Introduction
Colon cancer is one of the most common malignancies
and a leading cause of cancer deaths for both men and women
worldwide (1,2). Even today its prevalence is rising
and the 5-year survival rate is still poor. Although significant
advances in diagnosis and therapy have been achieved over the last
half century, it remains the second leading cause of cancer deaths
among American men and the third among American women (3). Nearly 50% of patients with advanced
tumors develop recurrent disease and die soon afterward (4). More powerful and safer
chemopreventive or chemotherapeutic approaches are urgently needed
to reduce mortality and garner better curative effects, since a
large number of patients with advanced disease fail to effectively
respond to current treatment regimens (5). In this regard, dietary supplements
that are capable of preventing carcinogenesis and inhibiting the
growth of colon carcinoma cells have generated intense interest
(6).
Epidemiologic studies suggest that dietary factors
are one of the major concerns in cancer etiology and may account
for ≤35% of the difference in cancer incidences (7,8). For
instance, in some countries, such as China, where the consumption
of soy isoflavone-containing foods is substantially higher than in
western countries, the risk of developing breast cancer has been
lower (9). Recent data have also
shown that continuous consumption of soy products is also
positively correlated with a reduction in other human malignancies,
such as prostate, stomach, colorectal and lung cancers (9). Most isoflavones are present in a
glycosidic form in nature and can be converted to a corresponding
aglycon form under certain conditions (10). Genistein and daidzein, which are
aglycon isoflavones, have already been tested as potential cancer
preventative agents. Genistein is currently considered to be the
primary anticancer component from soybeans, based on its dependable
inhibitory effect on tumors compared to daidzein. In vitro
and in vivo studies showed that genistein suppressed
angiogenesis and induced apoptosis and cell differentiation by
inhibiting protein tyrosine phosphorylation and topoisomerase
activity, implying that genistein could be a potential cancer
chemopreventive agent (11).
Cancer cells lack normal growth controls, exhibit
loss of cell cycle control, have unlimited reproductive potential
and have growth-signal self-sufficiency (12). Any compound aimed at controlling
these processes would be beneficial in suppressing the progression
of tumors. Epigenetic studies have confirmed that cell
overproliferation and loss of normal cell cycle regulation are
involved in colon cancer growth and progression (13). Current studies have shown that a
complicated cluster of regulatory factors, such as extracellular
signal-regulated kinases (ERKs), cell cycle regulators and the
tumor suppressor gene p53, play a pivotal role in the process of
colon cancer progression (13,14).
Substantial research is focused on exploring novel compounds that
can regulate cell proliferation, cell cycle progression and
apoptosis in order to elucidate new candidates for cancer therapy
(15). The inhibitory effect of
genistein on carcinogenesis and tumor growth has been known for
years, but the clear molecular mechanism is still not fully
understood. The involvement of estrogen receptors (ERs), tyrosine
kinases and the oxidative and angiogenesis pathways have been
reported (16,17), providing some insight into the
development of the mechanism of the anticancer effect derived from
genistein. In the present study, we investigated three isoflavones:
genistein, daidzein and biochanin A, to identify a safer, more
effective and reliable candidate compound for colon tumor therapy.
We examined the effects of each compound on HCT-116 and SW-480 cell
growth, apoptosis and cell cycle arrest. Although p53 was regarded
as an important defense mechanism that regulates apoptosis and cell
cycle arrest during multiple tumor development (18), reports related to the interaction
of p53 and genistein in cell cycle controlling are rare. Here we
hypothesized that activation of p53/ATM-p21, which is induced by
genistein treatment, plays a critical role in the modulation of
apoptosis and cell cycle arrest and helps to elucidate the
molecular mechanism. Our data demonstrated that genistein induced
specific G2/M cell cycle arrest via p53-dependent way by the
ATM/p53-p21 cross-talk regulatory pathway, which provided novel
evidence in the colon cancer chemoprevention of natural
flavones.
Materials and methods
Chemicals and reagents
Genistein, daidzein and biochanin A were obtained
from Sigma-Aldrich (St. Louis, MO), diluted to 2.5, 5, 10, 25, 50
and 100 mM in DMSO (Fisher Chemicals, Fair Lawn, NJ) and stored in
small aliquots at −20°C.
Cell culture conditions
Human colon cancer cell lines HCT-116 and SW-480
were obtained from the American Type Tissue Collection (Rockville,
MD) and maintained in McCoy’s 5A or L-15 medium (Hyclone, Logan,
UT). HCT-116 (p53+/+) and HCT-116 (p53−/−)
cells were manipulated and maintained in McCoy’s 5A medium as
previously described (19). All
media were supplemented with 10% fetal bovine serum (FBS),
penicillin (100 IU/ml) and streptomycin (100 μg/ml). The
cells were seeded twice a week and incubated at 37°C, 95% humidity,
5% CO2.
MTS assays
For cell proliferation assays, the HCT-116 and
SW-480 cells were seeded in 96-well plates at a concentration of
5000/well, allowed to adhere for 24 h and subsequently exposed to
different concentrations of compounds (2.5, 5, 10, 25, 50 and 100
μM). After 24, 48 and 72 h, survival and growth were
measured by the CellTiter 96 Aqueous MTS Reagent (Promega, Madison,
WI) according to the manufacturer’s instructions. The absorbance
value was measured by an automatic microplate reader (Epoch;
Bio-Tek Instruments, Winooski, VT) at 490 nm. Results are expressed
as a percentage versus control (vehicle set at 100%).
Apoptosis assay
HCT-116 and SW-480 cells were seeded in 24-well
plates. After 24 h, the medium was changed and chemicals were added
with indicated concentrations. After treatment for 48 h, all the
adherent cells were collected with 0.05% trypsin, including the
floating cells in the medium. Annexin-V-(FITC) and propidium iodide
(PI, Becton-Dickinson, San Diego, CA) were used for staining
according to the manufacturer’s instructions. Vehicle-treated cells
were set for control. The double-stained cells were subsequently
analyzed by a FACSCanto flow cytometer (Becton-Dickinson, Mountain
View, CA). All experiments were processed independently three
times. At least 10,000 cells were counted each time.
Cell cycle assay
For cell cycle arrest analysis, HCT-116, SW-480,
HCT-116 p53+/+ or p53−/− cells were seeded in
a 12-well plate. On the second day, the compounds were administered
at different concentrations. After 48 h, cells were dispensed and
fixed with 80% ethanol and frozen for >2 h at −20°C. With a
treatment of 0.25% Triton X-100 for 5 min, the cells were
resuspended in 150 μl of PI/RNase staining buffer
(Becton-Dickinson, San Diego, CA), incubated in the dark for 20
min, followed by counting with a FACSCanto flow cytometer. At least
10,000 cells were collected for each measurement in a triplicate
experiment.
Real-time PCR array of human cell cycle
related genes
Total RNA was extracted 48 h after exposure to
genistein using RNeasy mini kit (Qiagen, Valencia, CA) and
quantified by Nanodrop (Thermo, Wilmington, DE). The cDNA was
created with RT2 first strand kit (SAbioscience,
Frederick, MD). Then the first strand of transcription product was
applied as a template by using Human cell cycle RT2 Profiler PCR
array plate (cat no. PAHS-020E, 84 genes covered, SAbioscience)
following the manufacturer’s instructions. Experiments were run
three times. Relative genes expression quantification was
determined using the 2−ΔΔct method.
Immunoblot assay
The HCT-116 p53+/+ and p53−/−
cells were lysed in ice-cold radio immunoprecipitation assay (RIPA)
buffer supplemented with 1% (v/v) protease inhibitor cocktail and
PMSF. Then the lysates were collected and the clear supernatant was
stored in aliquots at −80°C for further analysis. The protein
concentration of the lysates was determined by a BCA protein assay
kit (Pierce, Rockford, IL). Aliquots of the lysates (50 μg
of total protein) were denatured with loading buffer for 5 min at
95°C and resolved by 4–15% Mini-PROTEAN TGX precast gel (Bio-Rad,
Hercules, CA). The assorted proteins were transferred to a PVDF
membrane (Bio-Rad) and blocked in PBST buffer (PBS with 0.05%
Tween-20) containing 5% non-fat dried milk. The transferred blots
were incubated with various primary antibodies (p53,
p21waf1/cip1, GADD45α, ATM, cdc2, cdc25A and
β-actin from Cell Signaling, Danvers, MA) overnight at 4°C,
followed by 1-h incubation with appropriate secondary antibodies
conjugated to horseradish peroxidase. β-actin expression was used
as the loading control. The intensity of the specific
immunoreactive bands was detected by SuperSignal West Pico
Substrate (Thermo-pierce, Rockford, IL) and quantified by
densitometry using ImageJ 1.45 software. Data were supplied as a
ratio of the β-actin for analyzing and plotting.
Statistical analysis
Data are presented as mean ± standard deviation.
Comparisons of 2 groups were made by Student’s t-test. A p=0.05 was
used to determine significant differences. All analyses were
performed using SPSS 14.0 (IBM Corp., Somers, NY).
Results
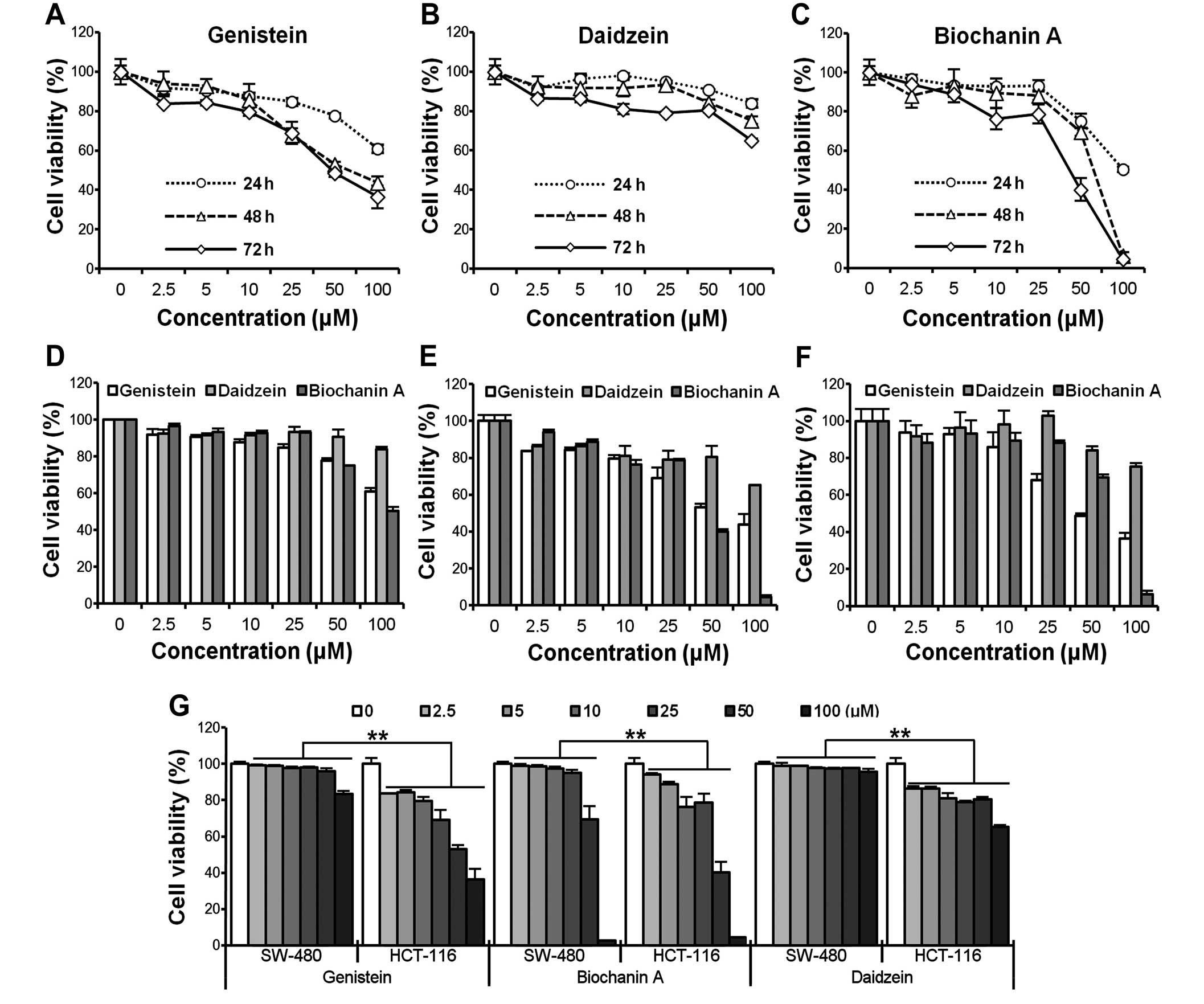
Genistein, daidzein and biochanin A
inhibits HCT-116/SW-480 cell proliferation
To investigate the growth inhibitory effect of
genistein, daidzein and biochanin A, HCT-116/SW-480 cells were
treated with different concentrations (2.5, 5, 10, 25, 50, 100
μM) of compounds for different periods of time (24, 48 and
72 h). Cell viability was assessed by the MTS assay. As shown in
Fig. 1A–C, the viability of
HCT-116 cells decreased at indicated time points in a
time-dependent manner. However, cell growth inhibition caused by
genistein was relatively remarkable compared to the other two
compounds at the logical doses (<50 μM, IC50
of genistein and biochanin A were ∼50 μM, HCT-116 cells were
more resistant to daidzein compared to the other two compounds). In
addition, HCT-116 also acted in a clearly dose-dependent way after
genistein administration at 24, 48 and 72 h (Fig. 1D–F). To further compare the
cytotoxicity induced by the compounds on cells with different
original p53 status, HCT-116 (p53 wild-type) and SW-480 (p53
mutated) cells were simultaneously exposed to the compounds. The
results are presented in Fig. 1G.
HCT-116 cells were more sensitive than SW-480 cells among the three
compounds, suggesting that the p53 status might contribute to the
different outcomes after the exposure (**p<0.01).
Genistein, biochanin A and daidzein
promote apoptosis
HCT-116/SW-480 cells were treated with different
concentrations (10, 25, 50 and 100 μM) of genistein,
daidzein and biochanin A for 48 h. Apoptotic cells were determined
by flow cytometry using Annexin V/propidium iodide (PI) double
labeling. The ratio of the apoptotic cells (Annexin
V-FITC-positive) in HCT-116 was significantly increased in a
dose-dependent manner when treated with genistein or biochanin A.
However, no explicit effects were observed after daidzein exposure
(Fig. 2A). Biochanin A showed a
relatively more intense proapoptotic effect than genistein at
higher concentrations (total apoptotic cell ratio 46.4 vs. 33.4% at
50 μM, 85 vs. 59.2% at 100 μM). Compared with the
proapoptotic impact promoted by these compounds in SW-480 cells it
also showed that HCT-116 cells were more sensitive when exposed to
genistein than SW-480 cells (Fig.
2B, **p<0.01), which suggested that the p53
status is crucial for stimulating programmed cell death after
genistein administration.
Genistein, biochanin A and daidzein
induce cell cycle arrest in HCT-116/SW-480 cells
The cell cycle assay was also examined by flow
cytometry. HCT-116/SW-480 cells were treated with 10, 25, 50
μM of the compounds for 48 h. As shown in Fig. 3A, genistein induced G2/M cell cycle
arrest in a dose-dependent manner in both HCT-116 and SW-480 cells
(p<0.01). Biochanin A showed an interesting result in that it
only expressed G2/M arrest in SW-480 cells but not in HCT-116 cells
(Fig. 3B, p<0.01). Daidzein did
not have a strong effect in arresting cell progression at the G2/M
phase in HCT-116 cells. Due to the above effects induced by
genistein, it was selected as a typical isoflavone for use in later
experiments.
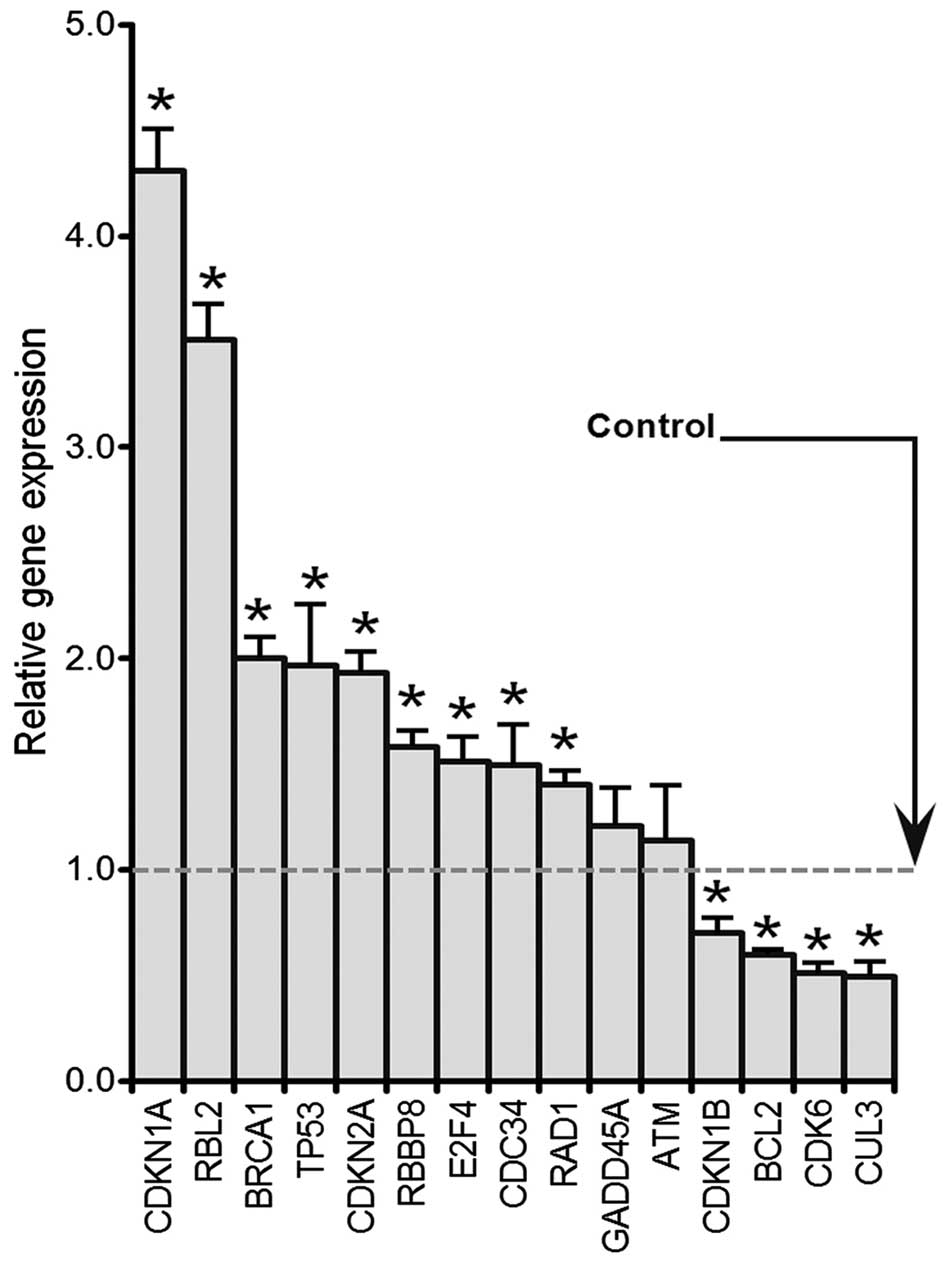
Genistein induces genes expression
involved in G2 to M transition by RT-PCR
HCT-116 cells were treated with 50 μM
genistein for 48 h. A human cell cycle RT2 Profiler PCR array kit
(cat no. PAHS-020E, 84 genes contained, SAbioscience) was used to
determine the expression levels of cell cycle-related genes.
Selected genes are presented in Fig.
4 that were either up or downregulated. p53, p21(CDKN1A), BRCA1
and E2F4 were significantly increased compared with the control
(p<0.05). In contrast, Bcl2, CDK6 and CUL3 levels were
significantly decreased after genistein exposure (p<0.05).
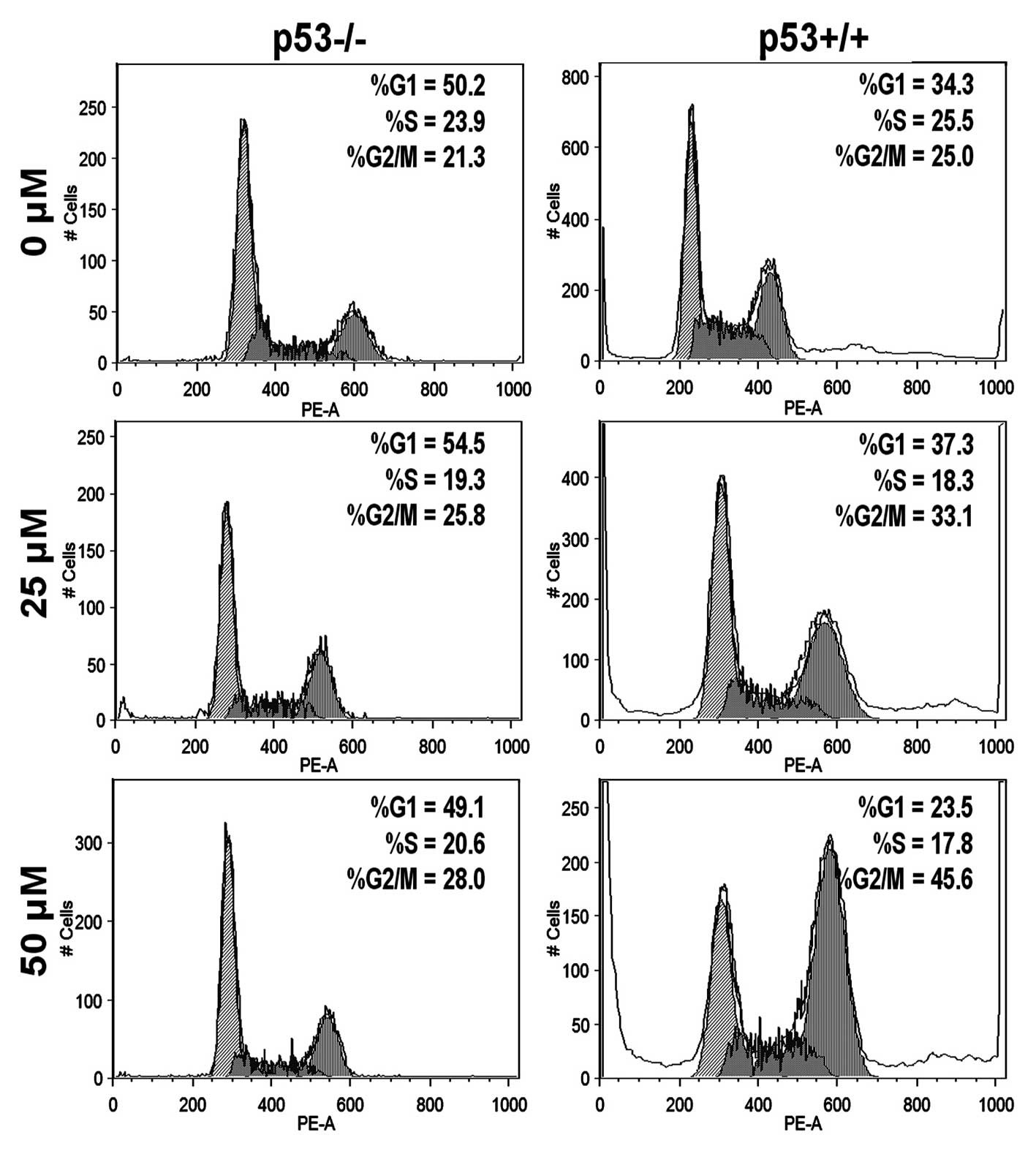
Genistein induces G2/M cell cycle arrest
partially in p53-dependent manner in HCT-116 cells
To evaluate whether p53 is a key factor in G2/M cell
arrest induced by genistein, we performed a cell cycle assay to
investigate both HCT-116 p53+/+ and p53−/−
cells. Fig. 5 shows that genistein
arrested more p53+/+ cells in the G2/M phase than
p53−/− cells at 25 and 50 μM (p<0.01,
statistically determined by 3 independent experiments), which
suggests that p53 plays a pivotal role in promoting G2/M cell cycle
capture in the HCT-116 cell line.
Genistein enhances ATM/p53-p21 expression
and suppresses cdc2, cdc25A expression in p53+/+ HCT-116
cells
HCT-116 p53+/+ and p53−/−
cells were exposed to different concentrations of genistein (25,
50, 100 μM) for 72 h and the expression of p53 and its
targets, p21waf1/cip1, GADD45α, were determined
by western blotting. β-actin was used as a control. p53 increased
in a dose-dependent manner in HCT-116 p53+/+ cells
(Fig. 6). The expression of
p21waf1/cip1 and GADD45α was enhanced in both
HCT-116 p53+/+ and p53−/− cells (Fig. 6). It should be noted that
p21waf1/cip1 was increased notably in
p53+/+ cells compared to p53−/− cells, which
suggested that p53 is an important transcriptional regulator of
p21waf1/cip. Ataxia telangiectasia mutated (ATM),
which acted as an upstream regulator and co-regulator of p53 in
cell cycle processing, also increased along with p53 and
p21waf1/cip1 in both cell types. cdc2 and cdc25A,
the effectors of p53/p21waf1/cip1, were
downregulated in our experiments. These two molecules dramatically
deceased in p53+/+ cells compared with the
p53−/− cells, which might contribute to the fact that
genistein induced a stronger cell cycle capture effect in HCT-116
cells with a stable p53 status.
Discussion
Plant-derived polyphenolic compounds containing
isoflavones have attracted considerable interest for their
anticancer properties. However, the mechanism of their anticancer
effects remains to be fully understood. In this study, we showed
that the soy isoflavones genistein, daidzein and biochanin A had
significant inhibitory effects on cell growth and promoted
apoptosis in human colon cancer cells. Among them, genistein was
the most attractive agent for G2/M phase cell cycle arrest. As one
of the major components of soy isoflavones, genistein clearly
exhibited anticancer effects in a time- and dose-dependent manner,
which might probably make it a competitive candidate for anticancer
research. Furthermore, we observed that genistein activated the
ATM/p53-p21waf1/cip1 cross-talk regulatory
network, a pathway implicated in G2/M cell arrest and apoptosis. A
microarray-based qPCR array presented that several genes involved
in cell cycle and apoptosis, such as CDKN1b, BRCA1 and BRCA2 and
Bcl2, were examined and underwent transcriptional activation after
genistein exposure. This suggests that genistein regulates multiple
regulatory factors, including the cyclin families and Bcl families,
to modulate the complicated cancer development and progress.
Kim et al reported that genistein and
daidzein suppressed cell proliferation and induced apoptosis in
HT-29 cells and that these events were related to the inhibition of
insulin-like growth factor-1 (IGF-1) receptor signaling and the
PI3k/AKT pathway, also including the involvement of the MAPK
pathway (20). The MAPK pathway,
which includes ERK-1/2 and AKT as key regulators, has been reported
to be crucial in cell proliferation and growth (21). Specifically, the ERK-1/2 signaling
pathway has been implicated in the regulation of cell cycle phases
such as G1 and G2/M. With regard to colon cancer, soy isoflavones
have been shown to inhibit HT-29 cell (22) and Colo320 cell (23) growth by an accumulation of cells at
the G2/M phase. p53, acting as an important tumor suppressor gene,
regulates cancer cell progression through multiple mechanisms,
including the induction of apoptosis and cell cycle arrest, which
has been well reviewed (24). It
is encouraging that, consistent with previous studies, we observed
that genistein exerted anticancer effects partially in a
p53-dependent manner. Genistein arrested HCT-116 cells at the G2/M
phase in a dose-dependent manner. This was correlated to its effect
of triggering the upregulation of a tumor suppressor gene, p53, as
well as the CDK inhibitor p21waf1/cip1. The
results are further substantiated by the accompanied decreases in
the expression of the cyclin family complex proteins, cdc2 and
cdc25A. p53 status is crucial to the apoptosis and cell cycle
arrest on human colorectal cancer cell line HCT-116.
It should be noted that genistein did not increase
the expression of p27kip1 by q-PCR assay in our
study, which implied that p21waf1/cip1 played a
more important factor in G2/M cell capture induced by genistein
exposure. A recent report suggested that genistein inhibited
EGF-induced proliferation, which favors dephosphorylation and
nuclear retention of FOXO3 in colon cancer cells. Upstream of
FOXO3, genistein acts via the PI3K/Akt pathway to inhibit
EGF-stimulated FOXO3 phosphorylation. Downstream, EGF induced
disassociation of FOXO3 from mutated tumor suppressor p53, but not
wild-type p53, which is inhibited by genistein favoring
FOXO3-p53(mut) interactions with the promoter of the cell cycle
inhibitor p27kip1 in colon cancer cells. The
FOXO3-p53(mut) complex leads to elevated p27kip1
expression and promotes cell cycle arrest (25). This might be interpreted that the
partially p53-dependent cell cycle has an arresting mechanism in
p53-wild-type or p53-mutanted human cancer cell lines. Our
experiments mainly aimed at studying the contributions of p53 on
G2/M cell arrest. Other pathways involved in the anticancer
properties of the compounds, including those that mediate cell
death and apoptosis, are still in need of further
investigation.
Eukaryotic cells employ multiple mechanisms to
ensure accurate transmission of genetic information between
generations. Critical surveillance of this transmission is provided
by the DNA damage response (DDR) (26). Disruption or attenuation of DDR
plays an essential role in promoting tumorigenesis (27–29).
This system not only functions in the process of DNA damage repair,
but is also integrated with other processes including the cell
cycle, apoptosis and transcription (26). DNA damage was able to activate the
MEK-ERK pathway in a p53-dependent (30) and independent manner (31). In our study, genistein induced G2/M
cell arrest via the p53/ATM pathway in a dependent way, which
suggested that p53 has a more crucial role in controlling cell
expansion. As demonstrated by numerous studies, p53 is a key tumor
suppressor gene and is one of the most important mainstays of our
body’s anticancer defense (32).
In response to multiple cellular stresses, such as DNA damage and
hyperproliferation (18,33), p53 is activated or stabilized. Once
induced, p53 can function as a transcription factor to regulate the
several genes leading to apoptosis, senescence and cell cycle
arrest (34–36). In the early stages of tumor
development, genomic instability and DNA damage lead to p53
activation and mediate tumor suppression (37,38).
Our data showed that ATM/p53-p21waf1/cip1 was
activated when human colon cancer cells were treated by genistein.
Similar to previous studies, genistein exposure introduced DNA
damage or genotoxic stress, which could lead to activation of ATM
and subsequently induce p53 expression. Through phosphorylation,
p53 induces the expression of p21waf/cip1 as well
as GADD45α. Accordingly, cdc2 and cdc25A activity is inhibited,
which results in the capture of G2/M phase cells. The G2/M
checkpoint prevents cells from initiating mitosis in the presence
of DNA damage (26,39). Interestingly, p53 was regarded as
the most important regulatory factor in the G2/M cell cycle
checkpoint, but we observed that it is not the only triggering
factor for controlling cell cycle transit, which suggests that a
complicated regulatory network is involved in the cell cycle
progression in response to DNA damage in HCT-116 p53 wild or p53
mutant cells. Our findings presented here suggest that
p53-p21waf1/cip1/cdc2 cross-talk co-regulates
cell cycle distribution at different phases, which could possibly
be an important mechanism for understanding the cell cycle progress
induced by genistein in human colon malignancies.
Taken together, our data showed that isoflavones,
such as genistein, inhibit human colon cell proliferation and
growth by causing cell arrest at the G2/M phase and promoting
substantial apoptosis. These processes are primarily mediated by
the upregulation of p53/p21waf1/cip1, GADD45α and
down-regulation of cdc2 and cdc25A. In response to DNA damage, the
cells trigger the checkpoint signaling cascades to regulate cell
cycle progression and elicit DNA repair mechanisms in need of
maintaining genomic stability and integrity. High doses of
genistein not only induce DNA damage and block the cell cycle, but
they also initiate programmed cell death. These findings may
provide some new insights into the genotoxic effects and antitumor
mechanisms of genistein and other isoflavonoids in human colon
cancers.
Acknowledgements
This study was supported in part by
the NIH/NCCAM grants P01 AT004418 and K01 AT005362.
References
|
1.
|
Arber N and Levin B: Chemoprevention of
colorectal cancer: ready for routine use? Recent Results Cancer
Res. 166:213–230. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Rodriguez M, Du GJ, Wang CZ and Yuan CS:
Letter to the editor: Panaxadiol’s anticancer activity is enhanced
by epicatechin. Am J Chin Med. 38:1233–1235. 2010.PubMed/NCBI
|
|
3.
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar
|
|
4.
|
Wils J, O’Dwyer P and Labianca R: Adjuvant
treatment of colorectal cancer at the turn of the century: European
and US perspectives. Ann Oncol. 12:13–22. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Segal NH and Saltz LB: Evolving treatment
of advanced colon cancer. Annu Rev Med. 60:207–219. 2009.
View Article : Google Scholar
|
|
6.
|
Thomasset SC, Berry DP, Garcea G, Marczylo
T, Steward WP and Gescher AJ: Dietary polyphenolic phytochemicals -
promising cancer chemopreventive agents in humans? A review of
their clinical properties. Int J Cancer. 120:451–458. 2007.
View Article : Google Scholar
|
|
7.
|
Doll R and Peto R: The causes of cancer:
quantitative estimates of avoidable risks of cancer in the United
States today. J Natl Cancer Inst. 66:1191–1308. 1981.PubMed/NCBI
|
|
8.
|
Umthong S, Puthong S and Chanchao C:
Trigona laeviceps propolis from Thailand: antimicrobial,
antiproliferative and cytotoxic activities. Am J Chin Med.
37:855–865. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Fournier DB, Erdman JW Jr and Gordon GB:
Soy, its components and cancer prevention: a review of the in
vitro, animal and human data. Cancer Epidemiol Biomarkers Prev.
7:1055–1065. 1998.PubMed/NCBI
|
|
10.
|
Kao TH, Chien JT and Chen BH: Extraction
yield of isoflavones from soybean cake as affected by sovlent and
supercritical carbon dioxide. Food Chem. 107:1728–1736. 2008.
View Article : Google Scholar
|
|
11.
|
Banerjee S, Li Y, Wang Z and Sarkar FH:
Multi-targeted therapy of cancer by genistein. Cancer Lett.
269:226–242. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Hartwell LH and Kastan MB: Cell cycle
control and cancer. Science. 266:1821–1828. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Igney FH and Krammer PH: Death and
anti-death: tumour resistance to apoptosis. Nat Rev Cancer.
2:277–288. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Agarwal MK, Hastak K, Jackson MW, Breit
SN, Stark GR and Agarwal ML: Macrophage inhibitory cytokine 1
mediates a p53-dependent protective arrest in S phase in response
to starvation for DNA precursors. Proc Natl Acad Sci USA.
103:16278–16283. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Liu BP, Chong EY, Cheung FW, Duan JA, Che
CT and Liu WK: Tangutorine induces p21 expression and abnormal
mitosis in human colon cancer HT-29 cells. Biochem Pharmacol.
70:287–299. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Taylor CK, Levy RM, Elliott JC and Burnett
BP: The effect of genistein aglycone on cancer and cancer risk: a
review of in vitro, preclinical and clinical studies. Nutr Rev.
67:398–415. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Schleipen B, Hertrampf T, Fritzemeier KH,
et al: ERbeta-specific agonists and genistein inhibit proliferation
and induce apoptosis in the large and small intestine.
Carcinogenesis. 32:1675–1683. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Vousden KH and Lane DP: p53 in health and
disease. Nat Rev Mol Cell Biol. 8:275–283. 2007. View Article : Google Scholar
|
|
19.
|
Luo X, Chen J, Song WX, et al: Osteogenic
BMPs promote tumor growth of human osteosarcomas that harbor
differentiation defects. Lab Invest. 88:1264–1277. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Kim EJ, Shin HK and Park JH: Genistein
inhibits insulin-like growth factor-I receptor signaling in HT-29
human colon cancer cells: a possible mechanism of the growth
inhibitory effect of Genistein. J Med Food. 8:431–438. 2005.
View Article : Google Scholar
|
|
21.
|
Meloche S and Pouyssegur J: The ERK1/2
mitogen-activated protein kinase pathway as a master regulator of
the G1- to S-phase transition. Oncogene. 26:3227–3239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Yu ZL, Li WJ and Liu FY: Inhibition of
proliferation and induction of apoptosis by genistein in colon
cancer HT-29 cells. Cancer Lett. 215:159–166. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Park JH, Oh EJ, Choi YH, et al:
Synergistic effects of dexamethasone and genistein on the
expression of Cdk inhibitor p21(WAF1/CIP1) in human hepatocellular
and colorectal carcinoma cells. Int J Oncol. 18:997–1002.
2001.PubMed/NCBI
|
|
24.
|
Vousden KH and Lu X: Live or let die: the
cell’s response to p53. Nat Rev Cancer. 2:594–604. 2002.
|
|
25.
|
Qi W, Weber CR, Wasland K and Savkovic SD:
Genistein inhibits proliferation of colon cancer cells by
attenuating a negative effect of epidermal growth factor on tumor
suppressor FOXO3 activity. BMC Cancer. 11:2192011. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Zhou BB and Elledge SJ: The DNA damage
response: putting checkpoints in perspective. Nature. 408:433–439.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Schar P: Spontaneous DNA damage, genome
instability and cancer--when DNA replication escapes control. Cell.
104:329–332. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Hoeijmakers JH: Genome maintenance
mechanisms for preventing cancer. Nature. 411:366–374. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Rouse J and Jackson SP: Interfaces between
the detection, signaling and repair of DNA damage. Science.
297:547–551. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Lee SW, Fang L, Igarashi M, Ouchi T, Lu KP
and Aaronson SA: Sustained activation of Ras/Raf/mitogen-activated
protein kinase cascade by the tumor suppressor p53. Proc Natl Acad
Sci USA. 97:8302–8305. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Tang J and Chu G: Xeroderma pigmentosum
complementation group E and UV-damaged DNA-binding protein. DNA
Repair. 1:601–616. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Levine AJ and Oren M: The first 30 years
of p53: growing ever more complex. Nat Rev Cancer. 9:749–758.
2009.PubMed/NCBI
|
|
33.
|
Toledo F and Wahl GM: Regulating the p53
pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer.
6:909–923. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Riley T, Sontag E, Chen P and Levine A:
Transcriptional control of human p53-regulated genes. Nat Rev Mol
Cell Biol. 9:402–412. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Chen Z, Trotman LC, Shaffer D, et al:
Crucial role of p53-dependent cellular senescence in suppression of
Pten-deficient tumorigenesis. Nature. 436:725–730. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Fridman JS and Lowe SW: Control of
apoptosis by p53. Oncogene. 22:9030–9040. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Hoglund P: DNA damage and tumor
surveillance: one trigger for two pathways. Sci STKE.
2006:pe22006.PubMed/NCBI
|
|
38.
|
Wang D and Lippard SJ: Cellular processing
of platinum anti-cancer drugs. Nat Rev Drug Discov. 4:307–320.
2005. View Article : Google Scholar
|
|
39.
|
Abraham RT: Cell cycle checkpoint
signaling through the ATM and ATR kinases. Genes Dev. 15:2177–2196.
2001. View Article : Google Scholar : PubMed/NCBI
|