Introduction
Colorectal carcinoma (CRC) is a serious global
health problem with over one million new cases and half a million
deaths worldwide each year (1). To
date, chemotherapy is the main therapeutic approach for invasive
and metastatic CRC. Despite recent advances in this area,
5-fluorouracil (5-FU)-based regimens continue to be the standard
therapeutic approach for patients with advanced CRC (2). However, due to drug resistance and an
unacceptable level of toxicity, 5-FU-based regimens produce
objective response rates of <40% (3,4).
The pathogenic mechanisms underlying CRC development
are complex and heterogeneous and involve multiple cellular
signaling transduction pathways including signal transducer and
activator of transcription 3 (STAT3), extracellular regulated
protein kinases (ERK), c-Jun NH(2)-terminal protein kinases (JNK) and p38
kinase. STAT3 is a transcription factor that plays an essential
role in cell survival and proliferation (5). After activation via phosphorylation
by receptor and non-receptor protein tyrosine kinases, STAT3
proteins in the cytoplasm dimerize and translocate to the nucleus
where they regulate the expression of critical genes involved in
cell proliferation and survival (5,6).
Constitutive activation of STAT3 is highly correlated with the
development of numerous types of cancer including CRC and commonly
suggests a poor prognosis (7–10).
MAPK signaling is one of the major cell-survival and proliferation
pathways. In mammals, there are three major subfamilies of MAPK,
including ERK, JNK and p38. Activation of all MAPK pathways is
regulated by a central three-tiered kinase core consisting of MAPK
kinase kinase (MAP3K), MAPK kinase (MAP2K) and MAPK; wherein MAP3K
phosphorylates MAP2K and activated MAP2K in turn phosphorylates and
activates MAPK (11,12). MAPK signaling is important in
intestinal epithelial differentiation (13); however, aberrant activation of MAPK
pathway is involved in colon carcinogenesis (14–17).
The molecular pathways described above modulate the
expression of key genes involved in the regulation of cell
proliferation, apoptosis and angiogenesis and are participants in
the processes of induction, progression and metastasis of
colorectal carcinoma. Thus, each serves as a potential target for
novel chemotherapeutics. It is noteworthy that these signaling
pathways usually have functional redundancy. In addition, there is
crosstalk between these pathways, forming a complicated and robust
cellular signal transduction network that is regulated by
compensatory mechanisms. Therefore, inhibitors that target only a
single pathway might not be effective initially and their long-term
use might generate drug resistance (18). These problems highlight the need
for the identification of novel cancer chemotherapies. Natural
products, such as traditional Chinese medicines (TCMs), have been
used clinically to treat various types of diseases including cancer
(19). Their effect, in general,
is believed to be the result of targeting multiple molecular
pathways. Moreover, TCMs have relatively fewer side effects as
compared to modern chemotherapeutics. Therefore, TCMs have gained
recent attention as potential chemotherapeutics for CRC.
Many traditional Chinese medicinal herbs, such as
Hedyotic diffusa, Spica prunella and Scutellaria
barbata, have long been used in China for the clinical
treatment of CRC (20–23). Ursolic acid (UA), a pentacyclic
triterpene acid, is a biologically active compound present in these
herbs. Previous studies report that UA exhibits a broad range of
pharmacological properties such as anti-inflammatory, antiviral,
antioxidant, hepatoprotective, cytotoxic, antitumor,
anti-angiogenesis and anti-metastatic activities (24). Recent studies have shown that UA
inhibits the proliferation and induces apoptosis and/inhibits
proliferation of colon carcinoma cells (25,26).
In this study, we further evaluated the efficacy of UA as a
therapeutic agent for CRC in vivo and in vitro and
investigated the underlying molecular mechanisms of its action.
Materials and methods
Materials and reagents
Ursolic acid (UA) was purchased from Sigma-Aldrich
Chemical (St. Louis, MO, USA). Matrigel was provided by
Becton-Dickinson (San Jose, CA, USA). Dulbecco’s modified Eagle’s
medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin,
trypsin-EDTA and TRIzol reagent were purchased from Invitrogen
(Carlsbad, CA, USA). TUNEL assay kit (TumorTACS in situ) was
purchased from R&D Systems (Minneapolis, MN, USA). BCA Protein
Assay kit was purchased from Tiangen Biotech Co., Ltd. (Beijing,
China). All antibodies were purchased from Cell Signaling
Technology (CST, Beverly, MA, USA). Bio-Plex phosphoprotein assay
kits were purchased from Bio-Rad (Hercules, CA, USA). All other
chemicals, unless otherwise stated, were obtained from
Sigma-Aldrich.
Cell culture
Human colon carcinoma HT-29 cells were purchased
from American Type Culture Collection (ATCC, Manassas, VA, USA).
HT-29 cells were grown in DMEM. DMEM was supplemented with 10%
(v/v) FBS, 100 U/ml penicillin and 100 μg/ml streptomycin.
The cells were cultured at 37°C, 5% CO2 and in a
humidified environment.
Animals
Male BALB/c athymic (nude) mice (with an initial
body weight of 20–22 g) were obtained from Shanghai SLAC Laboratory
Animal Co., Ltd. (Shanghai, China) and housed under pathogen-free
conditions with controlled temperature (22°C), humidity and a 12-h
light/dark cycle. Food and water were given ad libitum
throughout the experiment. All animal treatments were performed
strictly in accordance with international ethical guidelines and
the National Institutes of Health Guide concerning the Care and Use
of Laboratory Animals. The experiments were approved by the
Institutional Animal Care and Use Committee of Fujian University of
Traditional Chinese Medicine.
In vivo nude mice xenograft study
CRC xenograft mice were produced with HT-29 cells as
described previously (21). At 5
days following xenograft implantation (tumor size ∼3 mm in
diameter), mice were randomized into two groups (n=10) and treated
with UA (dissolved in PBS) 12.5 mg/kg or saline by daily
intraperitoneal injection, 6 days a week for 16 days. Body weight
and tumor size were measured. Tumor size was determined by
measuring the major (L) and minor (W) diameter with a caliper. The
tumor volume was calculated according to the following formula:
tumor volume = π/6xLxW2. At the end of the experiment,
the animals were anaesthetized with pelltobarbitalum natricum and
tumors were excised and weighed.
Cell viability evaluation by MTT
assay
UA was dissolved in DMSO and diluted to working
concentrations with culture medium. The final concentration of DMSO
in the medium for all cell-based experiments was 0.1%. HT-29 cells
were seeded into 96-well plates at a density of 1.0×104
cells/well in 0.1 ml medium. The cells were treated with various
concentrations of UA for 24 h or with 40 μM of UA for
different periods of time. Treatment with 0.1% DMSO was included as
the vehicle control. At the end of the treatment, 10 μl MTT
[5 mg/ml in phosphate-buffered saline (PBS)] were added to each
well and the samples were incubated for an additional 4 h at 37°C.
The purple-blue MTT formazan precipitate was dissolved in 100
μl DMSO. Absorbance was measured at 570 nm using an ELISA
reader (BioTek, Model EXL800, USA).
Colony formation assay
HT-29 cells were seeded into 6-well plates at a
density of 2×105 cells/well and treated with various
concentrations of UA for 24 h. The cells were harvested,
resuspended in medium in the absence of UA and reseeded into 6-well
plates at a density of 1.5×103 cells/well. After
incubation for 7 days in a 37°C humidified incubator with 5%
CO2, colonies were counted under light microscopy. Cell
survival was calculated by normalization to the survival of control
(untreated) cells.
Cell cycle analysis
Cell cycle analysis was carried out by flow
cytometry using FACS analysis with propidium iodide (PI) staining.
HT-29 cells were treated with various concentrations of UA for 24
h, harvested, adjusted to a concentration of 1×106
cells/ml and fixed in 70% ethanol at 4°C overnight. The fixed cells
were washed twice with cold PBS and incubated for 30 min with RNase
(8 μg/ml) and PI (10 μg/ml). The fluorescent signal
was detected through the FL2 channel and the proportion of DNA in
different phases was analyzed using ModfitLT Version 3.0 (Verity
Software House, Topsham).
Apoptosis detection in CRC tumor tissues
by TUNEL staining
Formalin-fixed (12 h) and paraffin-embedded sections
of tumors (4-μm-thick) were analyzed by TUNEL staining using
TumorTACS in situ kit (R&D Systems). Apoptotic cells
were counted as DAB-positive cells (brown stained) at five
arbitrarily selected microscopic fields at a magnification of ×400.
TUNEL-positive cells were counted as a percentage of the total
cells.
Apoptosis detection in HT-29 cells by
flow cytometry analysis with Annexin V/PI staining
After incubation with various concentrations of UA,
apoptosis of HT-29 cells were determined by flow cytometry using a
fluorescence-activated cell sorting (FACS) caliber
(Becton-Dickinson) and Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) kit (Becton-Dickinson). Staining was
performed according to the manufacturer’s instructions. The
percentage of cells in early apoptosis was calculated by Annexin
V-positivity and PI-negativity and the percentage of cells in late
apoptosis was calculated by Annexin V-positivity and
PI-positivity.
DNA fragmentation analysis in HT-29 cells
by gel electrophoresis
DNA fragmentation was determined using a DNA ladder
detection kit (Invitrogen), following the manufacturer’s
instructions. Briefly, HT-29 cells were detached from the culture
plates by scraping and washed in PBS. DNA was isolated from
2×106 cells. The DNA was analyzed after separation by
gel electrophoresis. The DNA bands were examined using a gel
documentation system (Model Gel Doc 2000, Bio-Rad).
Immunohistochemistical analysis of CRC
tumor tissues
Six tumors were randomly selected from UA-treatment
or control groups. Tumor tissues were fixed in 10% formaldehyde for
12 h, paraffin-embedded, sectioned and placed on slides. The
immunohistochemistical analysis was carried out as described
previously (21). Briefly, the
slides were subjected to antigen retrieval and endogenous
peroxidase activity was quenched with hydrogen peroxide.
Non-specific binding was blocked with normal serum in PBS (0.1%
Tween-20). Rabbit polyclonal antibodies against PCNA (in 1:200
dilution, CST) was used to detect the proteins. After staining,
five high-power fields (at magnification of ×400) were randomly
selected in each slide. The proportion of positive cells in each
field was determined using the true color multi-functional cell
image analysis management system (Image-Pro Plus, Media
Cybernetics, USA). To control for non-specific staining, PBS was
used to replace the primary antibody as a negative control.
RT-PCR analysis
Total RNA was isolated from tumor tissues (three
tumors were randomly selected from UA-treatment or control groups)
or HT-29 cells with TRIzol reagent. Oligo(dT)-primed RNA (1
μg, isolated from tumor tissues or cells) was
reverse-transcribed with SuperScript II reverse transcriptase
(Promega, Madison, WI, USA) according to the manufacturer’s
instructions. The obtained cDNA was used to determine the level of
Bcl-2, Bax, Cyclin D1, CDK4 and p21 mRNA by PCR with Taq DNA
polymerase (Fermentas). GAPDH was used as an internal control.
Samples were analyzed by gel electrophoresis (1.5% agarose). The
DNA bands were examined using a gel documentation system (Model Gel
Doc 2000, Bio-Rad).
Western blot analysis
The western blot analysis was carried out as
described previously (21).
Briefly, tumors and HT-29 cells were separately lysed in mammalian
cell lysis buffer with different protein inhibitor. Equal amounts
of protein from each tumor or cell lysate was subjected to SDS-PAGE
and transferred onto PVDF membranes. The membranes were blocked for
2 h with 5% non-fat dry milk and incubated with the desired primary
antibody directed against p-STAT3, total-STAT3, Bcl-2, Bax, Cyclin
D1, CDK4, p21, or β-actin (all in 1:1,000 dilutions) overnight at
4°C. Appropriate HRP-conjugated secondary antibodies with
chemiluminescence detection were used to image the
antibody-detected proteins.
Bio-Plex phosphoprotein assay
Eight tumors were randomly selected from
UA-treatment or control groups and homogenized. HT-29 cells
(2.5×105) were seeded into 25 cm2 flasks in 5
ml medium and treated with 40 μM of UA for 24 h. Tumor
tissues and treated cells were lysed using a commercially available
lysis kit (Bio-Rad Laboratories) and centrifuged at 14,000 × g for
15 min. The protein extracts were quantified by BCA protein assay.
The presence of p-ERK1/2, p-JNK and p-p38 was detected using a
bead-based multiplex assay for phosphoproteins (Bio-Plex
Phosphoprotein assay, Bio-Rad Laboratories) according to the
manufacturer’s protocol. Data was collected and analyzed using the
Bio-Plex 200 suspension array system (Bio-Rad).
Statistical analysis
Data were presented as mean ± SD for the indicated
number of independently performed experiments. The data were
analyzed using the SPSS package for Windows (Version 17.0).
Statistical analysis was carried out with Student’s t-test and
ANOVA. Differences with P<0.05 were considered to be
statistically significant.
Results
UA inhibits tumor growth in colorectal
cancer (CRC) xenograft mice
The in vivo antitumor effect of UA was
evaluated through comparison of tumor weight and volume in treated
and control CRC xenograft mice, while its adverse effects were
determined in the same mice by measuring changes in body weight. As
shown in Fig. 1A, UA treatment
resulted in 55.9% less tumor volume as compared to control
(control, 0.93±0.21 cm3; UA-treatment, 0.41±0.13
cm3, P<0.01). In a consistent manner, the tumor
weights in the UA-treatment group was 65.7% less than in the
control group (control, 0.67±0.15 g; UA-treatment, 0.23±0.11 g,
P<0.01, Fig. 1B). However,
administration of UA had no effect on gain in body weight during
the course of the study (Fig. 1C),
suggesting that UA is potent in suppressing colorectal tumor growth
in vivo, without noticeable toxicity.
UA inhibits proliferation of colorectal
cancer cells via G1/S cell cycle arrest and modulation of Cyclin
D1, CDK4 and p21 expression
The in vivo effect of UA on CRC cell
proliferation was determined via immunohistochemical staining (IHS)
for PCNA, a proliferation marker that is specifically expressed in
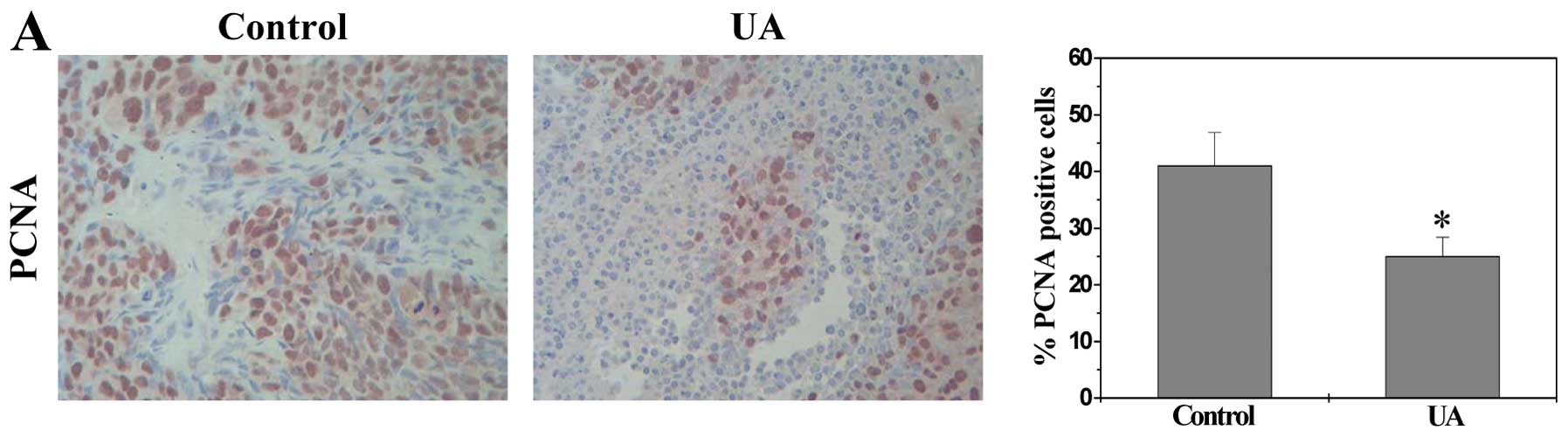
proliferating cell nuclei. As shown in Fig. 2A, the percentage of PCNA-positive
cells in tumor tissues from control and UA-treated CRC xenograft
mice was 41.0±5.9% and 25.0±3.4%, respectively (P<0.01),
suggesting that UA represses cell proliferation in tumor
tissues.
We next sought to examine the effect of UA on CRC
cell proliferation in vitro. We treated human colon
carcinoma HT-29 cells with various concentrations of UA and
measured the amount of surviving cells with the MTT assay. As shown
in Fig. 2B, treatment with 20–120
μM of UA for 24 h dose-dependently reduced the viability of
HT-29 cells (P<0.01). The estimated half maximal inhibitory
concentration (IC50) of UA in HT-29 cells was ∼35
μM. We next evaluated the effect of UA on cell viability as
a function of time. We found that treatment with UA (40 μM,
approximately the IC50) led to a gradual decrease in
cell viability with an increase in exposure time (P<0.01,
Fig. 2C). We further verified
these results using a colony formation assay. UA treatment
dose-dependently reduced the cell survival rate (P<0.01,
Fig. 2D). Thus, UA inhibits CRC
cell proliferation in vitro dose- and time-dependently.
G1/S transition is one of the two main cell cycle
checkpoints that are critical for regulation of cell proliferation,
which is strongly regulated by Cyclin D1, CDK4 and p21. We
therefore investigated the effect of UA on the G1 to S progression
and on the expression of these regulatory genes. Using PI staining
followed by FACS analysis we found that the percentage HT-29 cells
in S-phase following treatment with UA was decreased in a
dose-dependent manner (Fig. 2E,
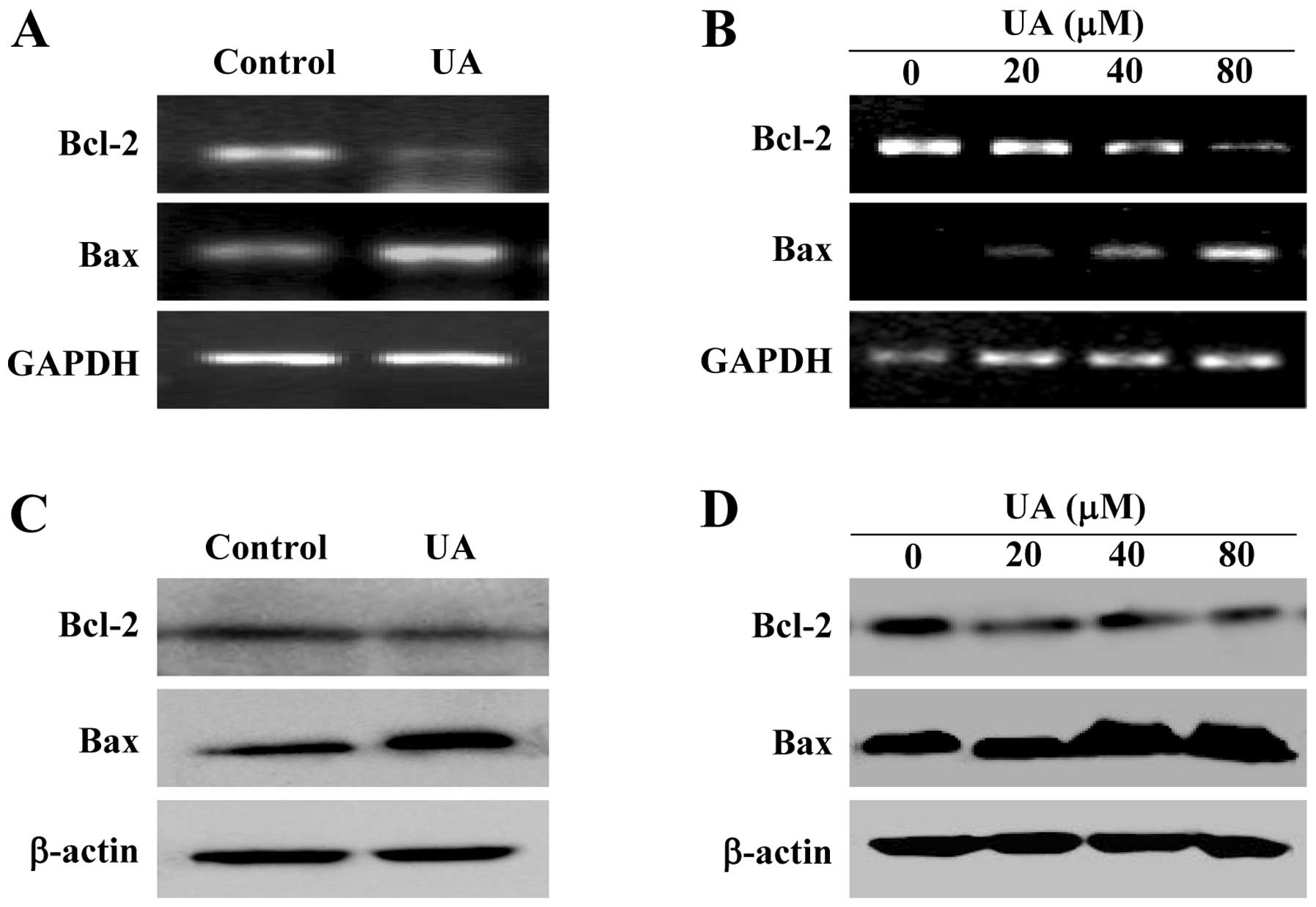
P<0.01). In addition, data from RT-PCR and western blot analysis
showed that UA treatment significantly reduced the mRNA and protein
levels of pro-proliferative Cyclin D1 and CKD4 both in CRC tumors
and HT-29 cells, whereas the mRNA and protein levels of
anti-proliferative p21 was significantly increased after UA
treatment (Fig. 3, P<0.01).
These data together suggest that UA inhibits CRC cell proliferation
through blockade of G1-S progression and the modulation of the
expression of cell cycle-regulatory genes.
UA induces colorectal cancer cell
apoptosis via alteration in the Bax/Bcl-2 ratio
To further evaluate the effect of UA on CRC cell
growth, we examined whether UA induces apoptosis. Apoptosis in
tumors was evaluated via TUNEL. As shown in Fig. 4A, the percentage of TUNEL-positive
cells was greater in tumors from UA-treated mice as compared to
controls (UA-treatment, 36.7±8.6%; control, 26.3±2.0%; P<0.01).
UA-induced apoptosis in vitro was examined using HT-29 cells
and Annexin V/PI staining followed by FACS analysis. As shown in
Fig. 4B and C, UA treatment
increased the percent of cells undergoing apoptosis [early
apoptosis found within the lower right (LR) and late apoptosis
upper right (UR) quadrants in the FACS diagram] in a dose-dependent
fashion (P<0.05). The in vitro pro-apoptotic activity of
UA was confirmed by its effect on cellular DNA fragmentation, a
typical feature of apoptosis. As shown in Fig. 4D, DNA extracted from HT-29 cells
treated with UA displayed a characteristic ladder pattern of
discontinuous DNA fragments.
To investigate the mechanism of the pro-apoptotic
activity of UA, we examined its effect on the expression of Bcl-2
family proteins that are important regulators of apoptosis. As
shown in Fig. 5A and B, UA
significantly reduced anti-apoptotic Bcl-2 mRNA level both in the
tumors of CRC mice and HT-29 cells, whereas the level of
pro-apoptotic Bax mRNA was significantly increased after UA
treatment. The protein expression patterns of Bcl-2 and Bax were
similar to the patterns observed for the respective mRNA (Fig. 5C and D). Taken together, these data
demonstrate that UA promotes colorectal cancer cell apoptosis both
in vivo and in vitro through an increase in the
pro-apoptotic Bax/Bcl-2 ratio.
UA suppresses multiple signaling pathways
in CRC cells
To further explore the underlying mechanisms of the
observed anticancer activities of UA, we determined its effect on
the activation of several CRC-related signal transduction cascades
including STAT3, ERK, JNK and p38. Activation of STAT3 is mediated
by its phosphorylation at tyrosine 705, we therefore investigated
the effect of UA on STAT3 activation in CRC by western blot
analysis using an antibody that recognizes STAT3 phosphorylation at
Tyr705. Since many cultured human cancer cell lines
including HT-29 do not express constitutively phosphorylated STAT3
in vitro, we stimulated STAT3 activation in HT-29 cells with
10 ng/ml of IL-6 for 15 min. As shown in Fig. 6A, UA profoundly inhibited IL-6
induced phosphorylation of STAT3 in HT-29 cells. In a consistent
fashion, UA treatment significantly decreased the level of
phosphorylated STAT3 in tumors of CRC xenograft mice (Fig. 6B). The levels of non-phosphorylated
STAT3 in tumor HT-29 cells and tumor tissues remained unchanged
with UA treatment.
The activation (phosphoralytion) of ERK1/2, JNK and
p38 in CRC xenograft tumor tissues and HT-29 cells was determined
by Bio-Plex Phosphoprotein assay. As shown in Fig. 6C and D, after UA treatment the
phosphorylation levels of ERK1/2, JNK and p38 in both tumors and
HT-29 cells were decreased as compared to controls (P<0.01).
Collectively, these data suggest that UA significantly suppresses
the activation of multiple CRC-related signaling pathways.
Discussion
Cancer development is strongly correlated with the
aberrant activation of multiple intracellular signal transduction
pathways which usually function redundantly. In addition, crosstalk
between these pathways generates a complicated and robust signaling
network that is regulated by compensatory mechanisms. Given the
complexity of cancer pathogenesis and progression, many of the
currently used antitumor agents, which typically target a single
intracellular pathway, might not always be effective on complex
tumor systems. In contrast, long-term use of these
single-target-based agents often generates drug resistance.
Moreover, most currently used chemotherapies contain intrinsic
toxicity against normal cells. Therefore, the development of novel
anticancer chemotherapies is urgently needed. Natural products have
received great interest since they have relatively fewer side
effects as compared to modern chemotherapeutics and have been shown
to display multiple therapeutic effects for various diseases
including cancer. Ursolic acid (UA), a major active compound of
many traditional Chinese medicinal herbs, has been shown to possess
anticancer activity. However, the precise mechanism of its
potential tumoricidal activity remains largely unclear. Therefore,
before UA can be further developed as an anticancer agent, the mode
of action for its antitumor effects should be fully elucidated.
Cancer cells are characterized by an uncontrolled
increase in cell proliferation and/or a reduction in cell
apoptosis. The mitochondrion-dependent pathway is the most common
apoptotic pathway in vertebrate animal cells, which is highly
regulated by Bcl-2 family proteins including anti-apoptotic members
such as Bcl-2 and pro-apoptotic members such as Bax (27). The ratio of active anti- and
pro-apoptotic Bcl-2 family members determines the fate of cells and
alteration of the ratio by aberrant expression of these proteins
impairs the normal apoptotic program contributing to various
apoptosis-related diseases including cancer (27,28).
Higher Bcl-2-to-Bax ratios due to the overexpression of Bcl-2 or
downregulation of Bax expression are commonly found in cancer,
which not only confers a survival advantage to the cancer cells but
also causes resistance to chemo- and radiotherapies. Eukaryotic
cell proliferation is primarily regulated by the cell cycle. G1/S
transition is one of the two main checkpoints of the cell cycle and
is responsible for initiation and completion of DNA replication
(29). G1/S progression is
strongly regulated by Cyclin D1 that exerts its function via
forming an active complex with its CDK major catalytic partners
(CDK4/6) (30). An unchecked or
hyperactivated Cyclin D1/CDK4 complex often leads to uncontrolled
cell division and malignancy (31,32).
As a proliferation inhibitor, p21 protein plays a role in G1 arrest
by binding to and inhibiting the activity of Cyclin-CDK complexes;
and the decrease of p21 is associated with the promotion of tumor
formation and a poor prognosis in many types of cancer (33). Therefore, re-balancing of cell
apoptosis and proliferation via regulation of the expression of
apoptosis- or cell cycle-related genes is a promising target for
cancer chemotherapies. Using a CRC mouse xenograft model and a
human colon carcinoma cell line, we demonstrated that UA inhibits
cancer growth both in vivo and in vitro, without
apparent toxicity. Additionally, UA suppresses cancer cell
proliferation through blocking G1/S arrest and promotes cell
apoptosis both in tumors of CRC mice and in colon cancer cell line.
The pro-apoptotic and anti-proliferative activities of UA were
mediated by its effects on the expression of relevant genes. UA
treatment profoundly increased the pro-apoptotic Bax/Bcl-2 ratio,
the expression of anti-proliferative p21 and downregulated the
expression of pro-proliferative Cyclin D1 and CDK4.
The processes of apoptosis and cellular
proliferation are regulated by multiple intracellular signals
including STAT3, ERK, JNK and p38 pathways. Aberrant activation of
these pathways is involved in cancer progression. In the present
study we demonstrated that UA significantly suppress the activation
of these CRC-related signaling pathways both in vivo and
in vitro.
In conclusion, we report for the first time that UA
possesses a broad range of anticancer activities due to its ability
to affect multiple intracellular targets. Our findings suggest that
UA could be a novel therapeutic agent for the treatment of
colorectal and possibly other cancers.
Abbreviations:
|
UA
|
ursolic acid;
|
|
CRC
|
colorectal cancer;
|
|
DMSO
|
dimethyl sulfoxide;
|
|
MTT
|
3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide;
|
|
IHS
|
immunohistochemical staining;
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated dUTP nick end-labeling;
|
|
PCNA
|
proliferating cell nuclear
antigen;
|
|
STAT3
|
signal transducer and activator of
transcription 3;
|
|
Erk1/2
|
extracellular signal-regulated protein
kinases 1 and 2;
|
|
JNK
|
c-Jun N-terminal kinase;
|
|
p38
|
p38 mitogen-activated protein
kinase
|
Acknowledgements
This study was sponsored by the
National Natural Science Foundation of China (no. 81073097), the
Developmental Fund of Chen Keji Integrative Medicine (no. CKJ
2011001), and the China Postdoctoral Science Foundation (no.
2012M511437).
References
|
1.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2.
|
Gustin DM and Brenner DE: Chemoprevention
of colon cancer: current status and future prospects. Cancer
Metastasis Rev. 21:323–348. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Gorlick R and Bertino JR: Drug resistance
in colon cancer. Semin Oncol. 26:606–611. 1999.
|
|
4.
|
Longley DB, Allen WL and Johnston PG: Drug
resistance, predictive markers and pharmacogenomics in colorectal
cancer. Biochim Biophys Acta. 1766:184–196. 2006.PubMed/NCBI
|
|
5.
|
Aggarwal BB, Kunnumakkara AB, Harikumar
KB, Gupta SR, Tharakan ST, Koca C, Dey S and Sung B: Signal
transducer and activator of transcription-3, inflammation, and
cancer: how intimate is the relationship? Ann NY Acad Sci.
1171:59–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Zhong Z, Wen Z and Darnell J: Stat3: a
STAT family member activated by tyrosine phosphorylation in
response to epidermal growth factor and interleukin-6. Science.
264:95–98. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Bromberg J and Wang TC: Inflammation and
cancer: IL-6 and STAT3 complete the link. Cancer Cell. 15:79–80.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Kusaba T, Nakayama T, Yamazumi K, Yakata
Y, Yoshizaki A, Inoue K, Nagayasu T and Sekine I: Activation of
STAT3 is a marker of poor prognosis in human colorectal cancer.
Oncol Rep. 15:1445–1451. 2006.PubMed/NCBI
|
|
9.
|
Lin Q, Lai R, Chirieac LR, Li C, Thomazy
VA, Grammatikakis I, Rassidakis GZ, Zhang W, Fujio Y, Kunisada K,
Hamilton SR and Amin HM: Constitutive activation of JAK3/STAT3 in
colon carcinoma tumors and cell lines: inhibition of JAK3/STAT3
signaling induces apoptosis and cell cycle arrest of colon
carcinoma cells. Am J Pathol. 167:969–980. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Xiong H, Zhang Z, Tian X, Sun D, Liang Q,
Zhang Y, Lu R, Chen Y and Fang J: Inhibition of JAK1, 2/STAT3
signaling induces apoptosis, cell cycle arrest, and reduces tumor
cell invasion in colorectal cancer cells. Neoplasia. 10:287–297.
2008.PubMed/NCBI
|
|
11.
|
Sebolt-Leopold JS: Development of
anticancer drugs targeting the MAP kinase pathway. Oncogene.
19:6594–6599. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Seger R and Krebs EG: The MAPK signaling
cascade. FASEB J. 9:726–735. 1995.PubMed/NCBI
|
|
13.
|
Taupin D and Podolski DK:
Mitogen-activated protein kinase activation regulates intestinal
epithelial differentiation. Gastroenterology. 116:1072–1080. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Wang X, Wang Q, Hu W and Evers BM:
Regulation of phorbol estermediated TRAF1 induction in human colon
cancer cells through a PKC/RAF/ERK/NF-kappaB-dependent pathway.
Oncogene. 23:1885–1895. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Schwartsmann G, Di Leone LP, Dal Pizzol F
and Roesler R: MAPK pathway activation in colorectal cancer: a
therapeutic opportunity for GRP receptor antagonists. Lancet Oncol.
6:444–445. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Fang JY and Richardson BC: The MAPK
signalling pathways and colorectal cancer. Lancet Oncol. 6:322–327.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Ma XH and Wang ZW: Anticancer drug
discovery in the future: an evolutionary perspective. Drug Discov
Today. 14:1136–1142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Gordaliza M: Natural products as leads to
anticancer drugs. Clin Transl Oncol. 9:767–776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Lin JM, Chen YQ, Wei LH, Chen XZ, Xu W,
Hong ZF, Sferra TJ and Peng J: Hedyotis Diffusa Willd
extract induces apoptosis via activation of the
mitochondrion-dependent pathway in human colon carcinoma cells. Int
J Oncol. 37:1331–1338. 2010.
|
|
21.
|
Cai QY, Lin JM, Wei LH, Zhang L, Wang LL,
Zhan YZ, Zeng JW, Xu W, Shen AL, Hong ZF and Peng J: Hedyotis
diffusa Willd inhibits colorectal cancer growth in vivo via
inhibition of STAT3 signaling pathway. Int J Mol Sci. 13:6117–6128.
2012. View Article : Google Scholar
|
|
22.
|
Wei LH, Chen YQ, Lin JM, Zhao JY, Chen XZ,
Xu W, Liu XX, Sferra TJ and Peng J: Scutellaria Barbata D.
Don induces apoptosis of human colon carcinoma cell via activation
of the mitochondrion-dependent pathway. J Med Plants Res.
5:1962–1970. 2011.
|
|
23.
|
Zheng LP, Chen YQ, Lin W, Zhuang QC, Chen
XZ, Xu W, Liu XX, Peng J and Sferra TJ: Spica Prunellae
extract promotes mitochondrion-dependent apoptosis in a human colon
carcinoma cell line. Afr J Pharm Pharmacol. 5:327–335. 2011.
View Article : Google Scholar
|
|
24.
|
Ikeda Y, Murakami A and Ohigashi H:
Ursolic acid: an anti- and pro-inflammatory triterpenoid. Mol Nutr
Food Res. 52:26–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Xavier CP, Lima CF, Preto A, Seruca R,
Fernandes-Ferreira M and Pereira-Wilson C: Luteolin, quercetin and
ursolic acid are potent inhibitors of proliferation and inducers of
apoptosis in both KRAS and BRAF mutated human colorectal cancer
cells. Cancer Lett. 281:162–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Andersson D, Liu JJ, Nilsson A and Duan
RD: Ursolic acid inhibits proliferation and stimulates apoptosis in
HT29 cells following activation of alkaline sphingomyelinase.
Anticancer Res. 23:3317–3322. 2003.PubMed/NCBI
|
|
27.
|
Adams J and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Youle RJ and Strasser A: The BCL-2 protein
family: opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Nurse P: Ordering S phase and M phase in
the cell cycle. Cell. 79:547–550. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Morgan DO: Principles of CDK regulation.
Nature. 374:131–134. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Zafonte BT, Hulit J, Amanatullah DF,
Albanese C, Wang C, Rosen E, Reutens A, Sparano JA, Lisanti MP and
Pestell RG: Cell-cycle dysregulation in breast cancer: breast
cancer therapies targeting the cell cycle. Front Biosci.
5:D938–D961. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Kouraklis G, Theocharis S, Vamvakas P,
Vagianos C, Glinavou A, Giaginis C and Sioka C: Cyclin D1 and Rb
protein expression and their correlation with prognosis in patients
with colon cancer. World J Surg Oncol. 4:52006. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Domagala W, Welcker M, Chosia M,
Karbowniczek M, Harezga B, Bartkova J, Bartek J and Osborn M:
p21/WAF1/Cip1 expression in invasive ductal breast carcinoma:
Relationship to p53, proliferation rate, and survival at 5 years.
Virchows Arch. 439:132–140. 2001.PubMed/NCBI
|