Introduction
Malignant fibrous histiocytoma (MFH), which has
recently been classified as undifferentiated pleomorphic sarcoma
(UPS), is the most common high-grade soft tissue sarcoma that
occurs in late adult life (1,2).
Advances in the treatment of MFH have led to multidisciplinary
treatment, including surgery, chemotherapy and radiation therapy,
resulting in great improvements in the quality of life of patients
with the disease, however, the chemotherapy and radiation therapy
strategies for MFH are not as effective as those for other
malignancies. After adequate local treatment and adjuvant therapy,
~50% of patients invariably relapse with local recurrence and
distant metastasis, hence the prognosis of patients with MFH is
poor (1,3,4).
Therefore, it is necessary to elucidate the factors contributing to
tumor progression and metastasis in MFH and to establish more
effective therapeutic strategies against MFH.
Decoy receptor 3 (DcR3) is a newly identified member
of the tumor necrosis factor receptor (TNFR) superfamily. DcR3
lacks a transmembrane domain and is thought to be a soluble
secreted protein. DcR3 is known to act as a decoy receptor for Fas
ligand (FasL) (5), LIGHT (6) and TL1A (7). Among these, FasL, which is produced
by activated T cells and natural killer cells, is the most
important regulator of cellular apoptosis through the death
receptor pathway. DcR3 competes with Fas receptor for binding to
FasL and inhibits the FasL/Fas apoptotic pathway. Fas receptor has
an intracellular death domain that triggers the extrinsic apoptotic
signaling pathway by activating caspase-8, which can induce
apoptosis through the activation of caspase-3, -6 and -7 and poly
(ADP-ribose) polymerase (PARP). The FasL/Fas apoptotic pathway is a
central regulator of apoptosis in mammals to eliminate malignant
tumor cells and resistance to this apoptotic pathway is thought to
be one of the hallmarks of malignant tumors (8,9).
Therefore, DcR3 has been thought to contribute to tumor progression
by inhibiting FasL-induced apoptosis of tumor cells.
Recent studies have demonstrated that DcR3 also
functions as an effector molecule independently of the FasL/Fas
apoptotic pathway. DcR3 overexpression promotes migration and
invasion of nasopharyngeal carcinoma cells (10). In breast cancer cells, DcR3
suppression decreases both migration and invasion (11). Moreover, several studies have
revealed that DcR3 promotes migration of HUVECs with an increased
expression of matrix metalloproteinase (MMP)-2 (12) and that DcR3 can activate various
signaling kinases such as Akt, extracellular signal-regulated
kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK) and p38
mitogen-activated protein kinase (p38) (13–17).
These reports strongly indicate that DcR3, in addition to its role
as a decoy receptor for FasL, may function as a modulator of
malignant progression in cancer cells.
Overexpression of DcR3 has been reported in various
malignancies such as lung and colon cancers (5), EBV or HTLV-1 associated lymphomas
(18), malignant gliomas (19) and pancreatic adenocarcinomas
(20). Furthermore, we have
previously reported that overexpression of DcR3 was observed in
bone and soft tissue sarcomas (21). Several studies have also revealed
that DcR3 overexpression is associated with distant metastasis and
overall survival in human cancers (22–27).
In the development of metastasis, MMPs are thought to play
important roles in tumor cells by degrading the extracellular
matrix (ECM) (28,29). The increased expression of certain
MMPs correlates with tumor expansion, invasiveness and poor
prognosis of patients with malignant tumors (30). Among the MMP subtypes, activation
of MMP-2 has been observed in MFH (31,32),
therefore, MMP-2 is thought to contribute to the metastatic
potential of human MFH.
Based on previous studies, DcR3 may contribute to
tumor progression by not only neutralizing FasL-induced apoptosis
but also by promoting tumor migration and invasion, which are
important for the development of metastasis. We hypothesized that
DcR3 may play important roles in tumor progression and metastatic
potential in human MFH through the inhibition of the apoptotic
pathway and the signaling pathway that is related to migration and
invasion. In the present study, we evaluated the effects of DcR3
inhibition on cell apoptosis, migration and invasion using human
MFH cell lines to elucidate the roles of DcR3 in human MFH.
Materials and methods
Human MFH cell lines
Two human MFH cell lines, TNMY1 and Nara-H, were
used in this study. TNMY1 was previously established in our
laboratory (33) and Nara-H was
obtained from ScienStuff Co. (Nara, Japan) (34). Cells were grown in culture medium
consisting of Dulbecco’s modified Eagle’s medium (DMEM;
Sigma-Aldrich Co., St. Louis, MO, USA) supplemented with 10% fetal
bovine serum (FBS; Sigma-Aldrich) and 100 U/ml
penicillin/streptomycin solution (Sigma-Aldrich). Cell lines were
routinely maintained at 37°C in a humidified 5% CO2
atmosphere. For all experiments, we used the DMEM containing 10%
FBS without the antibiotic solution.
Transfection of small interfering RNA
(siRNA)
To evaluate the effect of DcR3 knockdown, we
transfected MFH cell lines with DcR3-specific small interfering RNA
(siRNA). Briefly, 1 day before transfection, cells were seeded in
6-well culture plates in growth medium. Then, cells were
transfected with 60 nmol of either a specific siRNA against human
DcR3 (DcR3-si) (Invitrogen, Carlsbad, CA, USA) or a negative
control siRNA (Ctrl-si) (Invitrogen) using Lipofectamine 2000
transfection reagent according to the manufacturer’s protocol
(Invitrogen).
Recombinant Fc, FasL and PI3K inhibitor
treatment
After siRNA transfection, DcR3-si transfected cells
were incubated in medium with 0 (DcR3-si cells) or 3 μg/ml of
recombinant DcR3-Fc (DcR3-si+DcR3-Fc cells; R&D Systems,
Minneapolis, MN, USA), or 3 μg/ml of recombinant IgG-Fc
(DcR3-si+IgG-Fc cells, as a control; R&D Systems) for 24 h.
Ctrl-si transfected cells were incubated without any recombinant Fc
proteins (Ctrl-si cells).
To induce apoptosis, cells were treated with FasL
(100 ng/ml; Peprotech, Rocky Hill, NJ, USA) for 6 h after siRNA
transfection and Fc treatment.
To evaluate kinase activities, cells were pretreated
with 0 or 20 μM of LY294002 [a phosphatidylinositol 3 kinase (PI3K)
inhibitor; Cell Signaling Technology, Danvers, MA, USA] in DMSO
(Wako, Osaka, Japan) for 2 h followed by a 1-h treatment with
DcR3-Fc or IgG-Fc.
Quantitative real-time PCR
We isolated total RNAs from cell lines using an
RNeasy Mini kit according to the manufacturer’s protocol (Qiagen,
Valencia, CA, USA) and first strand cDNAs were transcribed.
Quantitative real-time PCR (qRT-PCR) was performed in a 20-μl
reaction mixture using the Power SYBR Green Master Mix reagent
(Applied Biosystems, Foster City, CA, USA) on an ABI PRISM 7500
sequence detection system (Applied Biosystems). The PCR conditions
were as follows: 1 cycle at 95°C for 10 min followed by 40 cycles
at 95°C for 15 sec and 60°C for 1 min. Primers for human
DcR3, MMP-2 and human β-actin (control) were
synthesized by Hokkaido System Science (Hokkaido, Japan). Primer
used were: DcR3: 5′-TCAATGTGCCAGGCTCTTC-3′ and 5′-AGCCACAAAG
TCGATGACG-3′; MMP-2: 5′-ACAGCAGGTCTCAGCC TCAT-3′ and
5′-TGCCTCTGGACAACACAGAC-3′; β-actin:
5′-GATGAGATTGGCATGGCTTT-3′ and 5′-CACCTTCA CCGTTCCAGTTT-3′. The
values were normalized with those for β-actin and relative
expression was analyzed using the ΔΔCt method.
Immunoblot analysis
Lysates were extracted from cells using a whole cell
lysis buffer (Mammalian Protein Extraction Reagent, Thermo
Scientific, Rockford, IL, USA) supplemented with a protease and
phosphatase inhibitor mix (Roche Applied Science, Indianapolis, IN,
USA). The protein content of lysates was then quantified using BCA
Protein Assay reagent (Bio-Rad, Richmond, CA, USA). Samples
containing equal amounts of protein were electrophoresed through
12% polyacrylamide gels and transferred onto PVDF membranes. After
blocking membranes were incubated overnight at 4°C with the
following antibodies in CanGet Signal Solution 1 (Toyobo Co., Ltd.,
Osaka, Japan): anti-human DcR3 (1:1,000), anti-human Fas (1:1,000),
anti-human PARP (1:1,000), anti-human cleaved PARP (1:1,000),
anti-human caspase-3 (1:1,000), anti-human cleaved caspase-3
(1:500), anti-human MMP-2 (1:1,000), anti-human Akt (1:2,000),
anti-human phosho-Akt (p-Akt; 1:1,000), anti-human ERK1/2
(1:2,000), anti-human phospho-ERK1/2 (p-ERK1/2; 1:1,500),
anti-human JNK (1:1,000), anti-human phospho-JNK (p-JNK; 1:1,000),
anti-human p38 (1:1,000), anti-human phospho-p38 (p-p38; 1:1,000).
All antibodies were purchased from Cell Signaling Technology.
Following washes, membranes were incubated with the appropriate
secondary antibody conjugated to horseradish peroxidase and were
exposed with ECL Plus western blot detection system reagent (GE
Healthcare Biosciences, Piscataway, NJ, USA). Antibody binding was
detected by Chemilumino analyzer LAS-3000 mini (Fujifilm, Tokyo,
Japan). Membranes were reprobed with anti-human α-tubulin antibody
(Sigma-Aldrich) to confirm equal protein loading.
Cell proliferation assays
To evaluate the involvement of DcR3 in MFH cell
proliferation, we performed WST-8 cell proliferation assay using
Cell Counting Kit-8 (CCK-8; Dojindo Inc., Kumamoto, Japan). Cells
were seeded in 96-well culture plates at a density of
5×103 cells/well in 100 μl culture medium. After siRNA
transfection and Fc treatment, cells were treated with or without
FasL to induce apoptosis. At the indicated incubation times (0, 24
and 48 h), 10 μl of the CCK-8 solution was added into each well and
incubated for 1 h. Optical density was measured at a wavelength of
450 nm using a Model 680 Microplate Reader (Bio-Rad, Hercules, CA,
USA). The relative number of viable cells in each well was
calculated.
Cell migration assays
To evaluate the effect of DcR3 on MFH cell
migration, we performed in vitro scratch wound healing
assays as previously described (35). Cells in 6-well culture plates were
transfected with siRNA and treated with recombinant Fc and then
incubated to form a confluent monolayer. A denuded area was created
by scraping with a sterile 200-μl pipette tip and each well was
washed three times with PBS to remove floating cells. Scratch
wounds were inspected with an inverted microscope (Zeiss,
Oberkochen, Germany) and captured by Motic Images Plus 2.2S
(Shimadzu, Kyoto, Japan) after 0, 12 and 24 h of wounding. The
distance between the opposing edges of the wound was measured at
three points and averaged on each image.
Cell invasion assays
The effect of DcR3 on cell invasion was assessed by
Transwell chamber invasion assays, as previously described
(36). After siRNA transfection
and Fc treatment, 5×104 cells were placed in the upper
wells of 24-well Transwell chambers (BioCoat Matrigel Invasion
Chamber, BD Biosciences, Bedford, MA, USA) and the lower wells were
filled with complete growth medium. The chambers were incubated for
30 h to allow cells to invade from the upper wells towards the
lower wells. After incubation, non-invading cells on the upper
surface of membranes were removed by scrubbing and invading cells
on the lower surface of the membranes were fixed, inspected with a
microscope and imaged. The number of invading cells was counted in
three random fields.
Gelatin zymography
To evaluate the enzyme activity of MMP-2, we
performed gelatin zymography as previously described (37). The cell culture supernatant in each
well was collected and concentrated through an Amicon Ultra-4
10,000 MWCO Centrifugal Filter Device (Millipore, Billerica, MA,
USA) and samples were electrophoresed through 10% gelatin gels
(Invitrogen). After electrophoresis, the gels were washed with
renaturing buffer (Invitrogen) for 30 min, followed by incubation
with developing buffer (Invitrogen) overnight at 37°C. The gels
were stained with Coomassie Brilliant Blue R-250 Staining Solution
(Bio-Rad) and clear bands of MMP-2 were visible against the dark
blue background.
Statistical analysis
Each experiment was performed independently at least
three times and data are presented as the mean ± standard deviation
(SD). The statistical significance of the differences among means
was evaluated by ANOVA with post hoc test. Results were considered
significant at P<0.05.
Results
DcR3 knockdown enhanced FasL-induced
apoptosis in human MFH cells
We performed transfection of DcR3-siRNA and DcR3-Fc
treatment to evaluate the effects of DcR3 in MFH cells. In both MFH
cell lines, DcR3-siRNA transfection strongly suppressed DcR3 mRNA
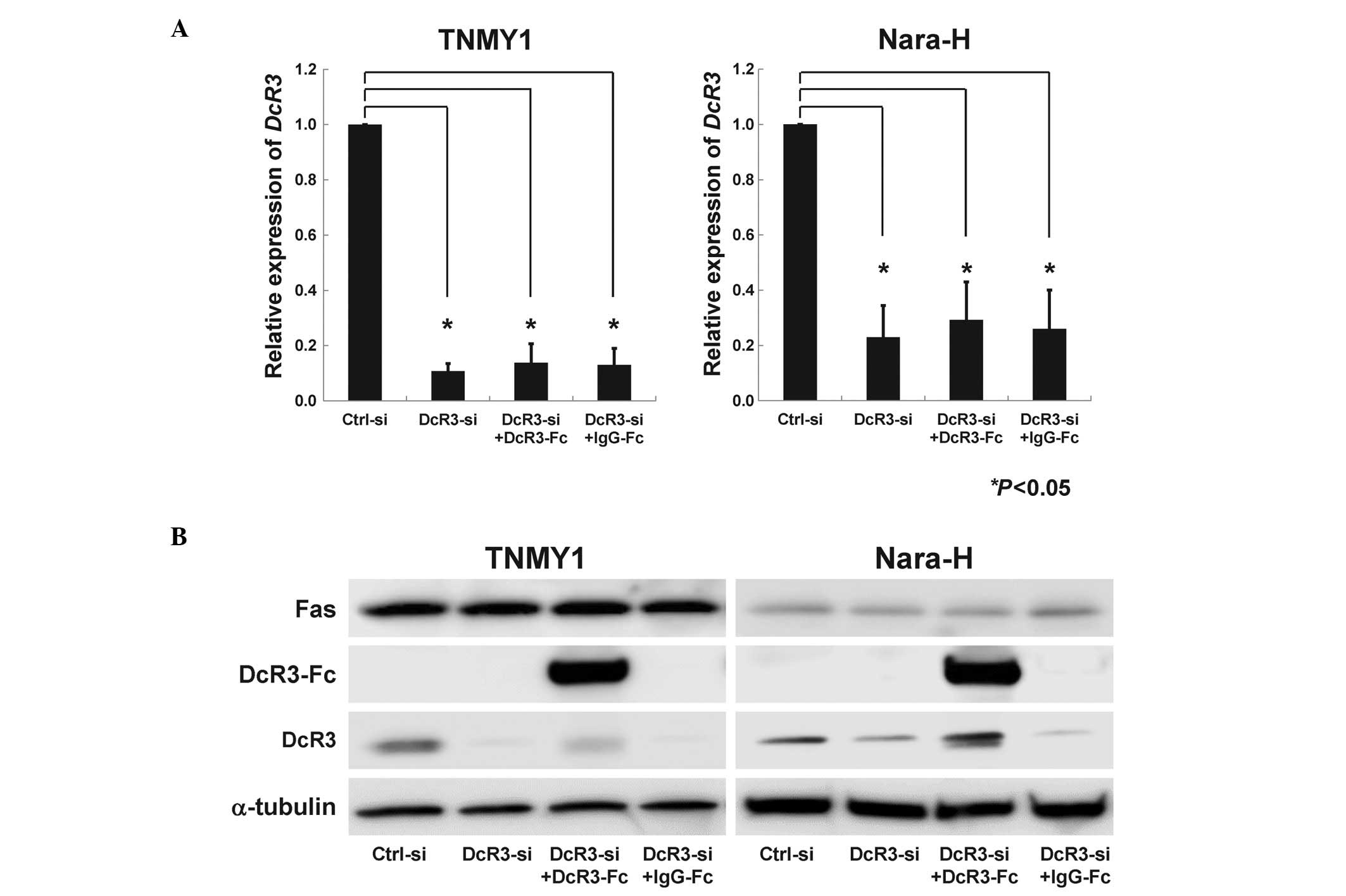
(*P<0.05, Fig. 1A)
and protein expression levels (Fig.
1B). DcR3-Fc treatment slightly, but not significantly,
increased DcR3 expression, while IgG-Fc treatment did not affect
DcR3 expression (Fig. 1B). Fas
expression was not affected by DcR3-siRNA transfection or DcR3-Fc
treatment (Fig. 1B).
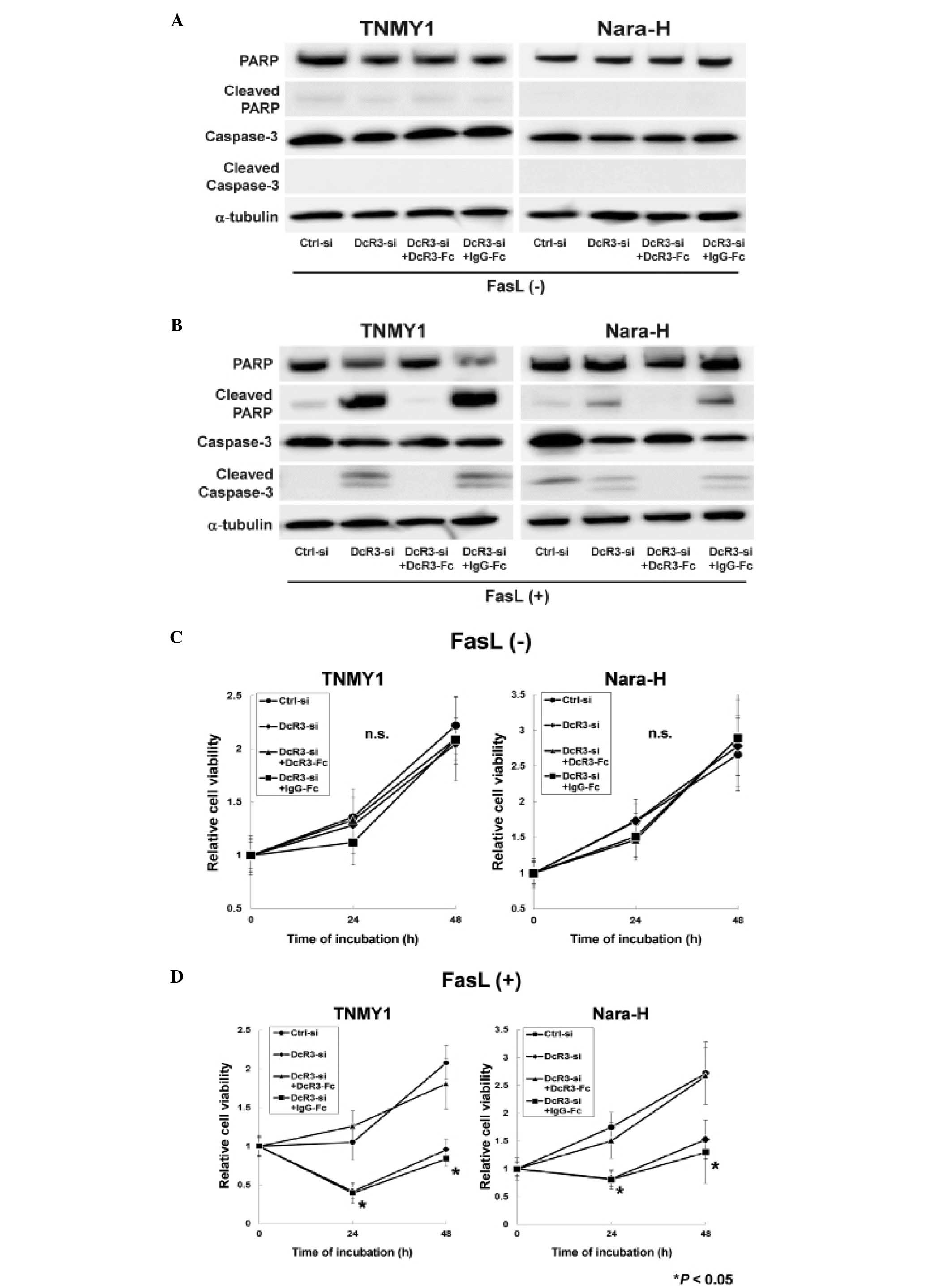
Immunoblot analysis revealed that DcR3 knockdown or
DcR3-Fc treatment without FasL treatment did not affect the
expressions of caspase-3, PARP and their cleaved forms in either
cell line (Fig. 2A). FasL
treatment strongly induced the cleavage of caspase-3 and PARP in
DcR3-si and DcR3-si+IgG-Fc cells, while the cleaved forms of
capase-3 and PARP were barely detected in Ctrl-si cells (Fig. 2B). The increased expressions of
cleaved capase-3 and cleaved PARP were suppressed by DcR3-Fc
treatment in both MFH cell lines (DcR3-si+DcR3-Fc cells; Fig. 2B).
A previous report demonstrated that DcR3 knockdown
with FasL treatment significantly suppressed cell proliferation in
human pancreatic adenocarcinoma cells (38), therefore, we evaluated the effect
of DcR3 inhibition with FasL treatment on MFH cell proliferation.
siRNA transfection and recombinant Fc treatment without FasL
treatment did not affect cell proliferation in either cell line
(Fig. 2C), whereas, cell
proliferation was significantly decreased in DcR3-si and
DcR3-si+IgG-Fc cells with FasL treatment (*P<0.05,
Fig. 2D). Moreover, DcR3-Fc
treatment increased cell proliferation in DcR3-si cells to the same
levels as in Ctrl-si cells (DcR3-si+DcR3-Fc cells; Fig. 2D).
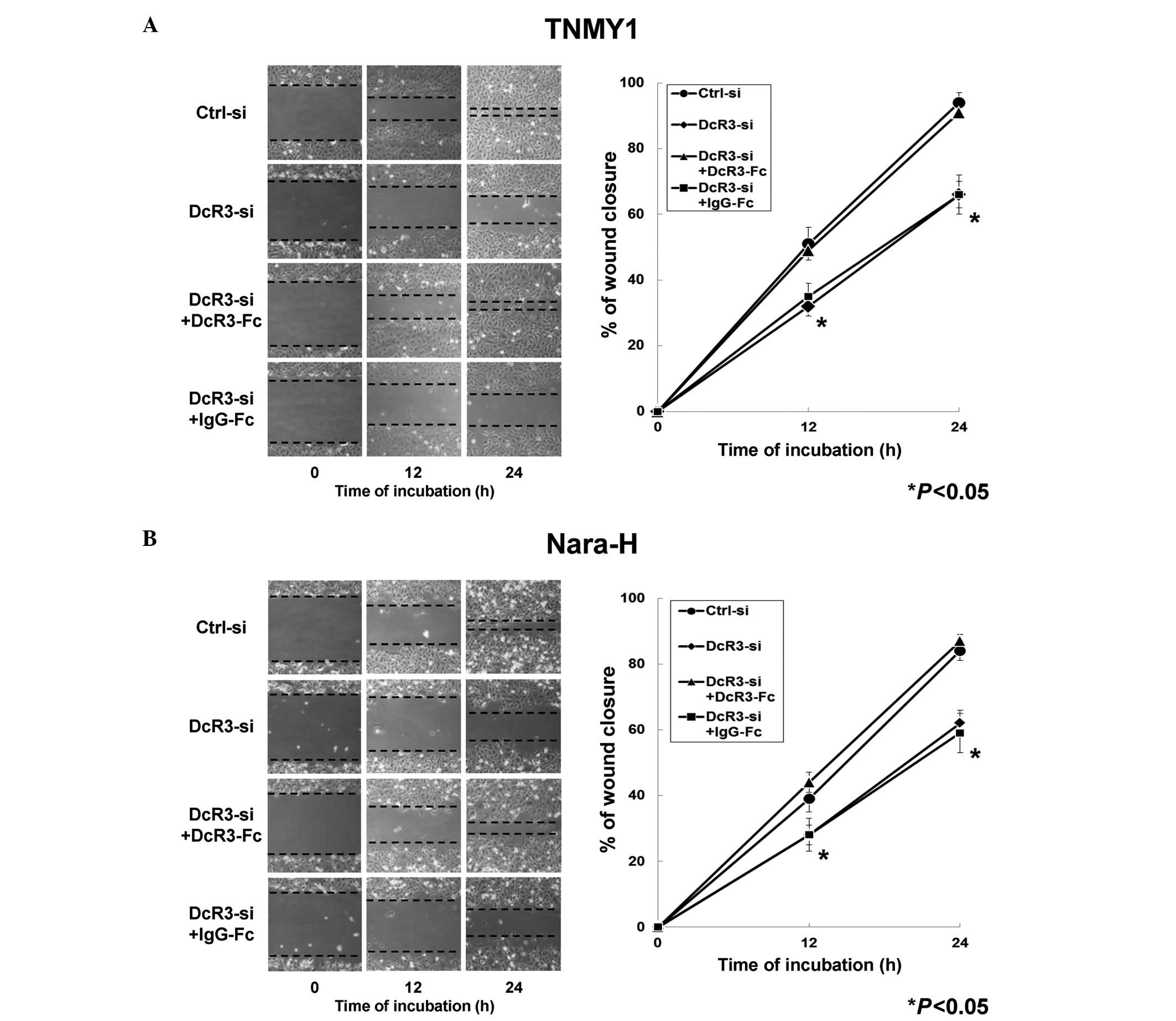
DcR3 knockdown significantly suppressed
MFH cell migration and invasion
We evaluated the effects of DcR3 on MFH cell
migration and invasion to investigate the non-decoy functions of
DcR3 in human MFH. In the in vitro scratch wound healing
assays, DcR3 knockdown significantly decreased cell migration in
both MFH cell lines at 12 and 24 h after wounding and DcR3-Fc
treatment significantly restored the migration ability compared
with control cells, respectively (*P<0.05, Fig. 3). Significant changes in cell
migration were not observed in either MFH cell line with IgG-Fc
treatment (Fig. 3).
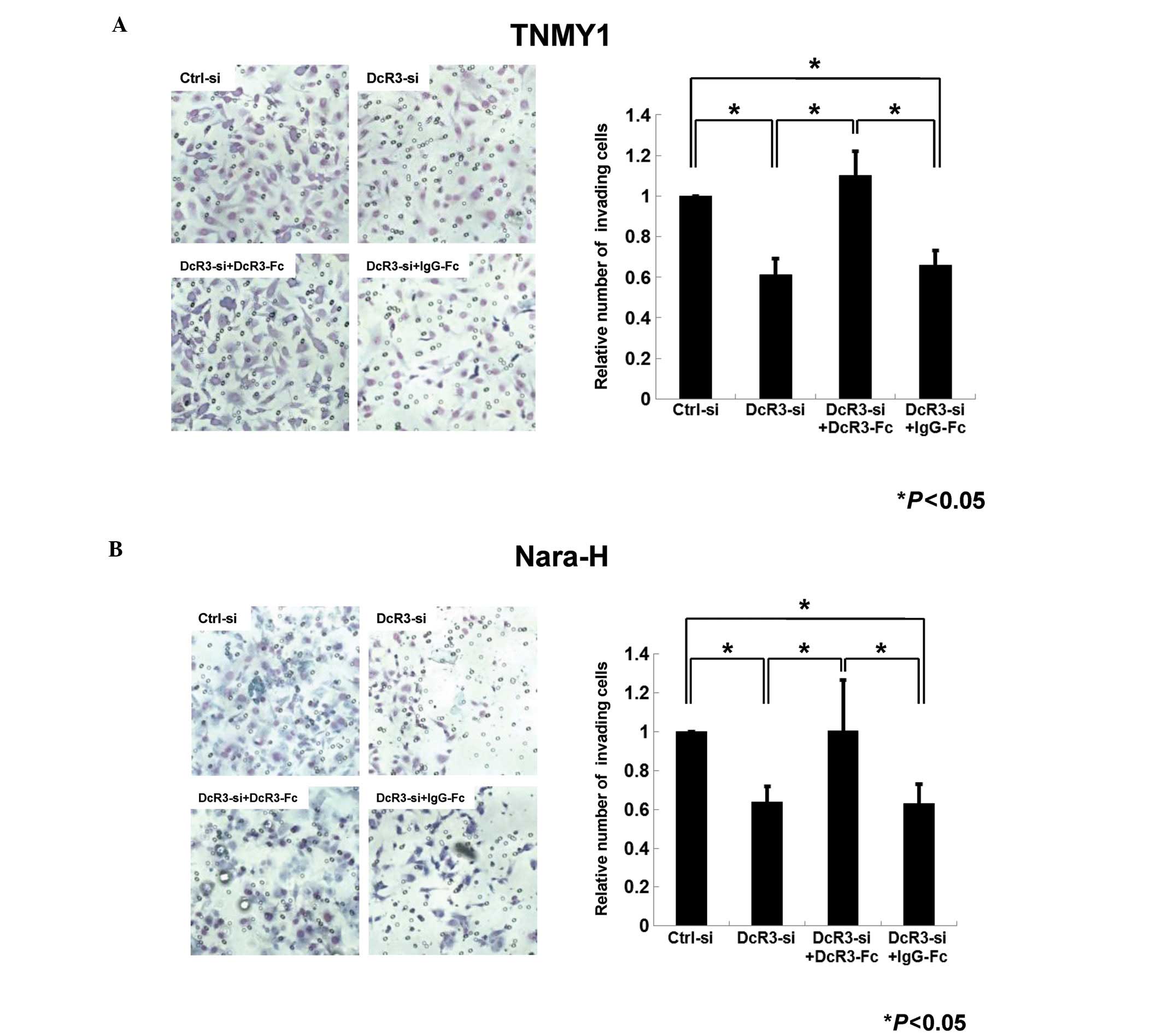
Next, we performed the transwell chamber invasion
assays to evaluate the role of DcR3 in MFH cell invasion. In both
cell lines, the number of invading cells in DcR3-siRNA transfected
cells was decreased to 61% (TNMY1, Fig. 4A) and 64% (Nara-H, Fig. 4B) of that in control cells
(*P<0.05). In addition, DcR3-Fc treatment after DcR3
knockdown significantly restored the invasiveness in both cell
lines (*P<0.05, Fig.
4).
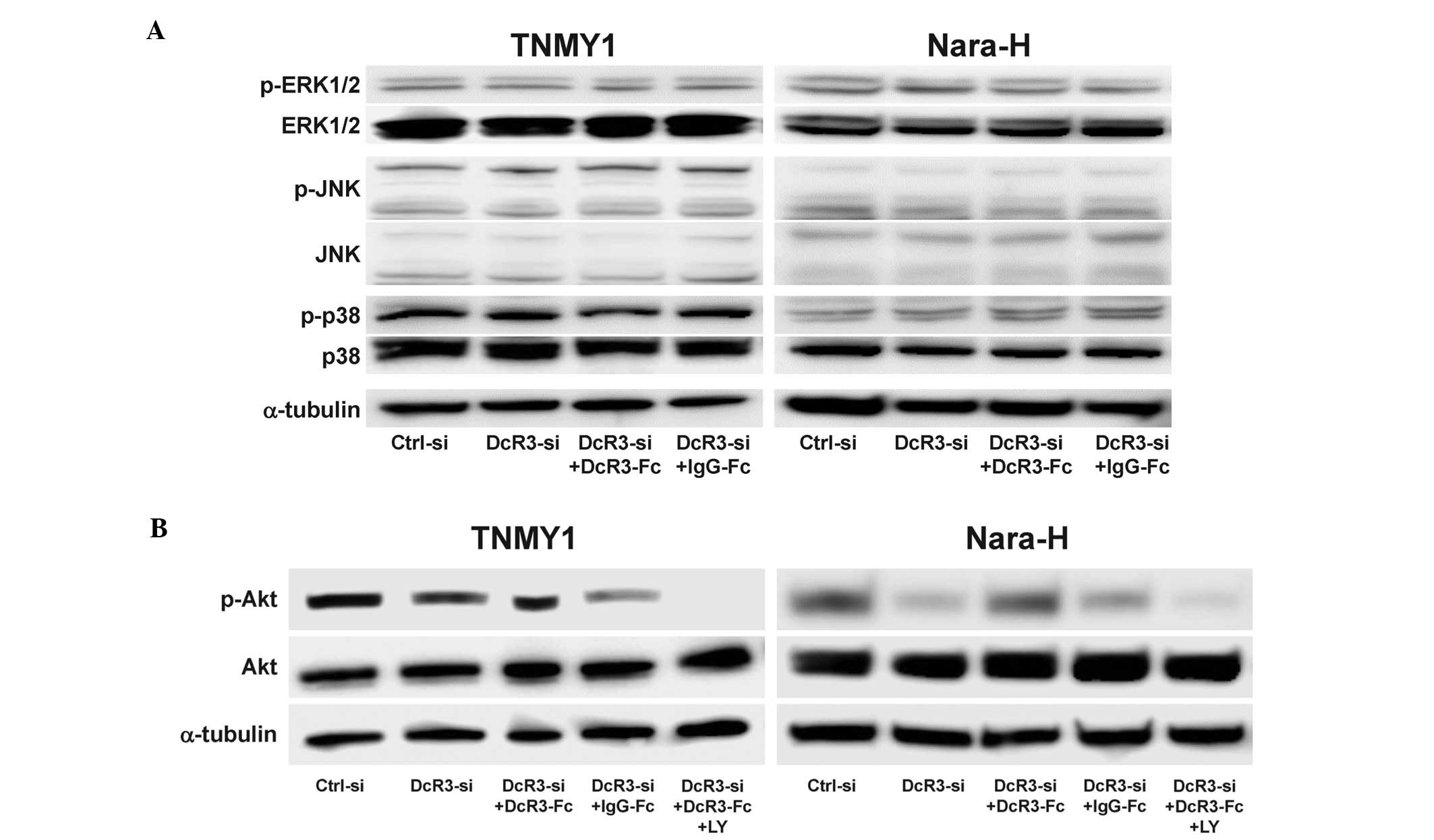
DcR3 activated the PI3K/Akt pathway in
MFH cells
Because DcR3 has been reported to be involved in the
activation of various kinases (13–17),
we evaluated the effect of DcR3 expression on the activation of
signaling kinases in MFH cells. DcR3 knockdown and/or DcR3-Fc
treatment did not affect the expressions of ERK1/2, JNK, p38 and
their phosphorylated forms (Fig.
5A) and interestingly, DcR3 knockdown significantly decreased
Akt phosphorylation in both MFH cell lines, which was restored by
DcR3-Fc treatment (Fig. 5B). The
expression of phosphorylated Akt was suppressed by DcR3-siRNA
and/or pretreatment with the PI3K inhibitor, LY294002 (LY)
(Fig. 5B).
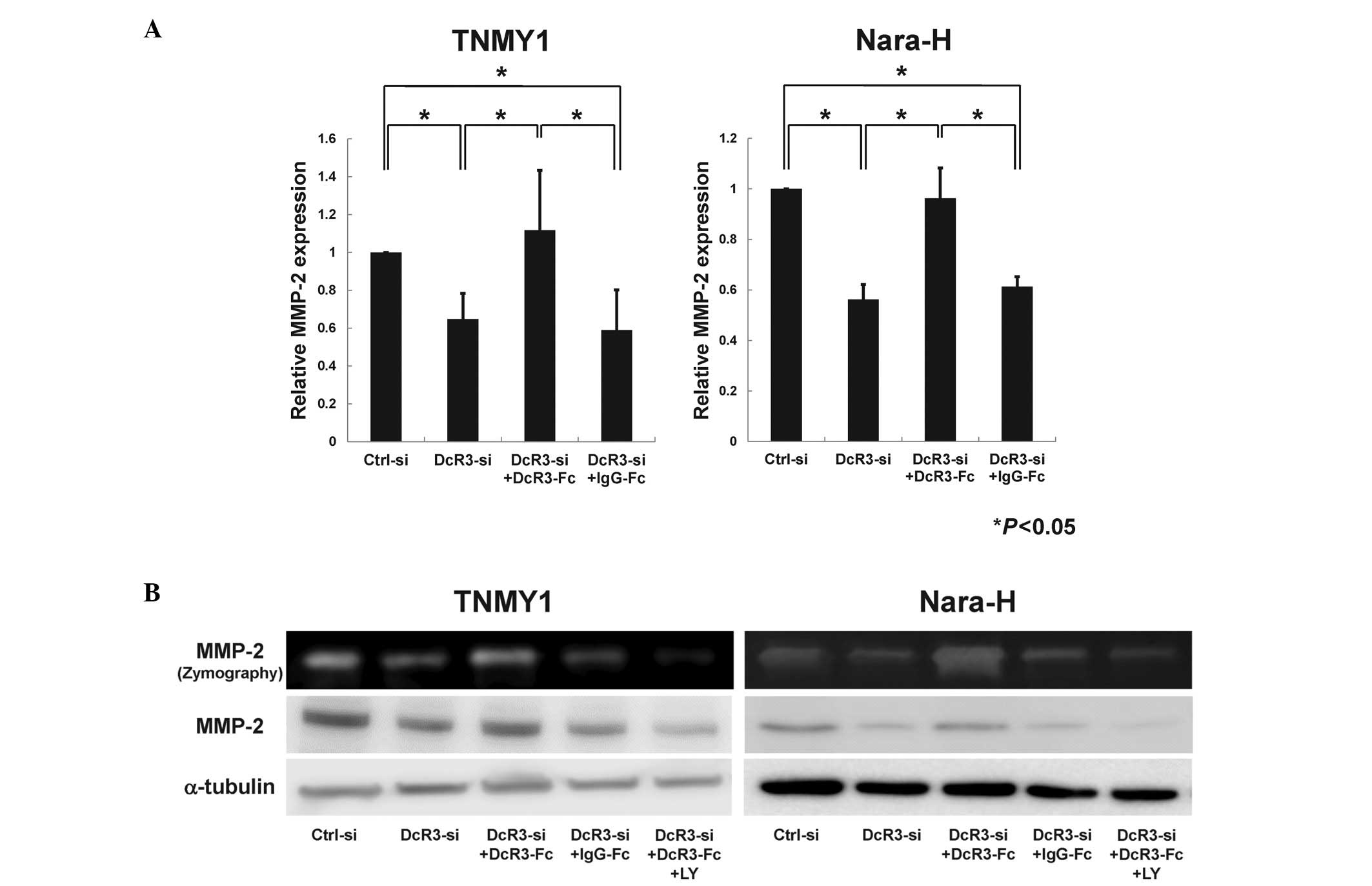
DcR3 regulated MMP-2 expression via the
PI3K/Akt pathway in human MFH cells
As previous reports suggest that DcR3 may affect
MMP-2 regulation via the PI3K/Akt pathway (12,17,39),
we evaluated the effect of DcR3 inhibition on MMP-2 expression
through the PI3K/Akt pathway in MFH cells. qRT-PCR analysis
revealed that MMP-2 mRNA expression was significantly
suppressed by DcR3 knockdown and was restored by DcR3-Fc treatment
in both MFH cell lines (*P<0.05, Fig. 6A). Immunoblot analysis and gelatin
zymography showed that the protein expression and enzyme activity
of MMP-2 were strongly reduced by siRNA knockdown of DcR3 (Fig. 6B). Consistent with the Akt
phosphorylation (Fig. 5B), both
the expression and enzyme activity of MMP-2 were increased by
DcR3-Fc treatment and were significantly suppressed by LY294002
pretreatment (Fig. 6B).
Discussion
DcR3 has been identified as a decoy receptor for
FasL in lung and colon cancers (5)
and recent studies have suggested that DcR3 acts as a modulator of
cellular functions such as migration and invasion (10,11).
It has been demonstrated that DcR3 overexpression is associated
with tumor metastasis and prognosis of patients with various
malignancies (5,18–26).
These findings strongly indicate that DcR3 may be an attractive
candidate as a novel therapeutic target for cancer treatment. We
have previously reported the overexpression of DcR3 in
musculoskeletal malignancies including MFH (21), however, the functional roles of
DcR3 in MFH have not been studied. Therefore, we investigated the
roles of DcR3 as a decoy receptor for FasL and as an effector
molecule in migration and invasion in human MFH cells.
Because resistance to apoptosis is a characteristic
property of malignant tumor cells to escape from immune attack by
host immune systems (8), we first
focused on the function of DcR3 as a decoy receptor for FasL. Many
cancers, in spite of their Fas expression, can be resistant to
FasL-induced apoptosis and several mechanisms may be responsible
for the decreased sensitivity to FasL-induced apoptosis, including
DcR3 (40). Yang et al
reported that siRNA knockdown of DcR3 increased FasL-induced
apoptosis in human pancreatic adenocarcinoma cells, which highly
express DcR3 (38). In this study,
we demonstrated that siRNA knockdown of DcR3 enhanced FasL-induced
apoptotic activity in human MFH cell lines and that FasL treatment
with DcR3 inhibition significantly suppressed MFH cell
proliferation. In addition, DcR3-Fc treatment after DcR3 knockdown
suppressed the increased FasL-induced apoptotic activity and
induced MFH cell proliferation. The expression of Fas receptor in
MFH cells was not affected by DcR3 knockdown or DcR3-Fc treatment.
These results strongly suggest that DcR3 may contribute to MFH cell
growth by inhibiting FasL-induced apoptosis as a decoy
receptor.
Moreover, several studies have demonstrated that
DcR3 functions as an effector molecule in various cells,
independently of the FasL/Fas pathway, by regulating migration and
invasion abilities (10,11), ERK stimulation in gastric cancers
(41), increasing adhesion in
monocytes via PI3K/Akt activation (17) and regulating proliferation and
migration of HUVECs via MMP-2 regulation (12). These reports suggested that DcR3
has non-decoy functions, which are independent of the FasL/Fas
apoptotic pathway. In the present study, we found that DcR3
inhibition significantly decreased cell migration and invasion in
human MFH cells and that DcR3-Fc treatment significantly increased
both abilities. These results indicate that DcR3 may regulate cell
migration and invasion in human MFH.
Previous studies have also suggested the involvement
of DcR3 in kinase phosphorylation (13–17).
Therefore, we further investigated the effect of DcR3 expression on
the activation of signaling kinases involved in migration and
invasion in MFH cells. We revealed that Akt phosphorylation, which
is inhibited by PI3K inhibition, was decreased by DcR3 knockdown
and increased by DcR3-Fc treatment. Akt is a major signal
transducer of the PI3K pathway, playing a pivotal role in the
maintenance of cellular processes including cell growth,
proliferation, survival and metabolism (42–44).
An increase in Akt activity has been detected in various cancers
(42–44). In addition, Akt signaling enhances
MMP-2 activity and promotes cell migration and invasion (42). Therefore, we investigated the
effect of DcR3 on MMP-2 activity in the PI3K/Akt pathway.
Consistent with the Akt phosphorylation, DcR3 knockdown decreased
MMP-2 expression and activity and DcR3-Fc treatment increased both
the expression and activity of MMP-2, which were inhibited by PI3K
inhibition. MMPs are thought to play a critical role in helping
cancer cells invade through ECM degradation and form metastatic
lesions (45). MMP-2, which is
regulated by various kinases, is the most abundant among all MMPs
(39,46) and has been reported to be
upregulated in MFH (31,32). Activation of both PI3K/Akt pathway
and MMP-2 are known to increase cell migration and invasion,
leading to metastasis in various cancers (42,45).
These findings indicate that DcR3 may regulate MMP-2 expression via
activation of the PI3K/Akt pathway and that this regulation may be
one of the important roles of DcR3 as an effector molecule
facilitating the progression or metastasis of MFH.
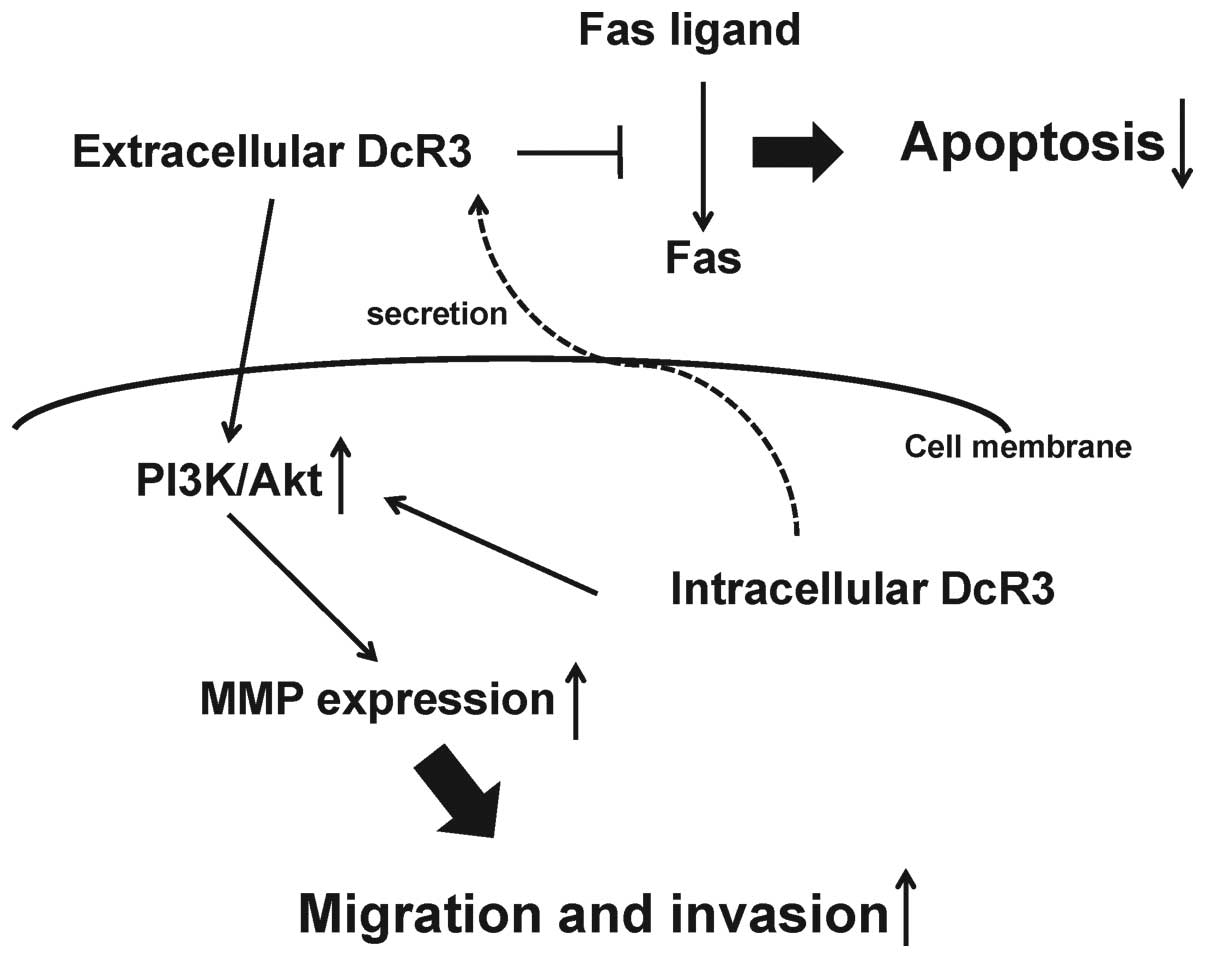
In conclusion, we demonstrated that in human MFH
cells DcR3 may increase tumor progression as a decoy, promoting
cell proliferation via inhibition of FasL-induced apoptosis and as
a non-decoy, regulating cell migration and invasion by MMP-2
activation via the PI3K/Akt pathway (Fig. 7). Although further studies are
needed to elucidate the roles of DcR3 in MFH tumor progression, our
findings strongly indicate that DcR3 may be a potential therapeutic
target in human MFH.
Acknowledgements
We thank Minako Nagata, Maya Yasuda and Kyoko Tanaka
for their expert technical assistance.
Abbreviations:
|
MFH
|
malignant fibrous histiocytoma
|
|
UPS
|
undifferentiated pleomorphic
sarcoma
|
|
DcR3
|
decoy receptor 3
|
|
TNFR
|
tumor necrosis factor receptor
|
|
FasL
|
Fas ligand
|
|
LIGHT
|
lymphotoxin-like, exhibits inducible
expression and competes with herpes simplex virus (HSV)
glycoprotein D (gD) for HVEM, a receptor expressed by T
lymphocytes
|
|
TL1A
|
TNF-like molecule 1A
|
|
PARP
|
poly (ADP-ribose) polymerase
|
|
HUVEC
|
human umbilical vein endothelial
cell
|
|
MMP
|
matrix metalloproteinase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
EBV
|
Epstein-Barr virus
|
|
HTLV-1
|
human T cell leukemia virus type 1
|
|
ECM
|
extracellular matrix
|
|
PI3K
|
phosphatidylinositol 3 kinase
|
References
|
1
|
Spira AI and Ettinger DS: The use of
chemotherapy in soft-tissue sarcomas. Oncologist. 7:348–359. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Matushansky I, Charytonowicz E, Mills J,
Siddiqi S, Hricik T and Cordon-Cardo C: MFH classification:
differentiating undifferentiated pleomorphic sarcoma in the 21st
Century. Expert Rev Anticancer Ther. 9:1135–1144. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Papai Z, Bodoky G, Szanto J, et al: The
efficacy of a combination of etoposide, ifosfamide and cisplatin in
the treatment of patients with soft tissue sarcoma. Cancer.
89:177–180. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jain A, Sajeevan K, Babu K and Lakshmaiah
K: Chemotherapy in adult soft tissue sarcoma. Indian J Cancer.
46:274–287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pitti RM, Marsters SA, Lawrence DA, et al:
Genomic amplification of a decoy receptor for Fas ligand in lung
and colon cancer. Nature. 396:699–703. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu KY, Kwon B, Ni J, Zhai Y, Ebner R and
Kwon BS: A newly identified member of tumor necrosis factor
receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. J
Biol Chem. 274:13733–13736. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Migone TS, Zhang J, Luo X, et al: TL1A is
a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell
costimulator. Immunity. 16:479–492. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar
|
|
9
|
Strasser A, Jost PJ and Nagata S: The many
roles of FAS receptor signaling in the immune system. Immunity.
30:180–192. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ho CH, Chen CL, Li WY and Chen CJ: Decoy
receptor 3, upregulated by Epstein-Barr virus latent membrane
protein 1, enhances nasopharyngeal carcinoma cell migration and
invasion. Carcinogenesis. 30:1443–1451. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ge Z, Sanders AJ, Ye L, Wang Y and Jiang
WG: Expression of death decoy receptor-3 (DcR3) in human breast
cancer and its functional effects on breast cancer cells in vitro.
J Exp Ther Oncol. 9:109–118. 2011.PubMed/NCBI
|
|
12
|
Yang CR, Hsieh SL, Teng CM, Ho FM, Su WL
and Lin WW: Soluble decoy receptor 3 induces angiogenesis by
neutralization of TL1A, a cytokine belonging to tumor necrosis
factor superfamily and exhibiting angiostatic action. Cancer Res.
64:1122–1129. 2004. View Article : Google Scholar
|
|
13
|
Yang CR, Wang JH, Hsieh SL, Wang SM, Hsu
TL and Lin WW: Decoy receptor 3 (DcR3) induces osteoclast formation
from monocyte/macrophage lineage precursor cells. Cell Death
Differ. 11(Suppl 1): S97–S107. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu SF, Liu TM, Lin YC, et al:
Immunomodulatory effect of decoy receptor 3 on the differentiation
and function of bone marrow-derived dendritic cells in nonobese
diabetic mice: from regulatory mechanism to clinical implication. J
Leukoc Biol. 75:293–306. 2004.
|
|
15
|
Hayashi S, Nishiyama T, Miura Y, et al:
DcR3 induces cell proliferation through MAPK signaling in
chondrocytes of osteoarthritis. Osteoarthritis Cartilage.
19:903–910. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
You RI, Chang YC, Chen PM, et al:
Apoptosis of dendritic cells induced by decoy receptor 3 (DcR3).
Blood. 111:1480–1488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hsu MJ, Lin WW, Tsao WC, et al: Enhanced
adhesion of monocytes via reverse signaling triggered by decoy
receptor 3. Exp Cell Res. 292:241–251. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ohshima K, Haraoka S, Sugihara M, et al:
Amplification and expression of a decoy receptor for fas ligand
(DcR3) in virus (EBV or HTLV-I) associated lymphomas. Cancer Lett.
160:89–97. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roth W, Isenmann S, Nakamura M, et al:
Soluble decoy receptor 3 is expressed by malignant gliomas and
suppresses CD95 ligand-induced apoptosis and chemotaxis. Cancer
Res. 61:2759–2765. 2001.PubMed/NCBI
|
|
20
|
Tsuji S, Hosotani R, Yonehara S, et al:
Endogenous decoy receptor 3 blocks the growth inhibition signals
mediated by Fas ligand in human pancreatic adenocarcinoma. Int J
Cancer. 106:17–25. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Imabori M, Akisue T, Kishimoto K, et al:
Expression of DcR3 in bone and soft tissue tumors. Cancer Ther.
7:43–48. 2009.
|
|
22
|
Macher-Goeppinger S, Aulmann S, Wagener N,
et al: Decoy receptor 3 is a prognostic factor in renal cell
cancer. Neoplasia. 10:1049–1056. 2008.PubMed/NCBI
|
|
23
|
Chen G and Luo D: Over-expression of decoy
receptor 3 in gastric precancerous lesions and carcinoma. Ups J Med
Sci. 113:297–304. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen G and Luo D: Expression of decoy
receptor 3 in liver tissue microarrays. Natl Med J India.
21:275–278. 2008.PubMed/NCBI
|
|
25
|
Chang PM, Chen PM, Hsieh SL, et al:
Expression of a soluble decoy receptor 3 in patients with diffuse
large B-cell lymphoma predicts clinical outcome. Int J Oncol.
33:549–554. 2008.PubMed/NCBI
|
|
26
|
Fujita Y, Sakakura C, Shimomura K, et al:
Chromosome arm 20q gains and other genomic alterations in
esophageal squamous cell carcinoma, as analyzed by comparative
genomic hybridization and fluorescence in situ hybridization.
Hepatogastroenterology. 50:1857–1863. 2003.
|
|
27
|
Takahama Y, Yamada Y, Emoto K, et al: The
prognostic significance of overexpression of the decoy receptor for
Fas ligand (DcR3) in patients with gastric carcinomas. Gastric
Cancer. 5:61–68. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Choong PFM: The molecular basis of
skeletal metastases. Clin Orthop Relat Res. 415:S192003. View Article : Google Scholar
|
|
29
|
Hidalgo M and Eckhardt SG: Development of
matrix metalloproteinase inhibitors in cancer therapy. J Natl
Cancer Inst. 93:178–193. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Egeblad M and Werb Z: New functions for
the matrix metalloproteinases in cancer progression. Nat Rev
Cancer. 2:163–176. 2002. View
Article : Google Scholar
|
|
31
|
Ohnishi Y, Ito Y, Tajima S, Ishibashi A
and Arai K: Immunohistochemical study of membrane type-matrix
metalloproteinases (MT-MMPs) and matrix metalloproteinase-2 (MMP-2)
in dermatofibroma and malignant fibrous histiocytoma. J Dermatol
Sci. 28:119–125. 2002. View Article : Google Scholar
|
|
32
|
Ahlen J, Enberg U, Larsson C, et al:
Malignant fibrous histiocytoma, aggressive fibromatosis and benign
fibrous tumors express mRNA for the metalloproteinase inducer
EMMPRIN and the metalloproteinases MMP-2 and MT1-MMP. Sarcoma.
5:143–149. 2001. View Article : Google Scholar
|
|
33
|
Nakatani T, Marui T, Yamamoto T, Kurosaka
M, Akisue T and Matsumoto K: Establishment and characterization of
cell line TNMY1 derived from human malignant fibrous histiocytoma.
Pathol Int. 51:595–602. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kiyozuka Y, Nakagawa H, Uemura Y, et al:
Novel cell lines established from a human myxoid malignant fibrous
histiocytoma arising in the uterus. Cancer Genet Cytogenet.
127:7–15. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liang CC, Park AY and Guan JL: In vitro
scratch assay: a convenient and inexpensive method for analysis of
cell migration in vitro. Nat Protoc. 2:329–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Albini A: Tumor and endothelial cell
invasion of basement membranes. The matrigel chemoinvasion assay as
a tool for dissecting molecular mechanisms. Pathol Oncol Res.
4:230–241. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Troeberg L and Nagase H: Zymography of
metalloproteinases. Curr Protoc Protein Sci. Chapter 21(Unit 21):
152004.PubMed/NCBI
|
|
38
|
Yang CR, Guh JH, Teng CM, Chen CC and Chen
PH: Combined treatment with denbinobin and Fas ligand has a
synergistic cytotoxic effect in human pancreatic adenocarcinoma
BxPC-3 cells. Br J Pharmacol. 157:1175–1185. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Reuben PM and Cheung HS: Regulation of
matrix metalloproteinase (MMP) gene expression by protein kinases.
Front Biosci. 11:1199–1215. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim R, Emi M and Tanabe K: Cancer
immunoediting from immune surveillance to immune escape.
Immunology. 121:1–14. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang D, Fan X, Yin P, et al: Significance
of decoy receptor 3 (Dcr3) and external-signal regulated kinase 1/2
(Erk1/2) in gastric cancer. BMC Immunol. 13:282012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chin YR and Toker A: Function of Akt/PKB
signaling to cell motility, invasion and the tumor stroma in
cancer. Cell Signal. 21:470–476. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sun M, Wang G, Paciga JE, et al:
AKT1/PKBalpha kinase is frequently elevated in human cancers and
its constitutive activation is required for oncogenic
transformation in NIH3T3 cells. Am J Pathol. 159:431–437. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Staal SP: Molecular cloning of the akt
oncogene and its human homologues AKT1 and AKT2: amplification of
AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci
USA. 84:5034–5037. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kessenbrock K, Plaks V and Werb Z: Matrix
metalloproteinases: regulators of the tumor microenvironment. Cell.
141:52–67. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Morrison CJ, Butler GS, Bigg HF, Roberts
CR, Soloway PD and Overall CM: Cellular activation of MMP-2
(gelatinase A) by MT2-MMP occurs via a TIMP-2-independent pathway.
J Biol Chem. 276:47402–47410. 2001. View Article : Google Scholar : PubMed/NCBI
|