Introduction
Pancreatic cancer is one of the most highly
aggressive malignancies in the world. Data from the GLOBOCAN series
in 2008, estimated 277,000 new cases and 266,000 deaths worldwide
(1). Despite developments in
diagnosis and treatment, the prognosis of pancreatic cancer remains
poor. Only 10–20% of pancreatic cancer patients undergo potentially
curative resection with a median survival of 17–23 months (2). Furthermore, pancreatic cancer
responds poorly to most chemotherapeutic agents and radiation,
resulting in little prolongation of the survival time in individual
groups. The overall 5-year survival rate is 5%, and more than 50%
of patients are at an advanced stage at the time of diagnosis
(3). This indicates that there is
a great need for novel modalities to assist in diagnostic and
therapeutic strategies for management of pancreatic cancer
patients.
Kindlin-1 is a novel focal adhesion protein that
belongs to the kindlin protein family, and is also known as
fermitin family member 1 (FERMT1). The kindlin proteins are
composed of a four-point-one, ezrin, radixin, moesin (FERM) domain
interrupted by a pleckstrin homology (PH) domain. Kindlin-1 was
discovered as a mutated gene related to Kindler’s syndrome, an
inherited skin disease characterized by skin blistering, atrophy,
photosensitivity and generalized poikiloderma (4,5).
Kindlin-1 is primarily found in epithelial cells such as
keratinocytes and intestinal epithelial cells (4,6,7),
while kindlin-2 is expressed ubiquitously and kindlin-3 is
exclusively detected in hematopoietic cells (8).
Integrins are key molecules for establishing
cell-extracellular matrix (ECM) adhesion, and are involved in the
mechanism of cancer progression (9). Several recent publications have
indicated the importance of kindlins for integrin regulation and
cytoskeletal reorganization (10–13).
To initiate intracellular signaling (‘inside-out’ signaling),
kindlin directly binds to β integrin tails, leading to integrin
activation and triggering increased adhesiveness in the
extracellular domain (14).
Likewise, in ‘outside-in’ signaling, kindlin-1 interacts with focal
adhesion proteins such as migfilin and FAK, and these interactions
link integrins and signaling for reorganization of the actin
cytoskeleton (15,16). Therefore, kindlin-1 may play a
crucial role in cancer progression via this signaling pathway.
Currently, kindlin-1 expression has been reported in colorectal,
lung (17) and breast (18) cancers with enhanced cell adhesion,
proliferation and motility. However, there have been no reports on
the expression of kindlin-1 in pancreatic cancer.
The aims of this study were to evaluate whether
kindlin-1 is expressed in pancreatic cancer cells and to determine
its biomolecular functions. We evaluated Kindlin-1 mRNA
expression in pancreatic cancer cell lines, a normal ductal
epithelial cell line and cancer-associated fibroblasts using
quantitative RT-PCR. We then investigated the molecular functions
of kindlin-1, such as its effects on cell proliferation, migration
and invasion, in in vitro experiments using pancreatic
cancer cell lines.
Materials and methods
Cells and culture conditions
The following 13 pancreatic cancer cell lines were
used: AsPC-1, KP-2, KP-3, Panc-1, SUIT-2 (Dr H. Iguchi, National
Shikoku Cancer Center, Matsuyama, Japan), MIA PaCa-2 (Japanese
Cancer Resource Bank, Tokyo, Japan), Capan-1, Capan-2, CFPAC-1,
H48N, Hs766T, SW 1990 (American Type Culture Collection, Manassas,
VA, USA) and NOR-P1 (established by Dr N. Sato in our laboratory).
Primary cultures of human normal pancreatic epithelial cells were
obtained from Cell Systems (Kirkland, WA, USA) and maintained in
Cell Systems Corporation (CS-C) medium containing 10% fetal bovine
serum (FBS), according to the supplier’s instructions. In addition,
primary cultures of cancer-associated fibroblasts (CAF) derived
from five patients with invasive pancreatic cancers were
established in our laboratory and used in this study. All cells
were maintained as previously described (19).
RNA isolation and qRT-PCR
Total-RNA was extracted from cultured cells using a
High Pure RNA Isolation Kit (Roche Diagnostics, Mannheim, Germany)
according to the manufacturer’s instructions. The RNA
concentrations were measured using a NanoDrop ND-1000
spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) at
260 and 280 nm (A260/280). qRT-PCR was performed using a Chromo4
real-time PCR Detection System (Bio-Rad Laboratories, Hercules, CA,
USA) for 40 cycles of 15 sec at 95°C and 1 min at 55°C with a
QuantiTect SYBR-Green Reverse Transcription-PCR Kit (Qiagen, Tokyo,
Japan) according to the manufacturer’s instructions. We designed
specific primer sequences as follows: FERMT1 (forward
primer, 5′-ttgggattcaggaagacagg-3′; reverse primer,
5′-ccctgaccagttgggataga-3′), β-actin (forward primer,
5′-tgagcgcggctacagctt-3′; reverse primer, 5′-tccttaatgtcacgcac
gattt-3′) and 18S rRNA (forward 5′-gtaacccgttgaaccccatt-3′;
reverse 5′-ccatccaatcggtagtagcg-3′. We performed BLAST searches to
ensure the specificity of these primers. All primers were purchased
from Sigma Genosys (Tokyo, Japan). The expression of each mRNA was
calculated from a standard curve constructed with total-RNA from
SUIT-2 cells and normalized by the expression of β-actin or
18S rRNA.
Immunohistochemistry for human pancreatic
cancer tissues
Pancreatic cancer tissues were obtained from
patients who underwent pancreatic resection for pancreatic cancer
at our institution. We also obtained normal pancreatic tissue
samples from intact pancreas resected for bile duct cancer as
control tissues. Serial 3 μm sections were prepared from the
selected paraffin blocks, deparaffinized in xylene and rehydrated
in ethanol. Endogenous peroxidase was blocked by incubation in 3%
hydrogen peroxide in methanol for 30 min. Antigen retrieval was
achieved by heating in a microwave in citrate buffer at pH 6.0. A
Histofine SAB-PO Kit (Nichirei, Tokyo, Japan) was used for
immunohistochemical labeling. The sections were incubated with a
rabbit polyclonal anti-kindlin-1 antibody (Millipore, Billerica,
CA, USA; 1:100 dilution) overnight at 4°C. The sections were then
incubated with a biotinylated anti-rabbit immunoglobulin solution
for 20 min followed by a 20-min incubation with peroxidase-labeled
streptavidin. The reaction products were visualized using
3,3′-diaminobenzidine as a chromogen, and the nuclei were
counterstained with hematoxylin. The study was approved by the
Ethics Committee of Kyushu University and conducted according to
the Ethical Guidelines for Human Genome/Gene Research enacted by
the Japanese Government and the Helsinki Declaration.
Silencing of kindlin-1 using small
interfering RNAs (siRNAs)
AsPC-1 and KP-2 cells were transfected with
kindlin-1 I (sense, 5′-cauguagauucuggacuaatt-3′; antisense,
5′-uuaguccagaaucuaca ugtt-3′) and kindlin-1 II (sense,
5′-gagaugugaccaugagaautt-3′; antisense,
5′-auucucauggucacaucuctt-3′) siRNAs (Sigma Genosys) by
electroporation using a Nucleofector system (Amaxa Biosystems,
Koln, Germany) according to the manufacturer’s instructions. To
verify the specificity of the knockdown effects, we used a control
siRNA (Qiagen). All cells were used in the subsequent experiments
at 24–96 h after transfection.
Matrigel invasion and migration
assays
The invasive ability of pancreatic cancer cells was
measured by the number of cells invading Matrigel-coated transwell
chambers. Briefly, 1×105 KP-2 or AsPC-1 cells were
suspended in 250 μl of Dulbecco’s modified Eagle’s medium
containing 10% FBS and placed in the upper transwell chamber (8
μm pore size; Becton Dickinson, Franklin Lakes, NJ, USA)
containing 100 μl of reconstituted Matrigel-coated membrane
(20 μg/well). The upper chamber was placed in a 24-well
culture plate containing 750 μl of the above-described
medium. After incubation for 48 or 72 h at 37°C, the numbers of
invaded KP-2 and AsPC-1 cells were counted, respectively.
Cell migration assay was performed in pancreatic
cancer cells under the same protocol as well as the invasion assay
without using Martigrel-coated membrane. Cells were allowed to
migrated and counted for 36 (KP-2) or 72 h (AsPC-1) after cell
seeding into the upper chamber. In both assays and at each time
point, the cells that migrated and invaded to the bottom side of
the inserted chamber were fixed with 70% ethanol, stained with
hematoxylin and eosin, and counted in 10 random fields at ×200
magnification under a light microscope. Each experiment was
performed in triplicate and repeated at least three times.
Propidium iodide (PI) assay
The proliferative phenotype of pancreatic cancer
cell lines was evaluated by PI assay. Cell numbers were counted by
measuring the fluorescence intensity of PI at specified times, as
described previously (20).
Briefly, pancreatic cancer cells were seeded in 24-well plates
(Becton Dickinson Labware, Bedford, MA, USA) at 1×104
cells/well. After 24, 48, 72 or 96 h, PI (30 μM) and
digitonin (600 μM) were added to each well to ensure that
all nuclei were labeled with PI. The fluorescence intensities
corresponding to the total cells were measured using a microplate
reader (Tecan, Männedorf, Switzerland).
Western blotting analysis
All cells were lysed in PRO-PREP™ (iNtRON
Biotechnology, Seongnam, Korea). The cell lysates were fractionated
in a Mini-Protean® TGM™ precast gel (Bio-Rad
Laboratories) and transferred to a Trans-Blot® Turbo™
Mini PVDF membrane (Bio-Rad Laboratories) using a Trans-Blot Turbo
transfer system (Bio-Rad Laboratories). The membrane was incubated
overnight at 4°C with a rabbit polyclonal anti-kindlin-1 antibody
(ab68041; Abcam, Cambridge, MA, USA; 1:1,000 dilution) and then
incubated with a horse-radish peroxidase-conjugated anti-rabbit IgG
antibody (Cell Signaling, Danvers, MA, USA; 1:5,000 dilution) for 1
h at room temperature. The bound antibodies were detected using an
Amersham™ ECL™ Prime Western blotting detection reagent (Amersham
Biosciences, Little Chalfont, UK) and visualized with a Molecular
Imager (Chemi-Doc XRS System; Bio-Rad Laboratories). The membranes
were stripped and probed with an anti-β-actin antibody (Santa Cruz
Biotechnology, Santa Cruz, CA, USA; 1:1,000 dilution) as an
internal control.
Statistical analysis
Statistical analyses and graph presentations were
carried out using JMP 8 software (SAS Institute, Cary, NC, USA).
Values were expressed as the mean ± SD. Comparisons between two
groups were performed using Student’s t-test. Statistical
significance was defined as P<0.05.
Results
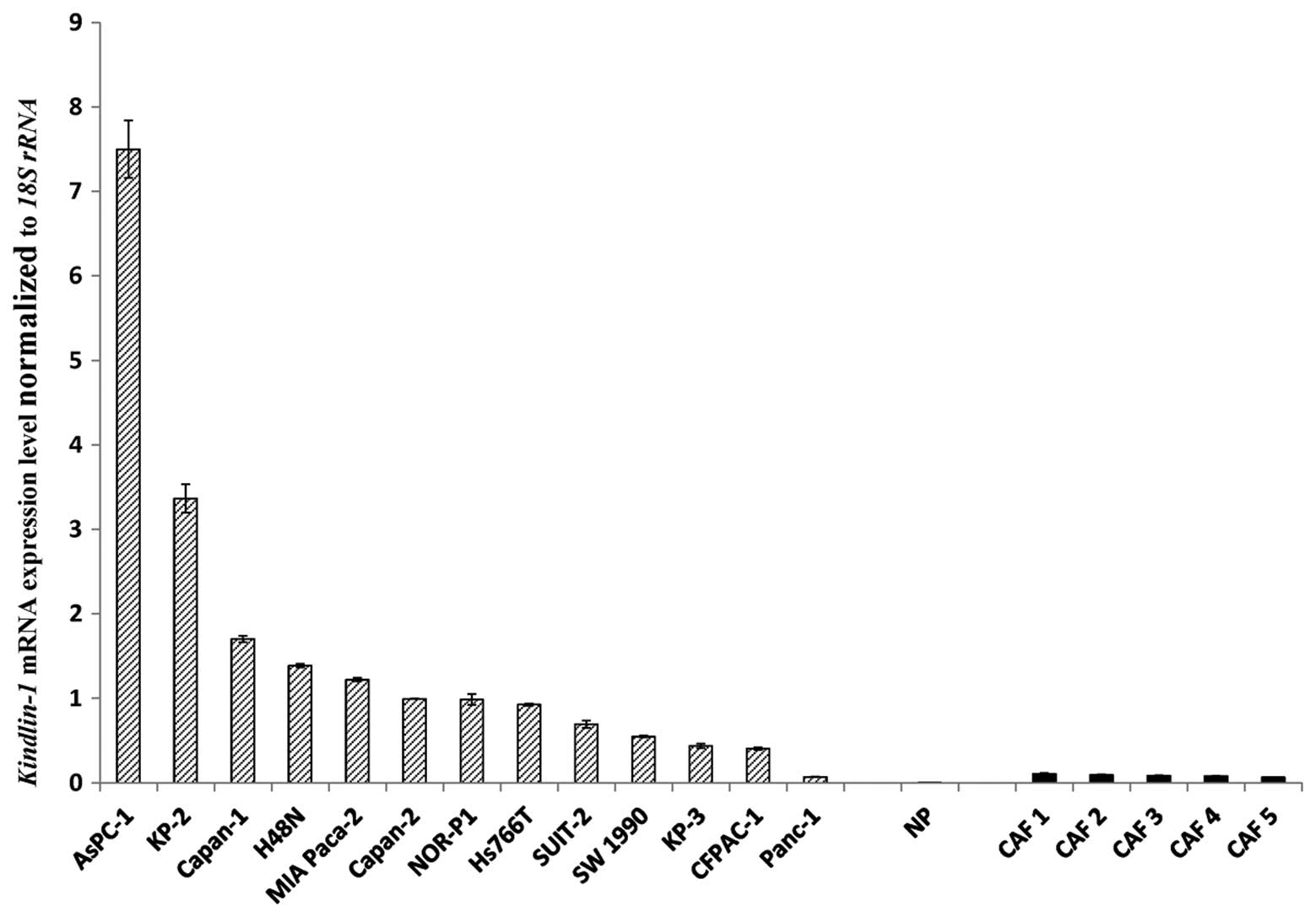
Kindlin-1 mRNA is highly expressed in
pancreatic cancer cell lines compared with normal pancreatic duct
cells and fibroblasts
We measured the expression levels of
Kindlin-1 mRNA in pancreatic cancer cell lines, human normal
pancreatic epithelial cells and primary cultures of
cancer-associated fibroblasts by qRT-PCR. There was a wide range of
relative Kindlin-1 mRNA expression levels among the
pancreatic cancer cell lines, whereas Kindlin-1 mRNA was
expressed at very low levels in cancer-associated fibroblasts and
normal pancreatic epithelial cells (Fig. 1).
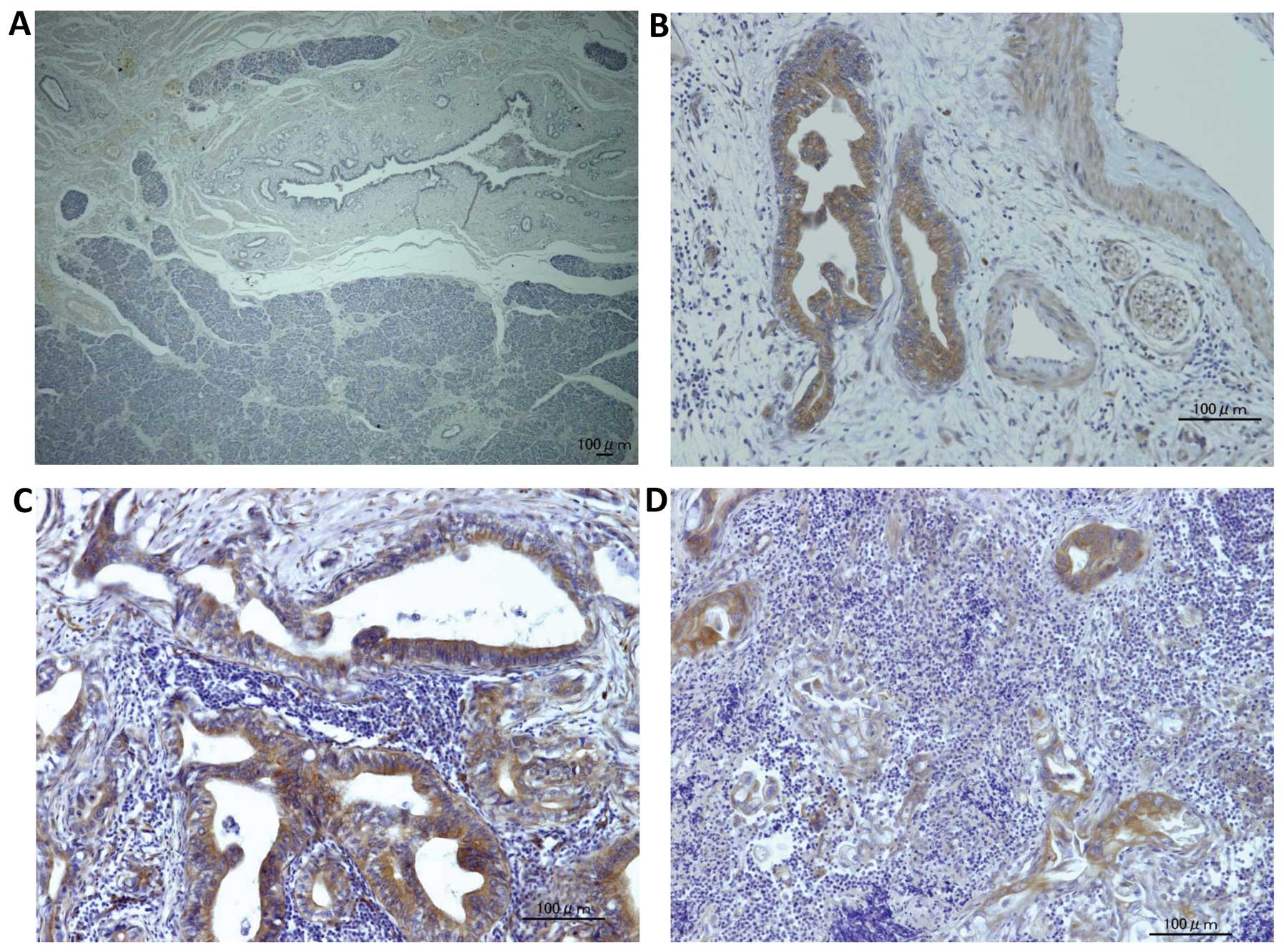
Kindlin-1 is exclusively expressed in
pancreatic cancer cells
To elucidate the expression of kindlin-1 in
pancreatic tissues, we examined kindlin-1 expression in human
samples including pancreatic ductal adenocarcinoma (PDAC) and
normal pancreatic tissues adjacent to resected bile duct cancers.
Kindlin-1 was exclusively expressed in PDAC, while normal ductal
epithelial cells and stromal cells showed no expression (Fig. 2).
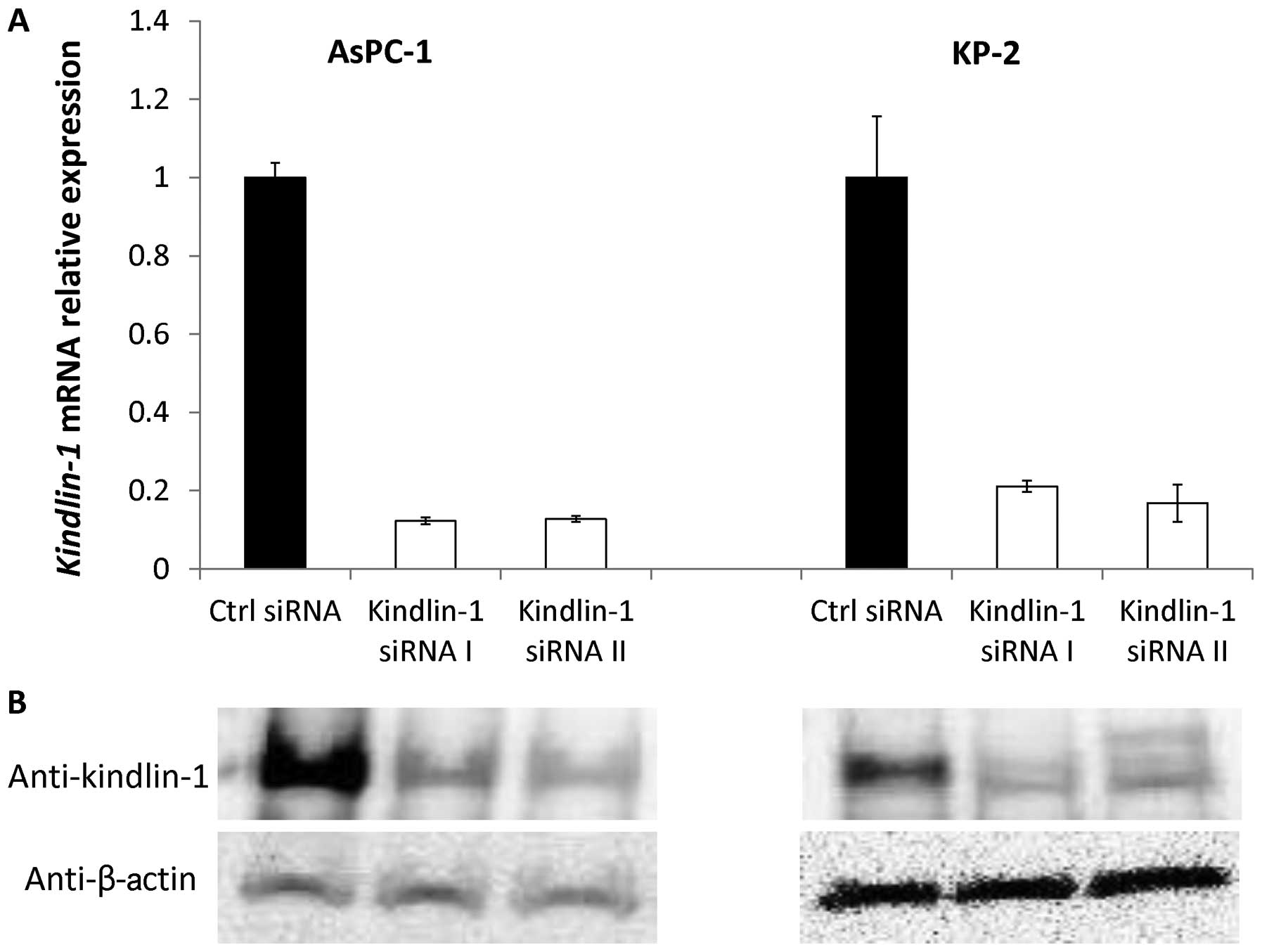
Efficacy of siRNAs targeting
Kindlin-1
We used AsPC-1 and KP-2 cells, which showed high
Kindlin-1 mRNA expression levels, to investigate the
biomolecular functions of kindlin-1 in pancreatic cancer cell
lines. We inhibited the expression of kindlin-1 using two different
siRNAs. At 24 h (day 1) after transfection with the control siRNA
or kindlin-1 siRNAs, the pancreatic cancer cells transfected with
the kindlin-1 siRNAs showed lower levels of Kindlin-1 mRNA
expression than those transfected with the control siRNA (Fig. 3). Consistently, immunoblot analyses
revealed that kindlin-1 protein was also decreased in
kindlin-1-knockdown cells at 48 h after transfection, suggesting
good efficacy of both siRNAs for use in the following
experiments.
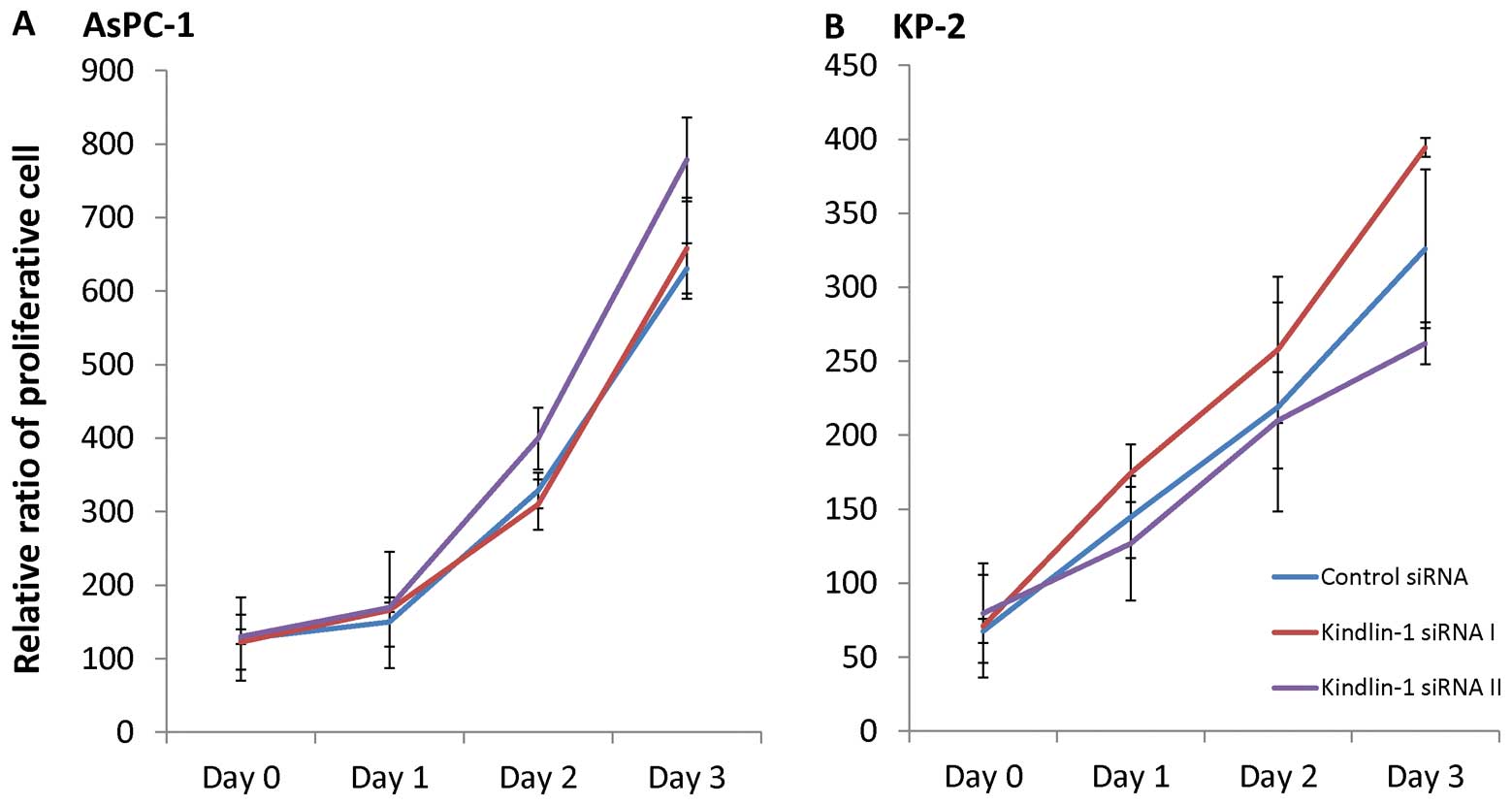
Kindlin-1 has no impact on the
proliferation of pancreatic cancer cells
AsPC-1 and KP-2 cells were transfected with the
kindlin-1 siRNAs and cultured for 24 h. The transfected cells were
then resuspended and seeded in 24-well plates to investigate the
effects of kindlin-1 on proliferation. Increasing cell numbers were
observed and counted on the indicated days, and the growth rates of
the kindlin-1-deficient cells were similar to those of cells
transfected with the control siRNA (Fig. 4). These findings suggested that
kindlin-1 had no effect on the proliferation of pancreatic cancer
cells.
Inhibition of kindlin-1 expression
decreases the abilities for migration and invasion in pancreatic
cancer cells
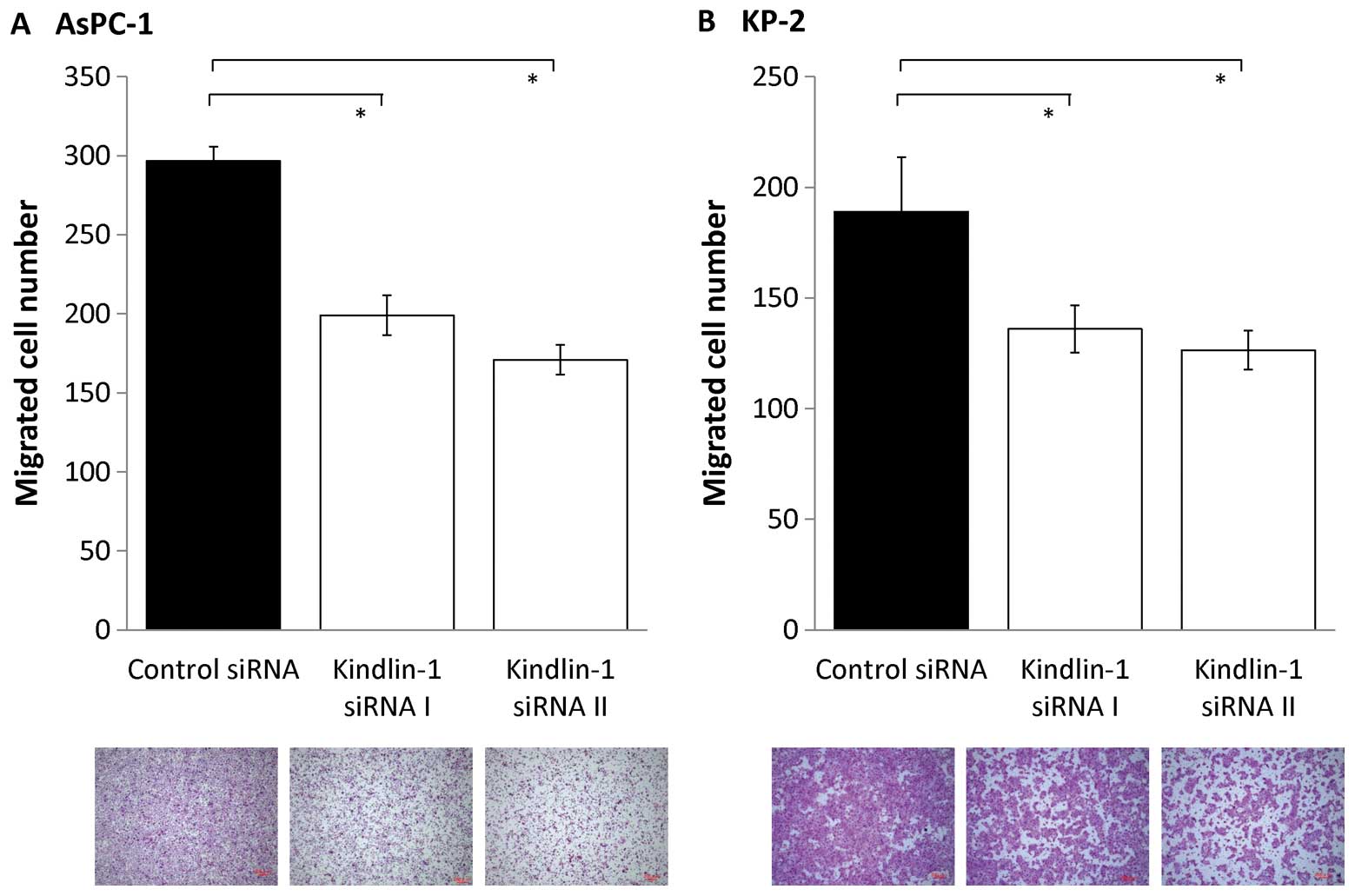
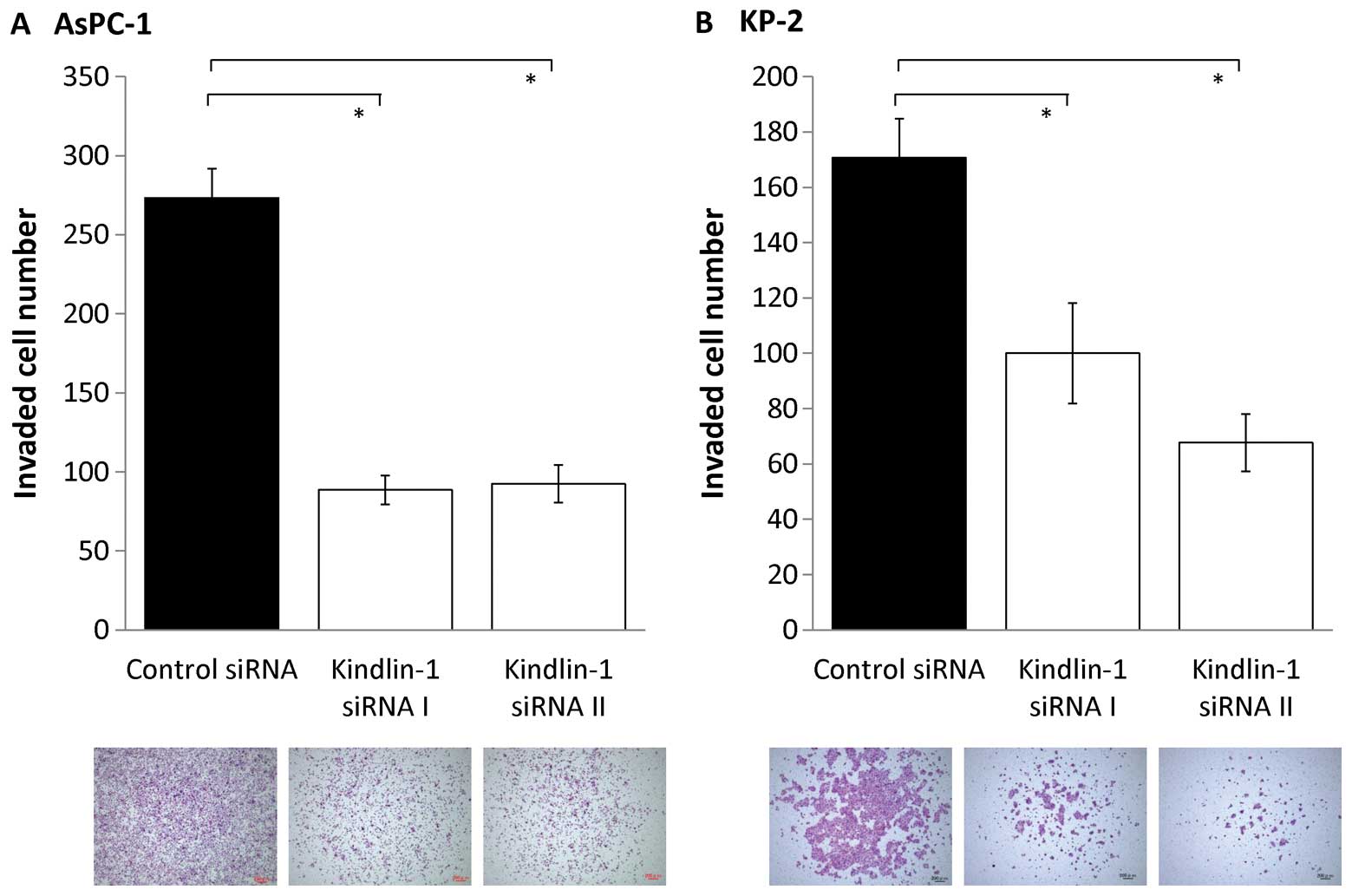
Next, we investigated the effects of inhibition of
kindlin-1 expression on the migratory and invasive abilities of
pancreatic cancer cells using a double-chamber assay. After
inhibition of kindlin-1 expression, the migratory abilities of
AsPC-1 and KP-2 cells were reduced to 60–70% (P<0.001; Fig. 5) and their invasive abilities were
reduced to 30–50% (P<0.001; Fig.
6). These findings indicate that kindlin-1 expression is
involved in both the migration and invasion of pancreatic cancer
cells and that its impact is greater on invasion than on
migration.
Discussion
This report describes a novel study on kindlin-1
expression and its biomolecular functions in pancreatic cancer.
There are a few publications regarding kindlin-1 expression and its
relationships with carcinomas. In 2003, Weinstein et
al(17) first found that
Kindlin-1 mRNA was overexpressed in 70% of colon carcinoma
tissues and 60% of lung carcinoma tissues by qRT-PCR. Papachristou
et al(21) further reported
the expression of kindlin-1 in leiomyoma and leiomyosarcoma of soft
tissue. They found that kindlin-1 immunoreactivity was exclusively
detected in the cytoplasm of tumor cells, although there were no
significant differences between high-grade and low-grade neoplasms
and no clinicopathological correlation between kindlin-1 expression
and leiomyosarcoma. In breast cancer, both mRNA and protein levels
of kindlin-1 have been detected (18). Immunohistochemical analyses further
demonstrated strong immunoreactivity in the primary tumors and lung
metastases, but not in the adjacent parenchyma or non-lung
metastasis sites, suggesting that kindlin-1 is potentially a
clinically relevant mediator of lung metastasis of breast cancer
and possibly other carcinomas metastasizing to the lung (18). In our study, we found that
Kindlin-1 mRNA was highly expressed in various pancreatic
cancer cells compared with normal pancreatic epithelial cells and
cancer-associated fibroblasts. In addition, immunohistochemical
analyses of PDAC and normal pancreatic tissues revealed that
kindlin-1 was exclusively expressed in PDAC cells, with no
immunoreactivities detected in normal duct cells and stromal cells
in pancreatic tissues. Consistent with the previous reports, the
present data suggest that expression of kindlin-1 is a promising
biomarker for carcinogenesis or progression of PDAC, although
investigations of large study groups are required to clarify the
clinical implications of kindlin-1.
It is now accepted that the binding between β-tail
integrin and kindlins, in concert with talin, is essential for
integrin regulation (10,22,23).
In particular, the cytoplasmic tail of β1 integrin was shown to be
the most important region for kindlin-1 in keratinocytes and
intestinal epithelial cells (6,11).
Arao et al(24)
demonstrated that pancreatic cancer cell lines exhibited high
expression of β1 integrin, and further showed that the level of
constitutive activity of β1 integrins was correlated with the
invasive ability of pancreatic cancer cell lines. In our study,
kindlin-1 expression had effects on both pancreatic cancer cell
invasion and migration, but a stronger effect was observed on the
invasive ability. However, kindlin-1 had no effect on pancreatic
cancer cell proliferation. Although the mechanism remains unclear,
these findings may be caused by binding of kindlin-1 to β1
integrins, thereby conferring a strong invasive property on the
pancreatic cancer cells. However, the partner ligands that bind to
kindlin-1, such as talin, may play some hidden roles in the effects
on pancreatic cancer cells. Further investigations are needed to
elucidate these issues.
It is possible that kindlin-1 may affect pancreatic
cancer cell invasion and migration via the TGF-β signaling pathway
and/or epithelial-mesenchymal transition (EMT), which is the
process allowing physiologic and genetic changes of cancer cells
from an epithelial phenotype to a mesenchymal phenotype and
considered to be an important step in cancer progression and
metastasis (25,26). Kloeker et al(7) demonstrated that kindlin-1 expression
in human mammary epithelial cell was increased in the presence of
TGF-β and might also act as a downstream mediator of
TGF-β-initiating EMT. Subsequent data in breast cancer tissues
revealed that kindlin-1-expressing cells showed increased cell
invasion, migration, clonogenicity and proliferation (18). The study also found that kindlin-1
initiates TGF-β-dependent EMT (18). Many studies have clearly shown the
role of TGF-β signaling and EMT in tumor initiation, progression
and metastasis, including pancreatic cancer (27,28).
Taken together with our data of in vitro experiments, the
decrease in cell invasion and migration after kindlin-1 knockdown
in pancreatic cancer cells may involve the TGF-β signaling pathway
and/or EMT. Elucidation of whether kindlin-1 expression contributes
to EMT in pancreatic cancer and other signaling pathways would be
beneficial for the development of diagnostic and therapeutic
interventions for this disease.
In conclusion, this study provides the first
evidence of kindlin-1 expression in pancreatic cancer.
Kindlin-1-deficient cells showed reduced invasion and migration,
but not proliferation. These findings suggest that kindlin-1 is
required for pancreatic cancer cell invasion and migration. Further
investigations to determine the biomolecular functions of kindlin-1
and its role in pancreatic cancer using both in vivo and
clinical studies are required.
Acknowledgements
This study was supported in part by a
Grant-in-Aid from the Ministry of Education, Culture, Sports,
Science and Technology of Japan. We are grateful to Nobuhiro
Torata, Emiko Manabe and Miyuki Omori (Department of Surgery and
Oncology, Kyushu University) for skillful technical assistance.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar
|
|
3
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel DH, Ashton GH, Penagos HG, et al:
Loss of kindlin-1, a human homolog of the Caenorhabditis
elegans actin-extracellular-matrix linker protein UNC-112,
causes Kindler syndrome. Am J Hum Genet. 73:174–187. 2003.
|
|
5
|
Jobard F, Bouadjar B, Caux F, et al:
Identification of mutations in a new gene encoding a FERM family
protein with a pleckstrin homology domain in Kindler syndrome. Hum
Mol Genet. 12:925–935. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ussar S, Moser M, Widmaier M, et al: Loss
of Kindlin-1 causes skin atrophy and lethal neonatal intestinal
epithelial dysfunction. PLoS Genet. 4:e10002892008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kloeker S, Major MB, Calderwood DA,
Ginsberg MH, Jones DA and Beckerle MC: The Kindler syndrome protein
is regulated by transforming growth factor-beta and involved in
integrin-mediated adhesion. J Biol Chem. 279:6824–6833. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ussar S, Wang HV, Linder S, Fassler R and
Moser M: The Kindlins: subcellular localization and expression
during murine development. Exp Cell Res. 312:3142–3151. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cabodi S, del Pilar Camacho-Leal M, Di
Stefano P and Defilippi P: Integrin signalling adaptors: not only
figurants in the cancer story. Nat Rev Cancer. 10:858–870. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moser M, Legate KR, Zent R and Fassler R:
The tail of integrins, talin, and kindlins. Science. 324:895–899.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harburger DS, Bouaouina M and Calderwood
DA: Kindlin-1 and -2 directly bind the C-terminal region of beta
integrin cytoplasmic tails and exert integrin-specific activation
effects. J Biol Chem. 284:11485–11497. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Plow EF, Qin J and Byzova T: Kindling the
flame of integrin activation and function with kindlins. Curr Opin
Hematol. 16:323–328. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Goult BT, Bouaouina M, Harburger DS, et
al: The structure of the N-terminus of kindlin-1: a domain
important for alphaiib-beta3 integrin activation. J Mol Biol.
394:944–956. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kuijpers TW, van de Vijver E, Weterman MA,
et al: LAD-1/variant syndrome is caused by mutations in FERMT3.
Blood. 113:4740–4746. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Has C, Herz C, Zimina E, et al: Kindlin-1
is required for RhoGTPase-mediated lamellipodia formation in
keratinocytes. Am J Pathol. 175:1442–1452. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Montanez E, Ussar S, Schifferer M, et al:
Kindlin-2 controls bidirectional signaling of integrins. Genes Dev.
22:1325–1330. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weinstein EJ, Bourner M, Head R, Zakeri H,
Bauer C and Mazzarella R: URP1: a member of a novel family of PH
and FERM domain-containing membrane-associated proteins is
significantly over-expressed in lung and colon carcinomas. Biochim
Biophys Acta. 1637:207–216. 2003. View Article : Google Scholar
|
|
18
|
Sin S, Bonin F, Petit V, et al: Role of
the focal adhesion protein kindlin-1 in breast cancer growth and
lung metastasis. J Natl Cancer Inst. 103:1323–1337. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ohuchida K, Mizumoto K, Murakami M, et al:
Radiation to stromal fibroblasts increases invasiveness of
pancreatic cancer cells through tumor-stromal interactions. Cancer
Res. 64:3215–3222. 2004. View Article : Google Scholar
|
|
20
|
Zhang L, Mizumoto K, Sato N, et al:
Quantitative determination of apoptotic death in cultured human
pancreatic cancer cells by propidium iodide and digitonin. Cancer
Lett. 142:129–137. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Papachristou DJ, Gkretsi V, Tu Y, et al:
Increased cytoplasmic level of migfilin is associated with higher
grades of human leiomyosarcoma. Histopathology. 51:499–508. 2007.
View Article : Google Scholar
|
|
22
|
Harburger DS and Calderwood DA: Integrin
signalling at a glance. J Cell Sci. 122:159–163. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ye F and Petrich BG: Kindlin: helper,
co-activator, or booster of talin in integrin activation? Curr Opin
Hematol. 18:356–360. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arao S, Masumoto A and Otsuki M: Beta1
integrins play an essential role in adhesion and invasion of
pancreatic carcinoma cells. Pancreas. 20:129–137. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Iwatsuki M, Mimori K, Yokobori T, et al:
Epithelialmesenchymal transition in cancer development and its
clinical significance. Cancer Sci. 101:293–299. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rane SG, Lee JH and Lin HM: Transforming
growth factor-beta pathway: role in pancreas development and
pancreatic disease. Cytokine Growth Factor Rev. 17:107–119. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bierie B and Moses HL: TGF-beta and
cancer. Cytokine Growth Factor Rev. 17:29–40. 2006. View Article : Google Scholar
|