Contents
Epidemiology
Pathogenesis
Diagnosis
Screening and surveillance
Treatment
Conclusion
Epidemiology
Colorectal cancer (CRC) is a tumor that develops
from the progression of acquired or hereditary premalignant
lesions. It arises from interactions among different risk factors
(environmental, dietary, familial and hereditary) that become
relevant during the different stages of colorectal carcinogenesis.
CRC is a major public health problem worldwide, although in
developed countries survival rates have significantly improved over
the past two decades, reflecting continuous progress in our
understanding of its biology, epidemiology, prevention, early
diagnosis and treatment. Nonetheless, CRC remains the third most
commonly diagnosed cancer and the third leading cause of cancer
death in both men and women, with over 1.2 million new cases and
over 600,000 deaths expected in 2013 (1,2).
As for esophageal and gastric cancer (3,4),
various pathological conditions or a positive personal or family
history that predisposes to CRC development are associated with a
particular frequency and lifetime risk of disease occurrence. Thus,
individuals are considered to be at average, increased or high
risk. About 70% of CRCs are sporadic, perhaps attributable to
unidentified genetic factors in the context of dietary and
environmental factors. Individuals 50 years of age or older are at
average risk, with a 5% lifetime risk of developing CRC. The risk
increases to 10–15% in individuals with a personal history of
adenoma/sessile serrated polyps, inflammatory bowel disease, or a
positive family history of CRC (5).
Colorectal adenoma
The precursors of almost all sporadic CRCs are
colorectal adenomas. These typically asymptomatic lesions are often
found incidentally during colonoscopy performed for unrelated
symptoms or for CRC screening. At least 25% of men and 15% of women
who undergo colonoscopic screening have one or more adenomas.
Colorectal adenomatous polyps develop in up to 40% of people over
the age of 60 (6). Although not
all colonic polyps are adenomas and more than 90% of adenomas do
not progress to cancer, a differential diagnosis that takes into
account the various types of colorectal polyps and the accurate
identification of those that will progress to cancer remain
challenging (Table I). Some easily
identifiable but wide-ranging pathological features, such as size,
architectural growth, type, and dysplastic grade and organization,
are predictive both of the natural history of these lesions and of
the time frame of their potential evolution from adenoma to
carcinoma.
 | Table IFeatures of colorectal polyps. |
Table I
Features of colorectal polyps.
| Polyp type | Side | Size | Histological
features | Risk of
carcinoma |
|---|
| Conventional
adenoma (85–90%) | | | | |
| Left colon and
rectum | Variable | Tubular
25%
Tubulo-villous 15%
Villous 5%
Variable grade of dysplasia | Low risk (1%):
<1cm/ ≤2polyps/tubular/low grade dysplasia
High risk (30–50%): ≥1cm/>3polyps/villous/high grade
dysplasia |
| Serrated adenoma
(10–15%) | | | | |
| Hyperplastic polyp
(80–90%) | Left colon and
rectum | <5mm | Slightly
protruding; no cytological atypia or architectural dysplasia | None |
| Traditional
serrated adenoma (1–5%) | Left colon and
rectum | Variable | Often pedunculated;
presence of dysplasia and/or foci of intraepithelial neoplasia | The same risk as
adenomatous polyps |
| Sessile serrated
adenoma (15–20%) | Right colon | >5mm | Sessile; no
cytologic dysplasia | Risk present, but
degree not known |
| Mixed polyp
(1%) | Right colon | Variable | Combinations of
conventional adenomas with different grades of dysplasia and
serrated lesions | Degree of risk
variable |
The transformation rate of adenomatous polyps into
carcinoma is about 0.25% per year. The size of the adenoma is a
relevant determinant, given that cancer devlops in 1% of adenomas
<1 cm, in 10% of adenomas >1 cm and <2 cm, and in 50% of
adenomas >2 cm. The histological features that determine the
malignant potential of an adenoma are its growth pattern and grade
of dysplasia. In adenomas with a mainly villous architectural
configuration (tubulovillous and villous) and high-grade dysplasia,
the risk of malignant transformation rises to 50% (5).
Between 10 and 15% of sporadic CRCs are likely to
originate from serrated polyps, which have a significant malignant
potential. Serrated polyps include hyperplastic polyps, which
account for 80–90% of cases, but also sessile serrated adenomas,
traditional serrated adenomas, and mixed polyps displaying features
of both serrated and ‘classical’ adenomas.
Adenomatous polyps, particularly those with villous
components, are regarded as precursors of CRC, unlike hyperplastic
polyps. The term ‘serrated adenoma’ was used to describe a group of
polyps sharing mixed features of hyperplastic polyps and adenomas.
Serrated adenocarcinoma is a distinct variant of CRC, accounting
for about 7.5% of all colorectal tumors and up to 17.5% of the most
proximal colorectal tumors. Sessile serrated adenomas have some
features of serrated adenomas, but their sessile configuration
distinguishes them from their more pedunculated counterparts, i.e.,
traditional serrated adenomas.
Sessile serrated adenomas, traditional serrated
adenomas, and conventional adenomas differ in their malignant
potential, reflecting differences in the molecular pathways of
carcinogenesis. Histological assessment has shown a significantly
lower degree of high-grade dysplasia and carcinoma in situ
in serrated adenomas than in traditional adenomas. Thus, serrated
adenomas are less likely to develop into CRC than traditional
adenomas, but the degree of risk is not yet known (7). However, recent studies have suggested
a close association between the peculiar molecular features of
mucinous carcinomas (i.e., higher diploidy index, lower expression
of p53, more frequent DNA replication errors leading to
microsatellite instability, specific codon 12 KRAS mutations) and
sessile serrated adenomas. Mucinous histological features are often
seen in sessile serrated polyps that progress to invasive
adenocarcinoma, both in sporadic cases of CRC and in individuals
with a well-defined genetic predisposition (8).
The risk of cancer from a colorectal adenoma is
eliminated with its complete removal even if discovery of the
adenoma indicates the possible risk of metachronous lesions with
variable malignant potential according to endoscopic and histologic
features. The presence of three or more colorectal adenomas or one
or more advanced adenomas is associated with a risk of metachronous
adenomas that is two to five times higher than in the general
population. Other characteristics of the baseline adenoma (e.g., a
location proximal to the splenic flexure) or of the patient (e.g.,
male sex, older age or a first-degree relative with CRC) are also
considered predictive of metachronous adenomas or CRC. The overall
risk of developing metachronous adenomas after the removal of an
adenoma is about 5–10% per year (9,10).
Chronic inflammatory bowel
disease
Patients with a chronic inflammatory bowel disease
(IBD), such as ulcerative colitis and Crohn’s disease, are at
greater risk of developing CRC. The risk of CRC increases with the
extent and duration of the IBD. Thus, 15% of patients with a
30-year history of ulcerative colitis will eventually develop CRC
(11).
Familiarity
Patients with a first-degree relative (parent,
sibling or offspring) who has had CRC have a two to three times
higher risk of developing CRC than individuals with no family
history. If the diagnosis in the relative was made at a young age
or if more than one relative is affected, the risk is three to six
times higher than in the general population. In general, about 25%
of all CRC patients have a close relative who was diagnosed with
the disease (12).
Genetic syndromes
Susceptibility to CRC is higher in individuals with
well-defined rare genetic syndromes, which comprise about 5% of all
cases of colorectal tumors. Thus, in patients with familial
adenomatous polyposis (FAP) not surgically treated, the lifetime
CRC risk is as high as 100% (Fig.
1). In addition, there is a greater risk of developing other
malignancies (13).
 | Figure 1Proposed model of colon cancer risk
susceptibility. Colon cancer risk can be stratified by the
estimated lifetime risk of colon cancer (y-axis) versus the
approximate frequency of colorectal cancer (CRC) cases (x-axis).
The well-defined rare genetic conditions include high lifetime risk
syndromes such as FAP (familial adenomatous polyposis coli), AFAP
(attenuated FAP), MAP (MUTYH-associated polyposis), LS (Lynch
syndrome), HPP (hyperplastic polyposis), and the hamartomatous
polyp syndromes PJS (Peutz-Jeghers syndrome) and JPS (juvenile
polyposis syndrome). The clinically defined ‘familial subset’
consists of individuals at increased risk based on a clear
hereditary component, as determined from the family history, but
with undefined causative genetic factors. The largest subset,
accounting for 70% of CRC cases, comprises individuals in the
general population with sporadic tumors. In this group, the
lifetime risk derives from combinations of unidentified rare and
common genetic factors in which environmental influences are likely
to play a role. |
Lynch syndrome. Also known as hereditary
non-polyposis CRC (HNPCC), Lynch syndrome accounts for 2–4% of all
CRC cases (14). Although
individuals with HNPCC are predisposed to several types of cancer,
the lifetime risk of CRC is the highest (∼75%). Colon cancers and
polyps arise in Lynch syndrome patients at a younger age than in
the general population with sporadic neoplasias, and the tumors
develop at a more proximal location. Histologically, the cancers
are often poorly differentiated, mucinous and are infiltrated by
large numbers of lymphocytes (15).
Familial adenomatous polyposis. With a
prevalence of 1 in 10,000 individuals, FAP is the second most
common genetic syndrome predisposing to CRC. For these individuals,
the lifetime risk of CRC without prophylactic colectomy approaches
100%. The characteristic features of FAP include the development of
hundreds to thousands of colonic adenomas beginning in early
adolescence. The average age of CRC diagnosis (if untreated) in FAP
patients is 40 years; 7% develop the tumor by the age of 20 and 95%
by the age of 50. Attenuated FAP is a less severe form of the
disease, with an average lifetime risk of CRC of 70%. In this
group, approximately 30 adenomatous polyps develop in the colon,
colonic neoplasms tend to be located in the proximal colon, and
cancer occurs at an older age. Other rare variants of FAP are
Gardner’s syndrome and Turcot’s syndrome. The former is
characterized by prominent extra-colonic features: epidermoid
cysts, osteomas, dental abnormalities and/or desmoid tumors. The
latter includes patients with colorectal adenomatous polyps; these
patients are prone to developing malignant tumors of the central
nervous system, above all medulloblastoma (16).
MUTYH-associated polyposis, Peutz-Jeghers
syndrome, juvenile polyposis syndrome. These frequent genetic
conditions have an incidence of <1% and their characteristics
include distinct cancer risks, clinical features and genetic
patterns. Patients with MUTYH-associated polyposis (MAP) develop
adenomatous polyposis of the colorectum and have an 80% risk of
CRC. The colonic phenotype of this syndrome is similar to that of
attenuated FAP, including a propensity for developing proximal
colonic neoplasms (17).
Peutz-Jeghers and juvenile polyposis syndromes are hamartomatous
conditions associated with an increased risk for colorectal and
other malignancies. In Peutz-Jeghers patients, the most consistent
extra-colonic feature is a muco-cutaneous pigmentation typically
occurring in childhood and seen on the lips, oral mucosa, and
periorbital area. The typical gastrointestinal lesions are
histologically distinctive hamartomatous polyps (96% of cases) that
arise in the small bowel. Gastric and colonic polyps develop in
approximately 25 and 30% of these patients, respectively. The
lifetime risk of gastrointestinal cancer is 40% (18). The main features of juvenile
polyposis syndrome are multiple polyps, most prominently in the
colon but also in the stomach, duodenum, and small bowel, that
develop in young patients. As in Peutz-Jeghers syndrome, the
lifetime risk of CRC is approximately 40% (19).
Hyperplastic polyposis
Little is known about the etiology, natural history,
and incidence of this rare condition, which is characterized by
multiple and/or large hyperplastic polyps of the colon. These
patients have a >50% risk of developing CRC before the age of
50–60 years. The tumors tend to develop in the proximal colon and
metachronous and synchronous cancers are frequently observed
(20).
Pathogenesis
Geographic and dietary factors
Geographic differences in CRC rates among immigrant
populations over time suggest that diet and lifestyle strongly
influence the CRC risk. High-level consumption of red and/or
processed meat increases the risk of both colon and rectal cancer
(21). CRC also has been linked to
even moderate alcohol consumption, with an increased risk of 23%
(22). However, the precise role
of specific dietary elements in colorectal tumorigenesis is poorly
understood.
Metabolic alterations
Obesity, especially abdominal obesity, is associated
with a higher risk of colorectal adenoma and CRC, especially in
men. This association implies that obesity promotes the early
stages of carcinogenesis but it may also play a role in the growth
of advanced adenomas, both of which favor adenoma recurrence. The
mechanistic relationship between obesity and colon cancer risk is
not well established but may include the mitogenic properties of
insulin, obesity-related insulin resistance, and associated
hyperinsulinemia. Insulin could also promote colorectal
carcinogenesis by increasing the levels of bioactive insulin-like
growth factor (IGF)-1, either directly or through a decrease of IGF
binding protein levels, which leads to increased free IGF-1.
Obesity may be a pro-inflammatory state, as demonstrated by the
high systemic levels of proinflammatory cytokines, chemokines, and
other acute phase proteins released from adipose tissue, which
consists not only of adipocytes but also of immune cells (23).
Individuals with diabetes mellitus have an increased
risk of CRC. This may be related to the metabolic consequences of
obesity, physical inactivity, and insulin resistance, but the
evidence supporting a link between hyperglycemia and CRC is
inconsistent (24).
Sex hormones
Differences in sex hormones might explain the lower
female/male ratio in the population under than above the age of 55
years (i.e., in pre-menopausal vs. post-menopausal women). This
likely reflects the inverse relation between tumor progression and
the expression of type β estrogen receptors on colon cancer cells
and thus the inhibitory effect of estrogens on tumor growth.
Estrogens have been suggested to alter bile acid composition,
modulate colonic transit, reduce the production of mitogenic IGF-1,
and stimulate the expression of the mismatch repair (MMR) gene MLH1
in colonic epithelial cells (25).
Chronic inflammation
As discussed above, chronic inflammation is a key
risk factor for CRC in patients with IBDs. The risk of colon cancer
increases not only with disease duration and the anatomic extent of
the colitis but also with the presence of other inflammatory
disorders (such as primary sclerosing cholangitis), whereas it
decreases in patients taking anti-inflammatory agents (such as
steroids). Inflammatory cells produce reactive oxygen and nitrogen
species, which can alter the expression of genes encoding
carcinogenesis-related factors (p53, DNA MMR proteins, and DNA base
excision-repair proteins), transcription factors (nuclear
factor-κB) or signaling proteins (cyclo-oxygenase-2, COX-2).
Moreover, individual components of the innate and adaptive immune
response (immune cells, cytokines, chemokines and the intestinal
bacterial flora) have been implicated in carcinogenesis, via
genetic or epigenetic alterations (26,27).
Thus, there is a close relationship between colonic inflammation
and neoplasia, which develops progressively: no dysplasia →
indefinite dysplasia → low-grade dysplasia → high-grade dysplasia →
carcinoma.
There are also similarities between the pathways of
sporadic cancers and colitis-associated cancers, including the
development of aneuploidy (chromosomal instabilities, CIN),
microsatellite instabilities (MSI), DNA methylation, activation of
the oncogene K-ras, activation of COX-2, and the mutation and
eventual loss of heterozygosity of p53, adenomatous polyposis coli
(APC), and two candidate tumor suppressor genes, namely deleted in
colorectal carcinoma and deleted in pancreatic carcinoma 4
(DCC/DPC4). However, the frequency and sequence of these events
differ between these CRC types (26).
Genetic and epigenetic
instabilities
Genetic and epigenetic instabilities are a hallmark
of colorectal carcinogenesis. The former comprises genomic
instabilities, i.e., CIN and MSI, and the latter the
cytosine-phosphate-guanine (CpG) island methylator phenotype (CIMP)
and global DNA hypo-methylation (28). Genomic instability characterizes
the early steps of malignant transformation from adenoma into
carcinoma, increasing the mutation rate and thereby facilitating
the progression to malignancy. The most common form of genomic
instability are CIN, which are found in about 85% of CRCs. CIN are
recognized by the presence of aneuploidy, i.e., numerical
chromosome changes, or multiple structural aberrations. They are an
efficient mechanism for causing the physical loss of a wild-type
copy of a tumor-suppressor gene such as APC, p53, and Short-name
Mothers Against Decapentaplegic (SMAD) family member 4 (29).
MSI, which account for 15% of sporadic CRCs, are an
epiphenomenon of the inactivation of genes involved in the repair
of base-base mismatches in DNA, defined as MMR genes (hMSH2, hMLH1,
hPMS1, hPMS2 or hMSH6). The loss of MMR function disrupts the
ability of the affected cell to repair strand slippage within
repetitive DNA sequence elements. Consequently, the sizes of the
mononucleotide or dinucleotide repeats (micro-satellites)
interspersed throughout the genome are altered and tumor-suppressor
genes whose functional regions contain these repeat sequences, such
as those encoding transforming growth factor-β (TGF-β) receptor
type II and Bcl2-associated X protein (BAX), are inactivated. In
MMR deficiency, the inactivation can be inherited, as occurs in
HNPCC (about 95% of the mutations involve hMSH2 or hMLH1), or
acquired, as observed in tumors with methylation-associated
silencing of a gene encoding an MMR protein, for example, biallelic
silencing of the promoter region of the MLH1 gene by promoter
methylation (13,30). Recently, germline deletions in the
EpCAM (epithelial cell adhesion molecule) gene, also known as
TACSTD1, were found in a subset of families with Lynch syndrome in
whom MMR gene mutations were absent because of the uncommon
hyper-methylation of the hMSH2 promoter (31).
Microsatellite status has been divided into three
types, with different clinical and prognostic features with respect
to CRC: i) microsatellite stable, with no instability; ii)
low-level instability (MSI-low), with <30% instability; and iii)
high-level instability (MSI-high), with >40% of the
microsatellite loci showing instability. MSI-high occurs in more
than 90% of patients with an inherited predisposition for CRC but
in only 15% of those with sporadic CRCs, i.e., without familial
predisposition.
Sporadic MSI tumors are associated with the serrated
adenoma neoplasia pathway and frequently carry BRAF V600E
mutations, which are not seen in patients whose cancers result from
germline mutations in MMR genes (Lynch syndrome). Thus, the
presence of a BRAF mutation in an MSI tumor effectively excludes
the possibility of Lynch syndrome (32).
In patients with MAP, there is germline inactivation
of a base excision repair gene, the mutY homologue (MUTYH, also
called MYH). The MYH protein excises from DNA the 8-oxoguanine
product of oxidative damage to guanine. Two MAP-associated
mutations in MYH have been identified, Y179C and G396D, which
together account for 85% of the cases of this disease. By contrast,
somatic inactivation of MYH has not been detected in sporadic CRC
(17).
Epigenetic instabilities in precancerous lesions and
CRC are manifested as hyper-methylation of gene promoters
containing CpG islands (CIMP) and global DNA hypo-methylation. CIMP
is observed in about 50% of premalignant colonic adenomas and in
about 50% of CRCs, which suggests that it is an early event in
colorectal tumorigenesis. MSI are also detected in about 45% of
CIMP-positive CRCs but in 100% of CIMP-positive cancers in which
hMLH1 is methylated (33). The
strong association between BRAF V600E mutations and CIMP CRC points
to a role for activated BRAF in the pathogenesis of the methylator
phenotype as well as a link between sporadic MSI and CIMP (34).
In addition to genomic and epigenomic instabilities,
the accumulation of mutations in specific genes (tumor-suppressor
genes or proto-oncogenes) is another pathogenetic route in the
neoplastic progression of precancerous lesions to CRC. Thus, the
malignant transformation of colon epithelial cells and thus CRC can
arise from the loss/inactivation of tumor-suppressor genes that are
not involved in specific signaling pathways, such as p53, and from
recurrent cytogenetic aberrations, such as in the 18q loss of
heterozygosity gene (28).
Moreover, in CRC one or more cellular pathways may become
deregulated. Of these, the best studied are the Wnt-β-catenin,
TGF-β, epidermal growth factor receptor (EGFR),
Ras/Raf/mitogen-activated protein kinase (MAPK) and
phosphatidylinositol 3-kinase (PI3K) signaling pathways.
Mutations in the APC gene itself are predominantly
associated with the classic tubular adenoma pathway and with
CIN-type cancer (35). They occur
in up to 70% of sporadic CRCs and explain the predisposition of
patients with FAP to cancer development. The APC gene is located on
chromosome 5q21. Although germline-inactivating mutations can occur
throughout the gene, somatic mutations are clustered in the
mutation cluster region, between codons 1286 and 1513. The
conventional form of the APC gene contains 15 exons. Exon 15 is the
largest, comprising more than 75% of the 8,535 base pairs of coding
sequence. In the tumors of FAP patients, it is the site of most
germline and somatic mutations (36). In addition to the conventional form
of APC, alternatively expressed exons of the gene encode protein
isoforms, including truncated proteins. Interestingly, mutational
analyses in FAP patients have identified significant
genotype-phenotype correlations: i) severe polyposis (>5,000
polyps), associated with mutations between codons 1250 and 1464;
ii) attenuated polyposis (<100 polyps), in which mutations occur
at the extreme 5′ and 3′ ends of the APC gene; iii) congenital
hypertrophy of the retinal epithelium, associated with mutations
between codons 457 and 1444; and iv) desmoid tumors, in which
mutations are found between codons 1403 and 1578 (37).
In contrast to the truncating APC gene mutations
implicated in FAP, APC I1307K is a single-nucleotide substitution
that results in a non-truncating, missense mutation and thus to a
single amino acid difference in the approximately 3,000 amino acids
that constitute the APC protein. The APC I1307K variant is carried
by an estimated 6% of the Ashkenazi Jewish population and its
presence approximately doubles the risk of developing colorectal
polyps and CRC in heterozygous carriers (38). Hyper-methylation of the APC
promoter is an alternative mechanism for APC gene inactivation; it
occurs in 18% of primary colorectal carcinomas and adenomas
(39). Defects in the Wnt
signaling pathway are an initiating event in CRC and underlie many
preneoplastic lesions. Wnt signaling occurs when the oncoprotein
β-catenin binds to nuclear LEF-1 and so creates a transcription
factor that regulates genes involved in cellular activation. The
β-catenin degradation complex regulates β-catenin levels by
proteolysis. A component of this complex, the APC protein, not only
degrades β-catenin but also inhibits its nuclear localization. The
most common mutation in CRC inactivates the gene that encodes the
APC protein. In the absence of functional APC, which acts as a
brake on the β-catenin pathway, Wnt signaling is inappropriately
and constitutively activated (40). Activating mutations in the
β-catenin gene (CTNNB1) protect the encoded protein from
APC-mediated degradation. CTNNB1 mutations are found more
frequently in adenomas (12.5%) than in invasive cancer (1.4%),
suggesting that CTNNB1-mutant tumors do not frequently progress to
carcinoma (32).
Another early and critical step in adenoma
development is the activation of prostaglandin signaling,
especially the increased production of prostaglandin E2 (PGE2). In
addition, growth factors, cytokines, inflammatory mediators and
tumor promoters induce the overexpression of COX-2 in about 43% of
adenomas and 86% of carcinomas. COX-2 and PGE2 regulate
proliferation, survival, migration and invasion in colorectal
tumors. COX-2 also regulates angiogenesis, inducing the production
of pro-angiogenic factors such as vascular endothelial growth
factor (VEGF) and basic fibroblast growth factor, which may also
contribute to the growth and lethal potential of CRC (28). PGE2 activity is increased by the
loss of 15-prostaglandin dehydrogenase, the rate-limiting enzyme in
PG degradation (41).
In summary, the accumulation of several acquired
genetic and epigenetic changes transform normal glandular
epithelial cells into invasive colorectal carcinoma. This stepwise
transformation of normal epithelium into benign neoplasia
(adenoma), followed by invasive carcinoma and eventually metastatic
cancer, are described in the classic tumor progression model
proposed by Fearon and Vogelstein (42). The complex progression to
colorectal carcinogenesis also involves colon cancer-initiating
stem cells (43,44), located within the crypt unit, which
account for 0.25–2.5% of the total number of tumor cells. An
overview of the most important molecular alterations and the main
risk factors in colorectal carcinogenesis is provided in Fig. 2.
 | Figure 2Genes, growth factor pathways and
other possibile causes driving multistep colorectal cancer
progression. Colorectal carcinogenesis progresses by at least two
well-recognized pathways: i) the chromosomal instability (CIN)
pathway, which is observed in benign adenomas and increases in
tandem with tumor progression; and ii) the microsatellite
instability (MSI) pathway, which may be associated with the CpG
island methylator phenotype (CIMP) and serrated adenoma pathways.
Additionally, growth factor pathways (e.g., mutations in the EGFR
pathway, aberrant overexpression of COX-2) are commonly activated
in colorectal cancer (CRC). VEGF and TGF-β also contribute to colon
carcinogenesis, as do lifestyle and dietary factors. Among the
latter, obesity and hyperinsulinemia are of particular interest as
they increase the levels of bioactive insulin-like growth factor
(IGF)-1. Chronic inflammation, in which reactive oxygen and
nitrogen species as well as pro-inflammatory cytokines are
released, is an important risk factor for CRC in patients with
inflammatory bowel diseases. Colon cancers also express type β
estrogen receptors, which are thought to account for the inhibitory
effect of estrogens on tumor growth, based on the inverse
relationship between tumor progression and the expression of these
receptors. The complex pathogenesis of CRC may be regulated by
colon cancer stem cells located within the crypt unit, especially
during the early stages of colorectal carcinogenesis. |
Diagnosis
The ‘gold standard’ for the diagnosis of
precancerous lesions and carcinoma involving the colon is
colonoscopy, which allows direct visualization, a fairly accurate
localization, and the opportunity to obtain tissue samples for
histologic evaluation. Colonoscopy has a significant impact on CRC
incidence and mortality, preventing about 65% of CRC cases
(1). For every 1% increase in the
colonoscopy rate, the risk of death is reduced by 3% (45). When a complete colonoscopy extended
to the ileo-cecal valve is not possible, air-contrast barium enema
and virtual colonoscopy are suitable options.
Until the advent of modern colonoscopy, barium enema
was considered the mainstay for the detection of large colonic
polyps and colon cancer. However, barium enema is neither as
sensitive nor as specific as colonoscopy and should be chosen as
the initial screening test only for patients in whom colonoscopy
has either failed or who are at high risk for complications from
the procedure, or when colonoscopy is not available. Barium enema
has no role in determining the extent of colonic wall invasion by
colon cancer, nor does it provide information on lymph node
involvement or distant disease in patients who are at high risk for
metastases.
Computed tomographic (CT) colonography (also known
as virtual colonoscopy) was introduced into clinical practice in
the mid-1990s and is a promising technique for the diagnosis and
screening of CRC. As a minimally invasive imaging examination of
the entire colon and rectum, it allows polyp detection, the
characterization of tumor density and site, and in some settings
evaluation of the extra-colonic structures. The risk of
test-related complications is also very low, as the perforation
rate associated with CT colonography is 0.06–0.08%, compared with
0.1–0.2% for colonoscopy (46). In
terms of colon cancer detection and the measurement of advanced and
adenomatous polyps ≥10 mm, recent data indicate that CT
colonography is comparable to colonoscopy (47), whereas for lesions 5–9 mm in
diameter the accuracy of the CT examination is lower and for
lesions <5 mm in diameter it is unacceptable (48). Another potential disadvantage of CT
colonoscopy is its poor ability to detect non-polypoid, flat
lesions. In a screening population, this subset of sessile polyps
had an overall prevalence of around 5.8% (49). Moreover, virtual colonoscopy is
only a diagnostic test and patients with polyps of significant size
will require therapeutic colonoscopy for subsequent polypectomy.
Thus, at present, CT colonoscopy is not used as a colorectal
screening technique and the optimal screening intervals have yet to
be established (50).
Another endoscopic technique for colorectal
screening is flexible sigmoidoscopy, which is limited to the
examination of the lower colon tract but requires neither sedation
nor intense bowel preparation, both of which are mandatory for
colonoscopy. While polypectomy is possible during the examination,
patients with lesions >1 cm should be referred to colonoscopy
since additional adenomatous polyps cannot be excluded. The finding
at rigid sigmoidoscopy of one or more advanced adenomas is
associated with a rate of metachronous colon cancer, especially in
the proximal tract, that is about five times higher than in the
general population, whereas for individuals with only small,
recto-sigmoid tubular adenomas the risk is the same (6). Sigmoidoscopy, followed by colonoscopy
if a polyp or tumor is detected, can identify advanced lesions in
70–80% of the cases and is associated with a 60–80% reduction in
CRC-related mortality. A single sigmoidoscopy screening between the
ages of 55 and 64 years reduces the incidence of CRC by 33% and
mortality by 43% (51).
Several tools are available to support the clinical
diagnosis of genetic syndromes. The Amsterdam criteria I were
originally developed to identify families with Lynch syndrome but
more than 50% of these families failed to meet these criteria. To
increase the sensitivity, the Amsterdam criteria II and the
Bethesda guidelines were developed (52,53).
Two other approaches for identifying Lynch syndrome are based on
MSI evaluation and immunohistochemistry. In the latter, colorectal
tumors are evaluated for MMR deficiency using four antibodies,
specific for hMLH1, hMSH2, hMSH6 and hPMS2 proteins. The
sensitivity of the two methods is comparable (54).
FAP is diagnosed when at least 100 colonic adenomas
are identified, although younger individuals with fewer polyps
might also be considered positive for this disease. The diagnosis
is supported by the finding of extra-colonic lesions (upper
gastrointestinal tract polyposis, congenital hypertrophy of the
retinal pigment epithelium, epidermoid cysts, osteomas, dental
abnormalities, desmoid tumors) and confirmed by the identification
of APC mutations in a proband, allowing precise identification of
other relatives who are at risk. Attenuated FAP is suspected when
>10 but <100 adenomas are found in a person older than 40–50
years of age. A precise diagnosis is often difficult in a single
patient and the polyp numbers vary with this disorder. Attenuated
FAP can mimic the typical, fully expressed form, MUTH-associated
polyposis or even sporadic polyp development (16).
As discussed above, the typical gastrointestinal
lesions in Peutz-Jeghers syndrome are small-bowel, histologically
distinctive hamartomatous polyps (96% of patients). Gastric and
colonic Peutz-Jeghers polyps are found in approximately 25 and 30%
of cases, respectively. Generally, gastrointestinal symptoms first
occur in the teen-age years, including small-bowel obstruction,
intususceptions, and bleeding. In general, a clinical diagnosis of
Peutz-Jeghers syndrome is made when two or more of the following
features are detected: i) ≥2 Peutz-Jeghers polyps of the small
intestine; ii) typical muco-cutaneous hyper-pigmentation; and iii)
a family history of the disease (55).
Unlike Peutz-Jeghers syndrome, the physical findings
in juvenile polyposis syndrome are not necessarily diagnostic. The
key feature of the disease is the occurrence of multiple polyps,
most prominently in the colon but also in the stomach, duodenum,
and small bowel. A diagnosis of juvenile polyposis should be
considered for any young individual of about 20 years with at least
three polyps of the colon, multiple polyps throughout the
gastrointestinal tract, or any number of polyps and a positive
family history. Congenital defects occur in approximately 15% of
patients with juvenile polyposis syndrome. A subset of patients
also has hereditary hemorrhagic telangiectasia, often accompanied
by mucocutaneous telangiectasias as well as gastrointestinal and
pulmonary arteriovenous malformations (56).
A clinical diagnosis of hyperplastic polyposis
syndrome is based on at least one of the following criteria: i)
≥20–30 hyperplastic polyps throughout the colon; ii) ≥5
hyperplastic polyps proximal to the sigmoid colon, with two polyps
>1 cm in diameter; iii) >1 hyperplastic polyps proximal to
the sigmoid colon; and iv) a first-degree relative with
hyperplastic polyposis. Chromo-endoscopy and narrow-band imaging
may improve the detection rates of this syndrome (13).
Screening and surveillance
Both colorectal premalignant and overt malignant
lesions are usually asymptomatic and their development is highly
insidious. Consequently, screening is often necessary to detect
preneoplastic lesions and CRC in its early stages. CRC screening
tests are subdivided into two groups: those capable of detecting
both cancer and precancerous lesions (structural exams: flexible
sigmoidoscopy, colonoscopy, CT colonography, and double-contrast
barium enema) and those that primarily detect cancer (stool tests).
Colonoscopy remains the most important screening method and has the
longest rescreening interval of all diagnostic tests; if normal,
the exam does not need to be repeated for 10 years in average-risk
adults age 50 years and over (46). Fecal-based screening tests are not
invasive and no bowel preparation is necessary but they are less
likely to detect adenomas and thus do not contribute to cancer
prevention. In addition, they have a significantly lower
sensitivity than structural tests for inadequate specimen
collection or when processing or interpretation is suboptimal.
Moreover, if the stool test is positive, colonoscopy is always
necessary.
Two fecal tests for the detection of occult blood
are currently available: one is guaiac-based and the other is
immuno chemical. Guaiac-based tests detect blood in the stool
through the pseudo-peroxidase activity of heme or hemoglobin, while
immunochemical tests recognize human globin. Although cancers and
some large polyps bleed intermittently into the intestinal lumen
and occult fecal blood tests can detect very small quantities of
blood in stool, the reliability of the test results requires annual
testing of two to three samples per test. Patients who have a
positive occult fecal blood test are referred for colonoscopy to
rule out the presence of polyps or cancer. Studies have shown that
the regular use of these screening methods reduces the risk of
death from CRC by 15–33% (1),
while the incidence of this disease is reduced by approximately 20%
based on the detection of large polyps, which can subsequently be
removed by colonoscopy (57).
The stool DNA test is a screening method that takes
advantage of what is currently known about the molecular properties
of cancer. Cancer tissues and large polyps shed cells that contain
altered DNA into the large bowel; these gene mutations can be
detected in stool samples analyzed by DNA tests. Although only a
one-time collection is necessary, adequate evaluation requires the
entire stool specimen (30 g minimum). The first generation of stool
DNA tests assayed for the presense of CIN pathway markers such as
mutations in APC, KRAS and p53, and the MSI marker BAT-26 (58), while more recent versions are
capable of detecting hyper-methylated gene markers of the
epigenetic pathway. The first version of this test in asymptomatic
patients undergoing colonoscopy identified 51% of cancers and 18%
of advanced cancer-precursor lesions. In newer versions of the
test, the sensitivity is greater: 80 and 40%, respectively
(59). Therefore, DNA testing is
more accurate than occult fecal blood testing and it can be
performed at home; however, it is expensive, the appropriate time
interval for repeating the test is uncertain, and the potential for
cancer prevention is limited because of poor sensitivity in the
detection of colorectal adenomas. Moreover, patients with a
positive test are referred for colonoscopy (60).
In 2008, the American Cancer Society, in
collaboration with the American College of Radiology and the US
Multi-Society Task Force on CRC, published consensus guidelines for
CRC screening. The recommendations, in addition to emphasizing
cancer prevention as the primary goal of screening (46), recognize three categories of
patients.
For the first group, comprising individuals at
intermediate risk (adults 50 years of age and older), the currently
recommended options include colonoscopy every 10 years, an annual
fecal-based test, or flexible sigmoidoscopy or CT colonography
every 5 years. Among these tests, colonoscopy is the preferred
screening modality.
In the second group are individuals at increased
risk, i.e., those with a personal history of adenoma as they are at
greater risk of recurrent adenomas or CRC development. For low-risk
adenomatous polyps (tubular, two or fewer, <1 cm in size),
colonoscopy should be repeated within 5 years and, if normal, every
5–10 years. For high-risk adenomatous polyps (villous, 3–10 polyps,
≥1 cm, high-grade dysplasia), colonoscopy within 3 years and
subsequent surveillance colonoscopies within 5 years are
recommended. Individuals with more than 10 adenomatous polyps
should undergo evaluation for polyposis syndrome, especially if
they are under the age of 40 years and have a strong family
history. Despite polypectomy of large sessile adenoma, the
recurrence rates are high because residual adenoma tissue is often
unavoidable. In this group, colonoscopy should be repeated within
2–6 months.
Individuals with a personal history of CRC who have
undergone colonic resection with curative intent have a higher risk
of recurrence in the 4–5 years following surgery and should
therefore undergo repeat colonoscopy at shorter intervals (1–3
years). For those with a first-degree relative with a diagnosis of
CRC between 50 and 60 years, colonoscopy is recommended every 5
years starting at 40 years of age. If CRC is diagnosed in a
first-degree relative age 60 years or more, colonoscopy is
recommended every 5 years starting from 50 years. In patients with
IBD, colonoscopy should be performed 8–10 years after the onset of
symptoms (usually the time required for CRC development) and then
repeated every 1–2 years.
For individuals with high-risk inherited syndromes,
or a genetic or clinical diagnosis of HNPCC, or an increased risk
of HNPCC, colonoscopy should be started at age of 20–25 years, or
10 years before the youngest case of CRC occurs in immediate family
members, and then repeated every 1–2 years. In addition, genetic
testing should be offered to first-degree relatives. For FAP
patients, colonoscopy screening should be started at age 10–12
years and repeated annually. Patients with MAP should undergo
colonoscopy at the age of 30–35 years and every 3–5 years
thereafter. In those with Peutz-Jeghers syndrome, endoscopic
screening for colon cancer should be performed initially during the
late teen years and every 2–3 years thereafter, whereas for
juvenile polyposis syndrome screening is recommended beginning at
the age of 15 years, with annual repeat colonoscopy if polyps are
initially detected, otherwise every 2–3 years. Although precise
surveillance strategies have not been established in hyperplastic
polyposis syndrome, regular colonoscopy is recommended every 1–2
years (46).
Treatment
Colonoscopy with removal of adenomas is an effective
strategy for reducing the incidence of CRC and disease mortality by
as much as 70 and 60%, respectively, in the general population,
especially for patients with left-side tumors (61). However, standard polypectomy
techniques are ineffective in dealing with sessile and non-polypoid
colorectal lesions; instead, endoscopic mucosal resection is now
the treatment of choice for such cases. Endoscopic mucosal
resection relies on the detachment of the submucosal layer from the
muscularis propria followed by resection between these layers to
effectively remove the lesion. While there are limits to this en
bloc technique, as it is effective only for lesions with a maximum
diameter of 1.5–2 cm, it is a practical approach with low rates of
complications and local recurrences (about 7%) (62).
An improved understanding of the modifiable risk
factors may result in additional primary prevention strategies,
even if their efficacy has yet to be determined. A recent study
found that a healthy lifestyle, i.e., maintaining a normal weight,
being physically active at least 30 min per day, eating a healthy
diet, not smoking, and avoiding excessive amounts of alcohol, was
associated with a lower risk of CRC (incidence rate ratio 0.89; 95%
confidence interval: 0.82–0.96), with about 25% of CRCs considered
accordingly preventable (63). In
addition, several trials have suggested that calcium and vitamin D,
antioxidants (selenium, β-carotene, vitamins A, C and E), and folic
acid supplements can contribute to CRC prevention (64–66).
It is now well established that non-steroidal
anti-inflammatory drugs (NSAIDs) can cause adenoma regression in
FAP patients (67). Aspirin and
NSAIDs have also been shown to modulate several of the early
molecular events in the classic adenoma → carcinoma sequence, thus
preventing malignant transformation (16), as well as adenoma recurrence and
the incidence of CRC in the general population, although this
effect was only observed in studies in which at least 300 mg
aspirin/day was administered, with a follow-up duration longer than
10 years (68). In a recent study,
the long-term administration of lower doses (75–300 mg daily) of
aspirin on CRC incidence resulted in no overall risk reduction for
rectal cancer. Notably, the benefit was instead greatest for
cancers of the proximal colon, which are not effectively prevented
by sigmoidoscopy- or colonoscopy-based screening (69). In general, NSAIDs have a moderate
chemopreventive effect, decreasing the number of new adenomas by
about 40% and the number of large or histologically advanced
adenomas by up to 60%. Despite this level of effectiveness, NSAIDs
are not currently recommended for adenoma chemoprevention, largely
because of their well-known adverse effects. As an alternative, CRC
chemoprevention based on combination therapies in patients at high
risk of advanced adenomas has gained increasing interest (70).
Although post-menopausal hormone therapy appears to
be associated with a lower risk of CRC, it remains unclear which
preparations of estrogen-alone or estrogen plus progestin are most
effective. A longer duration of treatment is associated with
increased protection, although the risk returns to that obtained
with placebo within 3 years of hormone cessation. Moreover, because
post-menopausal hormones increase the risk of breast cancer and
cardiovascular events, the balance of risks and benefits does not
support their use as a means of preventing CRC (71). Statins have also been claimed to
reduce the risk of CRC, but the evidence has been inconsistent;
however, recent studies support the efficacy of statins in
preventing the development and progression of adenomatous polyps
(72).
Table II and
Fig. 3 summarize the impact of the
different prevention options on the incidence of colon cancer based
on the results of recent studies. Dietary components, lifestyle
factors, and medications have been proposed to act either directly
or indirectly via anti-inflammatory mechanisms that are largely
mediated by COX-2 inhibition, as shown in Fig. 4 (73,74).
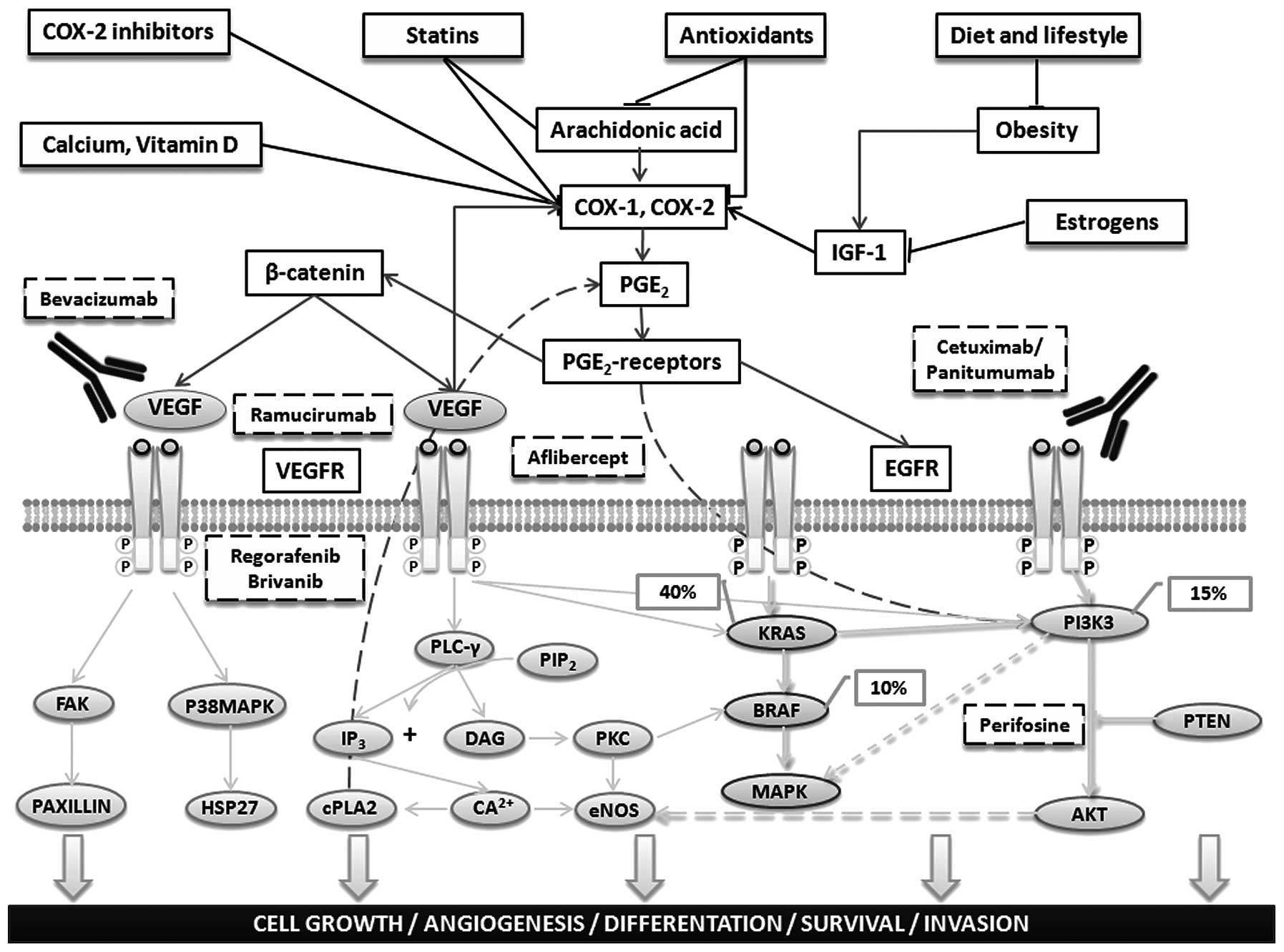 | Figure 4Main preventive and therapeutic
agents in colorectal cancer. Dietary components, lifestyle, and
medications may exert antineoplastic effects through direct or
indirect anti-inflammatory mechanisms that largely result in the
inhibition of COX-2. This enzyme is normally upregulated by
inflammatory or oncogenic stimuli via inflammatory cytokines, such
as interleukin-6 (IL-6), which induce nuclear factor κB (NF-κB).
COX-2 converts membrane-associated arachidonic acid to
prostaglandins (PGs) that, in turn, stimulate their respective
receptors, resulting in the upregulation of β-catenin
transcriptional activity and activation of the oncogene products
phosphatidylinositol-3-kinase (PI3K) and AKT kinase. PGs also
trigger phosphorylation of the epidermal growth factor receptor
(EGFR), thereby activating PI3K, AKT and the oncogenic
RAS-mitogen-activated protein kinase (MAPK) cascade. The EGFR
homodimer, formed after ligand activation of the receptor,
phosphorylates/activates the intracellular kinase domain and thus
also a cascade of downstream signaling that includes activation of
the Ras/Raf/MAPK and phosphatidylinositol 3-kinase (PI3K) pathways
associated with cell growth, differentiation, survival and
invasion. Anti-EGFR monoclonal antibodies (cetuximab, panitumumab)
are used to treat patients with metastatic colorectal cancer (CRC).
These drugs bind to the extracellular portion of the EGFR to
inhibit signaling. Drug resistance can occur via activating
mutations, such as in KRAS (∼40% of CRCs) or its direct downstream
effector BRAF (∼10%), that bypass the need for upstream EGFR
signals. Moreover, PGE2-mediated signaling pathways induce the
expression of other genes, including vascular endothelial growth
factor (VEGF), that have an important role in carcinogenesis by
altering normal cellular immunity and apoptosis or by increasing
proliferation, angiogenesis, migration, and invasion. Five novel
targeted agents for the treatment of CRC that are currently under
study in phase III trials are shown. |
| Table IIAnalysis of some important studies
regarding the impact on cancer incidence of the current preventive
options. |
Table II
Analysis of some important studies
regarding the impact on cancer incidence of the current preventive
options.
| Options for
prevention | No. of
patients | Follow-up
(years) | Reduction of
incidence (%) | P-value | OR/RR (95% CI) |
Authors/(Refs.) |
|---|
| Polypectomy | 4,344 | 10 | 77% | <0.001 | 0.23
(0.19–0.27) | Brenner et
al (62) |
| Lifestyle and
dietary factors | 55,487 | 9.9 | 13% | 0.2 | 0.89
(0.82–0.96) | Kirkegaard et
al (64) |
| Calcium plus
vitamin D | 36,282 | 7 | 10% | 0.73 | 1.08
(0.86–1.34) | Manson et al
(65) |
| Antioxidants
(selenium, β-carotene, vitamins A, C and E) | 676,141 | 20 | 10% | 0.97 | 0.88
(0.81–0.96) | Park et al
(66) |
| Folic acid | 120,852 | 13.3 | 20% | 0.18 | 0.85
(0.74–0.99) | Kennedy et
al (67) |
| COX-2
inhibitors | 14,033 | 20 | 45% | 0.01 | 0.76
(0.6–0.96) | Rothwell et
al (71) |
| Hormonal
therapy | 1,831 | 15 | 40% | 0.05 | 0.52
(0.38–0.72) | Long et al
(72) |
| Statins | 1,818 | 16 | 16% | 0.8 | 0.99
(0.86–1.14) | Baron et al
(73) |
In conclusion, only polypectomy currently offers
optimal treatment of colorectal preneoplastic lesions and it
remains a reliable strategy for CRC prevention, whereas
uncertainties remain regarding the effectiveness of lifestyle and
dietary factors. The potential toxicity of medications or
supplements as protective factors that can reduce CRC development
and progression is also a matter of concern.
In inherited syndromes, an awareness of an
individual’s genetic predisposition and the recognition of the
subset of patients at high lifetime risk of CRC allow for a
rational patient-tailored clinical management approach that in the
future might include effective chemoprevention and biologics-based
treatments. In the meantime, in Lynch syndrome patients, subtotal
colectomy with ileo-rectal anastomosis is the recommended approach
to colon cancer prevention (75).
Since the most common extra-colonic malignancy associated with
Lynch syndrome is endometrial cancer (incidence of 40–60%), with a
lifetime risk that is comparable to or even greater than the
estimated risk for CRC, prophylactic hysterectomy and bilateral
salpingo-oophorectomy are recommended for female patients after
completion of childbearing (76).
In patients with FAP, once adenomatous polyps emerge, an annual
follow-up colonoscopy is recommended. Colectomy should be
considered when more than 20 adenomas develop, when adenomas >1
cm in diameter are found, or when advanced histology is diagnosed.
Restorative proctocolectomy (also called total proctocolectomy)
with ileal pouch anal anastomosis is recommended for patients with
large numbers of rectal adenomas. If few or no rectal adenomas are
present, then preservation of the rectum with ileo-rectal
anastomosis is the preferred surgical therapeutic strategy. Annual
or more frequent endoscopic examinations must be performed if any
rectal tissue remains. However, up to 33% of patients with a
preserved rectum will eventually require a complete proctectomy
because of diffuse polyposis. If numerous adenomas are detected,
COX-2 inhibitors should be administered as they are effective in
achieving polyp regression, which in turn facilitates surveillance
(77). There are few data on the
use of NSAIDs in FAP patients, either to delay surgery or as a
primary treatment, and they remain to be tested in investigative
trials.
Patients with attenuated FAP should always undergo
colonoscopy screening because of the frequency of proximal colonic
polyps. Approximately 33% of these patients can be managed over the
long-term with colonoscopy and polypectomy because of the small
number of polyps. However, the majority will eventually require
colectomy, with rectal-sparing ileo-rectal anastomosis whenever
possible. Annual post-operative surveillance is required for polyp
ablation, but subsequent proctectomy is rarely needed. Over time,
more than 20% of patients will require therapeutic endoscopic
procedures or surgery to treat duodenal adenomas and adenomas at
the duodenal papilla. Gastric fundic gland polyps should be
sampled, especially if they are large or erythematous. Gastrectomy
is needed only if severe dysplasia appears (13).
In MAP, subtotal colectomy is advised for patients
who develop colon cancer but it should also be considered when
colonoscopic management becomes problematic or when polyps become
larger or exhibit high-grade dysplasia. Patients with Peutz-Jeghers
or juvenile polyposis syndrome should be referred to a specialized
center, because of the rarity of these conditions, the complexities
of their screening, diagnosis, and management, and the limited data
on the effectiveness of various screening modalities (78). In patients with hyper-plastic
polyposis syndrome, polyps >5 mm should be treated by
polypectomy. Subtotal colectomy should be considered if
colonoscopic treatment is inadequate or high-grade dysplasia occurs
(13).
Conclusion
The remarkable gains in our understanding of the
molecular basis of CRC and the transfer of this knowledge into
daily clinical practice have begun to reduce the burden of this
disease. The mortality rates for CRC are decreasing in several
Western countries, and above all in the United States, due to
increased awareness and more effective prevention of CRC, the
improved early detection of colorectal pre-cancerous lesions, and
the introduction of novel, personalized therapeutic options for
low- and high-risk individuals.
Abbreviations:
|
APC
|
adenomatous polyposis coli;
|
|
BAX
|
Bcl2-associated X protein;
|
|
CIMP
|
CpG island methylator phenotype;
|
|
CIN
|
chromosomal instability;
|
|
COX-2
|
cyclo-oxygenase-2;
|
|
CRC
|
colorectal cancer;
|
|
EGFR
|
epidermal growth factor receptor;
|
|
EpCAM
|
epithelial cell adhesion molecule;
|
|
FAP
|
familial adenomatous polyposis;
|
|
5-FU
|
5-fluorouracil;
|
|
HNPCC
|
hereditary non-polyposis colon
cancer;
|
|
IGF-1
|
insulin-like growth factor-1;
|
|
IL
|
interleukin;
|
|
JPS
|
juvenile polyposis syndrome;
|
|
MAPK
|
mitogen-activated protein kinase;
|
|
MMR
|
mismatch repair;
|
|
MSI
|
microsatellite instability;
|
|
MUTYH
|
mutY homologue;
|
|
MAP
|
MUTYH-associated polyposis;
|
|
PGE2
|
prostaglandin E2;
|
|
PI3K
|
phosphatidylinositol 3-kinase;
|
|
TGF-β
|
transforming growth factor-β;
|
|
VEGF
|
vascular endothelial growth factor
|
Acknowledgements
The study was supported by grants
from: Finalized project of the Apulia Region ‘Biotecnoter’; Italian
Association for Cancer Research; ‘Cassa di Risparmio di Puglia’
Foundation; University of Bari.
References
|
1.
|
American Cancer Society: Colorectal Cancer
Facts & Figures 2011–2013. American Cancer Society Inc.;
Atlanta, GA: 2011
|
|
2.
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 61:69–90. 2011. View Article : Google Scholar
|
|
3.
|
Conteduca V, Sansonno D, Ingravallo G,
Marangi S, Russi S, Lauletta G and Dammacco F: Barrett’s esophagus
and esophageal cancer: an overview. Int J Oncol. 41:414–424.
2012.
|
|
4.
|
Conteduca V, Sansonno D, Lauletta G, Russi
S, Ingravallo G and Dammacco F: H. pylori infection and
gastric cancer: state of the art (Review). Int J Oncol. 42:5–18.
2013.
|
|
5.
|
NCCN Clinical Practice Guidelines in
Oncology. Colorectal Cancer screening. 2013.
|
|
6.
|
Levine JS and Ahnen DJ: Adenomatous polyps
of the colon. N Engl J Med. 355:2551–2557. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Leggett B and Whitehall V: Role of the
serrated pathway in colorectal cancer pathogenesis.
Gastroenterology. 138:2088–2100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Fujita K, Yamamoto H, Matsumoto T, et al:
Sessile serrated adenoma with early neoplastic progression: a
clinicopathologic and molecular study. Am J Surg Pathol.
35:295–304. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Winawer SJ, Zauber AG, Fletcher RH, et al:
Guidelines for colonoscopy surveillance after polypectomy: a
consensus update by the US Multi Society Task Force on colorectal
cancer and the American Cancer Society. Gastroenterology.
130:1872–1885. 2006. View Article : Google Scholar
|
|
10.
|
Martínez ME, Baron JA, Lieberman DA, et
al: A pooled analysis of advanced colorectal neoplasia diagnoses
after colonoscopic polypectomy. Gastroenterology. 136:832–841.
2009.PubMed/NCBI
|
|
11.
|
Rizzo A, Pallone F, Monteleone G, et al:
Intestinal inflammation and colorectal cancer: a double-edged
sword? World J Gastroenterol. 17:3092–3100. 2011.PubMed/NCBI
|
|
12.
|
Butterworth AS, Higgins JP and Pharoah P:
Relative and absolute risk of colorectal cancer for individuals
with a family history: a meta-analysis. Eur J Cancer. 42:216–227.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Jasperson KW, Tuohy TM, Neklason DW, et
al: Hereditary and familial colon cancer. Gastroenterology.
138:2044–2058. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Lynch HT and de la Chapelle A: Hereditary
colorectal cancer. N Engl J Med. 348:919–932. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Stoffel E, Mukherjee B, Raymond VM, et al:
Calculation of risk of colorectal and endometrial cancer among
patients with Lynch syndrome. Gastroenterology. 137:1621–1627.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Galiatsatos P and Foulkes WD: Familial
adenomatous polyposis. Am J Gastroenterol. 101:385–398. 2006.
View Article : Google Scholar
|
|
17.
|
Lubbe SJ, Di Bernardo MC, Chandler IP, et
al: Clinical implications of the colorectal cancer risk associated
with MUTYH mutation. J Clin Oncol. 27:3975–3980. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
McGarrity TJ and Amos C: Peutz-Jeghers
syndrome: clinicopathology and molecular alterations. Cell Mol Life
Sci. 63:2135–2144. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Brosens LA, van Hattem A, Hylind LM, et
al: Risk of colorectal cancer in juvenile polyposis. Gut.
56:965–967. 2007. View Article : Google Scholar
|
|
20.
|
Boparai KS, Mathus-Vliegen EM, Koornstra
JJ, et al: Increased colorectal cancer risk during follow-up in
patients with hyper-plastic polyposis syndrome: a multicentre
cohort study. Gut. 59:1098–1100. 2010.PubMed/NCBI
|
|
21.
|
Alexander DD, Weed DL, Cushing CA, et al:
Meta-analysis of prospective studies of red meat consumption and
colorectal cancer. Eur J Cancer Prev. 20:293–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Park JY, Mitrou PN, Dahm CC, et al:
Baseline alcohol consumption, type of alcoholic beverage and risk
of colorectal cancer in the European Prospective Investigation into
Cancer and Nutrition-Norfolk study. Cancer Epidemiol. 33:347–354.
2009. View Article : Google Scholar
|
|
23.
|
Kant P and Hull MA: Excess body weight and
obesity - the link with gastrointestinal and hepatobiliary cancer.
Nat Rev Gastroenterol Hepatol. 8:224–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Giouleme O, Diamantidis MD and Katsaros
MG: Is diabetes a causal agent for colorectal cancer?
Pathophysiological and molecular mechanisms. World J Gastroenterol.
17:444–448. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Jin P, Lu XJ, Sheng JQ, et al: Estrogen
stimulates the expression of mismatch repair gene hMLH1 in colonic
epithelial cells. Cancer Prev Res. 3:910–916. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Ullman TA and Itzkowitz SH: Intestinal
inflammation and cancer. Gastroenterology. 140:1807–1816. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Gallimore AM and Godkin A: Epithelial
barriers, microbiota, and colorectal cancer. N Engl J Med.
368:32013. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Markowitz SD and Bertagnolli MM: Molecular
basis of colorectal cancer. N Engl J Med. 361:2449–2460. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Pino MS and Chung DC: The chromosomal
instability pathway in colon cancer. Gastroenterology.
138:2059–2072. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Hewish M, Lord CJ, Martin SA, et al:
Mismatch repair deficient colorectal cancer in the era of
personalized treatment. Nat Rev Clin Oncol. 7:197–208. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Ligtenberg MJ, Kuiper RD, Chan TL, et al:
Heritable somatic methylation and inactivation of MSH2 in families
with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat
Genet. 41:112–117. 2009.PubMed/NCBI
|
|
32.
|
Pritchard CC and Grady WM: Colorectal
cancer molecular biology moves into clinical practice. Gut.
60:116–129. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Weisenberger DJ, Siegmund KD, Campan M, et
al: CpG island methylator phenotype underlies sporadic
microsatellite instability and is tightly associated with BRAF
mutation in colorectal cancer. Nat Genet. 38:787–793. 2006.
View Article : Google Scholar
|
|
34.
|
Hinoue T, Weisenberger DJ, Pan F, et al:
Analysis of the association between CIMP and BRAF in colorectal
cancer by DNA methylation profiling. PLoS One. 4:e83572009.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Powell S, Zilz N, Beazer-Barclay Y, et al:
APC mutations occur early during colorectal tumorigenesis. Nature.
359:235–237. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Cottrell S, Bicknell D, Kaklamanis L, et
al: Molecular analysis of APC mutations in familial adenomatous
polyposis and sporadic colon carcinomas. Lancet. 340:626–630. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Nieuwenhuis MH and Vasen HF: Correlations
between mutation site in APC and phenotype of familial adenomatous
polyposis (FAP): a review of the literature. Crit Rev Oncol
Hematol. 61:153–161. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Rennert G, Almog R, Tomsho LP, et al:
Colorectal polyps in carriers of the APC I1307K polymorphism. Dis
Colon Rectum. 48:2317–2321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Kim YS and Deng G: Epigenetic changes
(aberrant DNA methylation) in colorectal neoplasia. Gut Liver.
1:1–11. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Goss KH and Groden J: Biology of the
adenomatous polyposis coli tumor suppressor. J Clin Oncol.
18:1967–1979. 2000.PubMed/NCBI
|
|
41.
|
Greenhough A, Smartt HJ, Moore AE, et al:
The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and
adaptation to the tumour microenvironment. Carcinogenesis.
30:377–386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Vogelstein B, Fearon ER, Hamilton SR, et
al: Genetic alterations during colorectal-tumor development. N Engl
J Med. 319:525–532. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Todaro M, Francipane MG, Medema JP, et al:
Colon cancer stem cells: promise of targeted therapy.
Gastroenterology. 138:2151–2162. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Zeki SS, Graham TA and Wright NA: Stem
cells and their implications for colorectal cancer. Nat Rev
Gastroenterol Hepatol. 8:90–100. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Rabeneck L, Paszat LF, Saskin R, et al:
Association between colonoscopy rates and colorectal cancer
mortality. Am J Gastroenterol. 105:1627–1632. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Levin B, Lieberman DA, McFarland B, et al:
Screening and surveillance for the early detection of colorectal
cancer and adenomatous polyps, 2008: a joint guideline from the
American Cancer Society, the US Multi-Society Task Force on
Colorectal Cancer, and the American College of Radiology.
Gastroenterology. 134:1570–1595. 2008. View Article : Google Scholar
|
|
47.
|
Johnson CD, Chen MH, Toledano AY, et al:
Accuracy of CT colonography for detection of large adenomas and
cancers. N Engl J Med. 359:1207–1217. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Laghi A, Iafrate F, Rengo M, et al:
Colorectal cancer screening: The role of CT colonography. World J
Gastroenterol. 16:3987–3994. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Pickhardt PJ and Kim DH: Performance of CT
colonography for detecting small, diminutive, and flat polyps.
Gastrointest Endosc Clin N Am. 20:209–226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Atkin WS, Edwards R, Kralj-Hans I, et al:
Once-only flexible sigmoidoscopy screening in prevention of
colorectal cancer: a multicentre randomised controlled trial.
Lancet. 375:1624–1633. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Hewitson P, Glasziou P, Watson E, et al:
Cochrane systematic review of colorectal cancer screening using the
fecal occult blood test (hemoccult): an update. Am J Gastroenterol.
103:1541–1549. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Vasen HF, Watson P, Mecklin JP, et al: New
clinical criteria for hereditary nonpolyposis colorectal cancer
(HNPCC, Lynch syndrome) proposed by the International Collaborative
group on HNPCC. Gastroenterology. 116:1453–1456. 1999. View Article : Google Scholar
|
|
53.
|
Umar A, Boland CR, Terdiman JP, et al:
Revised Bethesda guidelines for hereditary nonpolyposis colorectal
cancer (Lynch syndrome) and microsatellite instability. J Natl
Cancer Inst. 96:261–268. 2004. View Article : Google Scholar
|
|
54.
|
Berg AO, Armstrong K, Botkin J, et al:
Recommendations from the EGAPP Working Group: genetic testing
strategies in newly diagnosed individuals with colorectal cancer
aimed at reducing morbidity and mortality from Lynch syndrome in
relatives. Evaluation of Genomic Applications in Practice and
Prevention (EGAPP) Working Group. Genet Med. 11:35–41. 2009.
|
|
55.
|
Beggs AD, Latchford AR, Vasen HF, et al:
Peutz-Jeghers syndrome: a systematic review and recommendations for
management. Gut. 59:975–986. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Brosens LA, Langeveld D and van Hattem WA:
Juvenile polyposis syndrome. World J Gastroenterol. 17:4839–4844.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Imperiale TF, Ransohoff DF, Itzkowitz SH,
et al: Fecal DNA versus fecal occult blood for colorectal-cancer
screening in an average-risk population. N Engl J Med.
351:2704–2714. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Ahlquist DA: Next-generation stool DNA
testing: expanding the scope. Gastroenterology. 136:2068–2073.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Ahlquist DA, Sargent DJ, Loprinzi CL, et
al: Stool DNA and occult blood testing for screen detection of
colorectal neoplasia. Ann Intern Med. 149:441–450. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Van Gossum A, Munoz-Navas M,
Fernandez-Urien I, et al: Capsule endoscopy versus colonoscopy for
the detection of polyps and cancer. N Engl J Med. 361:264–270.
2009.
|
|
61.
|
Brenner H MD, Chang-Claude J, Seiler CM,
et al: Protection from colorectal cancer after colonoscopy. A
population-based, case-control study. Ann Intern Med. 154:22–30.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Ferrara F, Luigiano C, Ghersi S, et al:
Efficacy, safety and outcomes of ‘inject and cut’ endoscopic
mucosal resection for large sessile and flat colorectal polyps.
Digestion. 82:213–220. 2010.
|
|
63.
|
Kirkegaard H, Johnsen NF, Christensen J,
et al: Association of adherence to lifestyle recommendations and
risk of colorectal cancer: a prospective Danish cohort study. BMJ.
341:c55042010. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Manson JE, Mayne ST and Clinton SK:
Vitamin D and prevention of cancer-ready for prime time? N Engl J
Med. 364:1385–1387. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Park Y, Spiegelman D, Hunter DJ, et al:
Intakes of vitamins A, C, and E and use of multiple vitamin
supplements and risk of colon cancer: a pooled analysis of
prospective cohort studies. Cancer Causes Control. 21:1745–1757.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Kennedy DA, Stern SJ, Moretti M, et al:
Folate intake and the risk of colorectal cancer: a systematic
review and meta-analysis. Cancer Epidemiol. 35:2–10. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
67.
|
Chan AT: Aspirin and familial adenomatous
polyposis: coming full circle. Cancer Prev Res (Phila). 4:623–627.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
68.
|
Cole BF, Logan RF, Halabi S, et al:
Aspirin for chemoprevention of colorectal adenomas: meta-analysis
of the randomised trials. J Natl Cancer Inst. 101:256–266. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
69.
|
Tsujii M: Search for novel target
molecules for the effective treatment or prevention of colorectal
cancer. Digestion. 85:99–102. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70.
|
Rothwell PM, Wilson M, Elwin CE, et al:
Long-term effect of aspirin on colorectal cancer incidence and
mortality: 20-year follow-up of five randomised trials. Lancet.
376:1741–1750. 2010.PubMed/NCBI
|
|
71.
|
Long MD, Martin CF, Galanko JA, et al:
Hormone replacement therapy, oral contraceptive use and distal
large bowel cancer: a population-based case-control study. Am J
Gastroenterol. 105:1843–1850. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72.
|
Baron JA: Statins and the colorectum: hope
for chemoprevention? Cancer Prev Res. 3:573–575. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73.
|
Chan AT and Giovannucci EL: Primary
prevention of colorectal cancer. Gastroenterology. 138:2029–2043.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
74.
|
Chu E: An update on the current and
emerging targeted agents in metastatic colorectal cancer. Clin
Colorectal Cancer. 11:1–13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75.
|
Parry S, Win AK, Parry B, et al:
Metachronous colorectal cancer risk for mismatch repair gene
mutation carriers: the advantage of more extensive colon surgery.
Gut. 60:950–957. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
76.
|
Obermair A, Youlden DR, Young JP, et al:
Risk of endometrial cancer for women diagnosed with HNPCC-related
colorectal carcinoma. Int J Cancer. 127:2678–2684. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
77.
|
Arber N, Spicak J, Rácz I, et al:
Five-year analysis of the prevention of colorectal sporadic
adenomatous polyps trial. Am J Gastroenterol. 106:1135–1346.
2011.PubMed/NCBI
|
|
78.
|
Bolocan A, Ion D, Stoian RV, et al: MAP
syndrome (MYH associated polyposis) colorectal cancer,
etiopathological connections. J Med Life. 4:109–111.
2011.PubMed/NCBI
|















