodgkin and Reed-Sternberg cells by characterisation of homozygous
deletions in classical Hodgkin lymphoma cell lines. Br J Haematol.
142:916–924. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ray K, Kunsch C, Bonner LM and Robishaw
JD: Isolation of cDNA clones encoding eight different human G
protein gamma subunits, including three novel forms designated the
gamma-4, gamma-10, and gamma-11 subunits. J Biol Chem.
270:21765–21771. 1995. View Article : Google Scholar
|
|
19
|
Shibata K, Mori M, Tanaka S, Kitano S and
Akiyoshi T: Identification and cloning of human G-protein gamma 7,
down-regulated in pancreatic cancer. Biochem Biophys Res Commun.
246:205–209. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ohta M, Mimori K, Fukuyoshi Y, et al:
Clinical significance of the reduced expression of G protein gamma
7 (GNG7) in oesophageal cancer. Br J Cancer. 98:410–417. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang M, Gong B, Li Y and Wang Y: Human
G-protein gamma 7 in extrahepatic cholangiocarcinoma and its
clinicopathological significance. Hematol Oncol Stem Cell Ther.
3:66–70. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wollman EE, d’Auriol L, Rimsky L, et al:
Cloning and expression of a cDNA for human thioredoxin. J Biol
Chem. 263:15506–15512. 1988.PubMed/NCBI
|
|
23
|
Meng L, Wong JH, Feldman LJ, Lemaux PG and
Buchanan BB: A membrane-associated thioredoxin required for plant
growth moves from cell to cell, suggestive of a role in
intercellular communication. Proc Natl Acad Sci USA. 107:3900–3905.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nishiyama A, Matsui M, Iwata S, et al:
Identification of thioredoxin-binding protein-2/vitamin D(3)
up-regulated protein 1 as a negative regulator of thioredoxin
function and expression. J Biol Chem. 274:21645–21650. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han SH, Jeon JH, Ju HR, et al: VDUP1
upregulated by TGF-beta1 and 1,25-dihydroxyvitamin D3
inhibits tumor cell growth by blocking cell-cycle progression.
Oncogene. 22:4035–4046. 2003.PubMed/NCBI
|
|
26
|
Fischer WH and Schubert D:
Characterization of a novel platelet-derived growth
factor-associated protein. J Neurochem. 66:2213–2216. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kondo M, Ji L, Kamibayashi C, et al:
Overexpression of candidate tumor suppressor gene FUS1 isolated
from the 3p21.3 homozygous deletion region leads to G1 arrest and
growth inhibition of lung cancer cells. Oncogene. 20:6258–6262.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ivanova AV, Ivanov SV, Prudkin L, et al:
Mechanisms of FUS1/TUSC2 deficiency in mesothelioma and its
tumorigenic transcriptional effects. Mol Cancer. 8:912009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou YB, Huang ZX, Ren CP, Zhu B and Yao
KT: Screening and preliminary analysis of the apoptosis- and
proliferation-related genes in nasopharyngeal carcinoma. Nan Fang
Yi Ke Da Xue Xue Bao. 29:645–647. 2009.(In Chinese).
|
|
30
|
Lerman MI and Minna JD: The 630-kb lung
cancer homozygous deletion region on human chromosome 3p21.3:
identification and evaluation of the resident candidate tumor
suppressor genes. The international lung cancer chromosome 3p213
tumor suppressor gene consortium. Cancer Res. 60:6116–6133.
2000.
|
|
31
|
Uno F, Sasaki J, Nishizaki M, et al:
Myristoylation of the fus1 protein is required for tumor
suppression in human lung cancer cells. Cancer Res. 64:2969–2976.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Prudkin L, Behrens C, Liu DD, et al: Loss
and reduction of FUS1 protein expression is a frequent phenomenon
in the pathogenesis of lung cancer. Clin Cancer Res. 14:41–47.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hennessey PT, Ochs MF, Mydlarz WW, Hsueh
W, Cope L, Yu W and Califano JA: Promoter methylation in head and
neck squamous cell carcinoma cell lines is significantly different
than methylation in primary tumors and xenografts. PLoS One.
6:e205842011. View Article : Google Scholar : PubMed/NCBI
|