Introduction
Cisplatin (CDDP, cis-diaminedichloroplatinum) is a
widely used chemotherapeutic agent for the management of gastric
cancers. Resistance to CDDP based chemotherapy is a major cause of
treatment failure. Chemotherapy resistance is a multifactorial
phenomenon of the molecular mechanisms, many of which are poorly
understood. One mechanism of resistance may be mediated through
enhanced anti-apoptotic activity (1). In general, the target for CDDP is
DNA, to which it binds efficiently to form a variety of monoadducts
and cross links, either between adjacent bases on the same strand
of DNA or on opposing strands (2,3).
These DNA lesions contribute to the cytotoxicity of CDDP through
blocking DNA replication and stimulating signals for apoptosis
(4). In the CDDP-induced
apoptosis, mitochondria play an important role. CDDP can induce MMP
(ΔΨm), which leads to cytochrome c release
to the cytoplasm and subsequent activation of caspases (5,6). In
this process, pro-apoptotic and anti-apoptotic proteins serve as
determinants of the cell fate. In the induction of pro-apoptotic
proteins by DNA-damage-induced signaling, the role of tumor
suppressor p53 is indisputable. Upregulation of proapoptotic
proteins such as Bax and PUMA is mediated by post-translational
modifications, such as phosphorylation and acetylation of the p53
protein. However, the wild-type p53 alone is not a direct predictor
of the chemotherapeutic response (6). Regarding CDDP resistance, activation
of the phosphatidylinositol-3-kinase/Akt pathway also plays an
important role in chemotherapy resistance by inducing
anti-apoptotic proteins. We are still confronting this resistance
problem in treating cancer patients even though much effort has
been devoted to solve it. In this study, we screen the sensitivity
of cancer cells to CDDP and then looked into the CDDP-induced
apoptotic process to investigate CDDP resistance.
Materials and methods
Cell line and cell culture
Three of the gastric cancer cell lines, SNU-1,
SNU-5, SNU-16 cells were obtained from the Laboratory of Cell
Biology at the Cancer Research Institute in Seoul National
University College of Medicine. They were cultured in RPMI-1640
supplemented with 10% FBS (Gibco-BRL, Carlsbad, CA, USA), 100 units
of penicillin and 100 μg/ml of streptomycin at 37°C in the
humidified atmosphere of 95% air and 5% CO2 in an
incubator. Molecular mass markers for proteins were obtained from
Pharmacia Biotech (Saclay, France). Antibodies against phospho-Akt
(Ser473), Akt 1/2/3, XIAP, Bcl-2 (N-19), p53, phospho-p70 S6 kinase
α (Thr389), and GFP were purchased from Santa Cruz Biotechnology
Inc. (Santa Cruz, CA, USA). Antibodies against phospho-Akt
(Thr308), and phospho-p53 (ser15) were purchased from Cell
Signaling Technology Inc. (Beverly, MA, USA). Antibody against
β-actin was from Sigma (Beverly, MA, USA). Peroxidase-labeled
donkey anti-rabbit and sheep anti-mouse immunoglobulin, and an
enhanced chemiluminescence (ECL) kit were purchased from Amersham
(Arlington Heights, IL, USA). All other chemicals not specifically
cited here were purchased from Sigma Chemical Co. (St. Louis, MO,
USA).
TdT-mediated dUTP nick end labeling
staining
TdT-mediated dUTP nick end labeling (TUNEL) staining
was conducted using an in situ cell death detection kit, TMR
Red, according to the protocol supplied by the manufacturer (Roche
Molecular Biochemicals, Mannheim, Germany). Briefly, cells were
plated in 25-cm2 flasks at a density of 2×105
cells/ml. The following day cells were treated with 0–50
μg/ml of CDDP, harvested and fixed with 2% paraformaldehyde
solution and permeabilized with 0.1% Triton X-100 in 0.1% sodium
citrate. After washing twice with PBS, cells were incubated in a
TUNEL reaction mixture containing terminal deoxynucleotidyl
transferase and tetramethyl-rhodamine-dUTP. Cells were analyzed for
fluorescence intensity using a FACS flow cytometer
(Becton-Dickinson, Mountain View, CA, USA). When necessary, pan
caspase inhibitor z-VAD-fmk (0–100 μg/ml), caspase-3/7
inhibitor (0–100 μg/ml), caspase-9 inhibitor (0–100
μg/ml), caspase-8 inhibitor (0–100 μg/ml)
(Calbiochem, Darmstadt, Germany) were applied 1 h prior to CDDP
treatment and were kept in the medium until the cells were
analyzed.
Cell line selection
The relative CDDP cytotoxicity of the three cell
lines was evaluated using an MTT colorimetric assay. Briefly,
SNU-1, SNU-5 and SNU-16 cells were plated in triplicate at
1.2×104 cells/well in 96-well culture plates with
RPMI-1640. The following day, the cells were treated with CDDP at
concentrations ranging from 0–50 μg/ml. Following treatment,
cell viability was determined by MTT assays. Absorbance at 600 nm
(OD600) was determined for each well using an ELX 808 automated
microplate reader (Bio-Tek Instrument Inc., Winooski, VT, USA).
Measurement of mitochondrial membrane
potential (MMP, ΔΨm) and reactive oxygen species
generation
Cells (5×105/ml) were exposed to 12
μg/ml of CDDP for 20 h. ROS scavenger N-acetyl-L-cysteine
(NAC) (0.5 mM) (Sigma Chemical Co.) was applied 1 h prior to CDDP
treatment, and the cells were kept in the medium until they were
analyzed. The cells were then washed with PBS and harvested by
trypsinization. Early apoptosis was detected by Annexin V stain and
measured using a FACS flow cytometer (Becton-Dickinson, San Jose,
CA, USA). SNU-1 and SNU-16 cells were treated with the same
condition with CDDP. After exposure to CDDP, the cells were
incubated with 10 μM 2′,7′-dichlorofluorescein diacetate
(DCF-DA) for ROS levels and 30 nM 3′,3′-dihexyloxacarboxyanine
iodide [DiOC6(3)] (Sigma Chemical Co.) for
MMP(ΔΨm) at 37°C for 30 min. The cells were then
washed with ice-cold PBS and harvested by trypsinization.
Fluorescence was determined using a FACS flow cytometer.
Western blot analysis
Cells were washed twice with cold PBS, and total
cell lysates were obtained using lysis buffer containing 0.5% SDS,
1% NP-40, 1% sodium deoxycholate, 150 mM NaCl, 50 mM Tris-Cl (pH
7.5), and protease inhibitors. The concentrations of cell lysate
proteins were determined by means of the Bradford protein assay
(Bio-Rad Laboratories, Richmond, CA, USA) using bovine serum
albumin as the standard. Molecular mass markers for proteins were
obtained from Pharmacia Biotech. For western blot analysis, protein
(30 μg) was resolved by electrophoresis, electrotransferred
to polyvinylidene difluoride membranes (Millipore, Bedford, MA,
USA), and then incubated with primary antibodies followed by
incubation with a secondary antibody conjugated to peroxidase.
Blots were developed with an ECL detection system. Autoradiography
film was exposed at multiple time points to obtain the best
images.
Results
SNU-16 cells are relatively resistant to
CDDP whereas SNU-1 cells are sensitive and the difference is
derived from the difference in apoptosis
To investigate CDDP-induced apoptosis in gastric
cancer cells, we examined the CDDP sensitivity through MTT assay of
the three cell lines (SNU-1, SNU-5, SNU-16 cells). As shown in
Fig. 1A and B, the growth of the
three cell lines was inhibited by CDDP treatment in a
dose-dependent manner, and IC50 for the 24 h CDDP
treatment was less than 10 μg/ml in SNU-1 cells whereas in
SNU-16 cells IC50 was greater than 20 μg/ml.
SNU-16 was the most resistant to CDDP. To determine whether the
decrease in viability was related to apoptosis, we performed a
TUNEL assay. The discrepancy in the degree of apoptosis became
apparent after 12 h of treatment (Fig.
1C). These findings suggest that SNU-16 cells are relatively
resistant to CDDP whereas SNU-1 cells are sensitive and that the
difference is derived from the difference in apoptosis between the
two cell lines.
CDDP treatment induces caspase-dependent
apoptosis in SNU-1 cells
To confirm that CDDP-induced apoptosis was
caspase-dependent, we performed MTT assay with caspase inhibitors
(z-VAD-FMK, Z-LEHD-FMK, Z-IETD-FMK and Z-DEVD-FMK). The SNU-1 cells
were exposed to only 12 μg/ml of CDDP for 20 h which caused
70% apoptosis in the SNU-1 cells. These inhibitors significantly
suppressed the apoptosis induced by CDDP (Fig. 2). This result suggests that CDDP
induced-apoptosis is caspase-dependent.
Loss of mitochondrial membrane potential
[MMP(ΔΨm)] is critical in CDDP induced apoptosis in
SNU-1 and SNU-16 cells
Oxidative damage plays an important role in
CDDP-induced apoptosis (7). To
investigate the causes of the difference in sensitivity to CDDP
between SNU-1 cells and SNU-16 cells, we compared intracellular ROS
level in SNU-1 and SNU-16 cells after CDDP treatment. CDDP (12
μg/ml for 20 h) increased the ROS level in SNU-16 cells
(data not shown). Many SNU-1 cells were dead at the time of
measurement (data not shown). Hence we assessed the effects of the
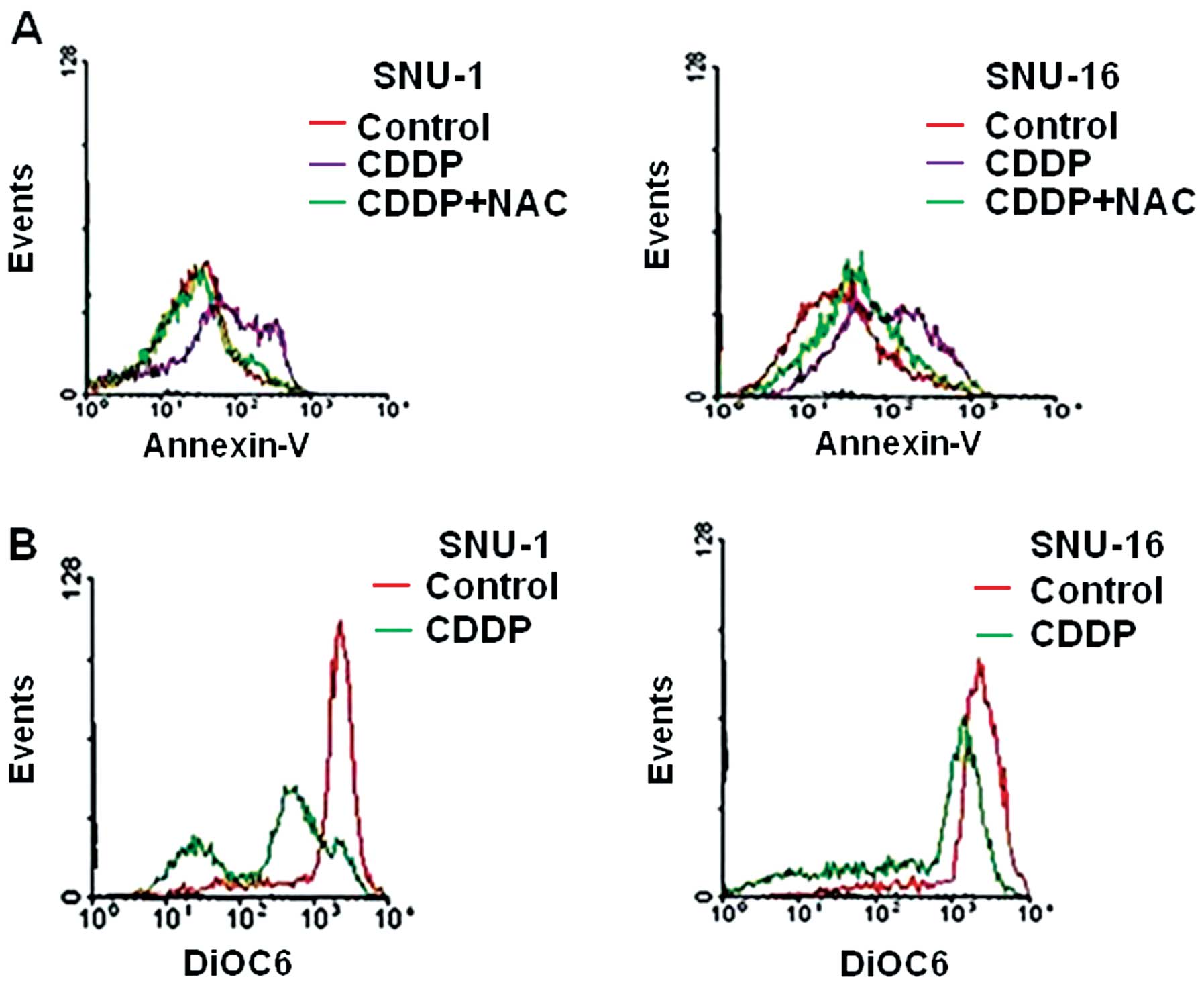
ROS scavenger, N-acetyl-L-cysteine (NAC) on CDDP-treated cells. The
effects were different; NAC treatment significantly reduced
apoptotic cell death of SNU-1 cells, but not in SNU-16 cells
(Fig. 3A). We also compared the
loss of MMP (ΔΨm) induced by CDDP between SNU-1
and SNU-16 cells. The loss of MMP level was significantly higher in
SNU-1 cells than in SNU-16 cells (Fig.
3B). These findings indicated that CDDP-induced reactive oxygen
species (ROS) generation significantly induced loss of MMP
(ΔΨm) in SNU-1 cells, but not in SNU-16 cells,
suggesting that the loss of MMP (ΔΨm) may
determine the level of apoptotic cell death.
The ratio of Bax to Bcl-2 protein is
different in CDDP-induced apoptosis between SNU-1 and SNU-16
cells
We investigated further proteins regulating MMP
(ΔΨm) such as Bcl-2 and Bax proteins. Western
blot analysis revealed that Bax protein level of SNU-1 was
significantly increased by CDDP compared to SNU-16 cells (Fig. 4A), while Bcl-2 protein expression
in SNU-1 treated by CDDP was significantly suppressed compared to
that in SNU-16 cells (Fig. 4B).
Hence the ratio of Bax to Bcl-2 protein increased after CDDP
treatment in SNU-1 cells, but not in SNU-16 cells. This result
suggests that the ratio of pro-apoptotic drive to anti-apoptotic
drive such as the ratio of Bax to Bcl-2 protein may determine the
difference in CDDP-induced apoptosis between SNU-1 and SNU-16
cells.
CDDP increased Akt phosphorylation but
the induced Akt activity did not prevent phosporylation of p53 in
SNU-16 cells
Increased Akt activity promotes CDDP resistance by
inhibiting pro-apoptotic drive as well as augmenting anti-apoptotic
drive (8–10). First, we assessed Akt expression in
SNU16 cells treated with CDDP over the time frame. Western blot
analysis revealed that CDDP increased Akt activity (Fig. 5A). Increased Akt activity promotes
CDDP resistance in cancer cells through inhibition of p53
phosphorylation and transcriptional activity (10). In p53-functioning cells, the
phosphorylation of p53 has also been reported to be an independent
determinant of transcriptional upregulation of pro-apoptotic
proteins such as Bax and PUMA in CDDP-induced apoptosis. However,
Bax proteins can be upregulated by other transcriptional factors
(11). Hence, we examined the
expression of p53 or p-p53 levels. CDDP increased the expression of
p53 and p-p53 (Ser15) in both SNU-1 and SNU-16 cells (Fig. 5B). This result suggests that
phosporylated p53 should not induce Bax protein in SNU-16 cells,
which means that SNU-16 cells should be p53-mutant or
p53-non-functioning cancer cells; the results also confirmed that
phosporylated p53 (Ser15) is an independent determinant for
CDDP-induced apoptosis only in p53 functioning cancer cells. These
findings suggest that Bax induction by CDDP in SNU-1 cells may be
derived from p53 activation, and that increased Akt may not
significantly suppress CDDP-induced phosphorylation of p53 in
SNU-16 cells.
PI3K/Akt inhibition enhanced CDDP-induced
apoptosis by suppression of anti-apoptotic proteins, but the
efficacy was minimal
To augment loss of MMP, ΔΨm in
SNU-16 by suppressing anti-apoptotic activity, and to investigate
the role of PI3K/Akt pathway in CDDP-induced apoptosis in SNU-16
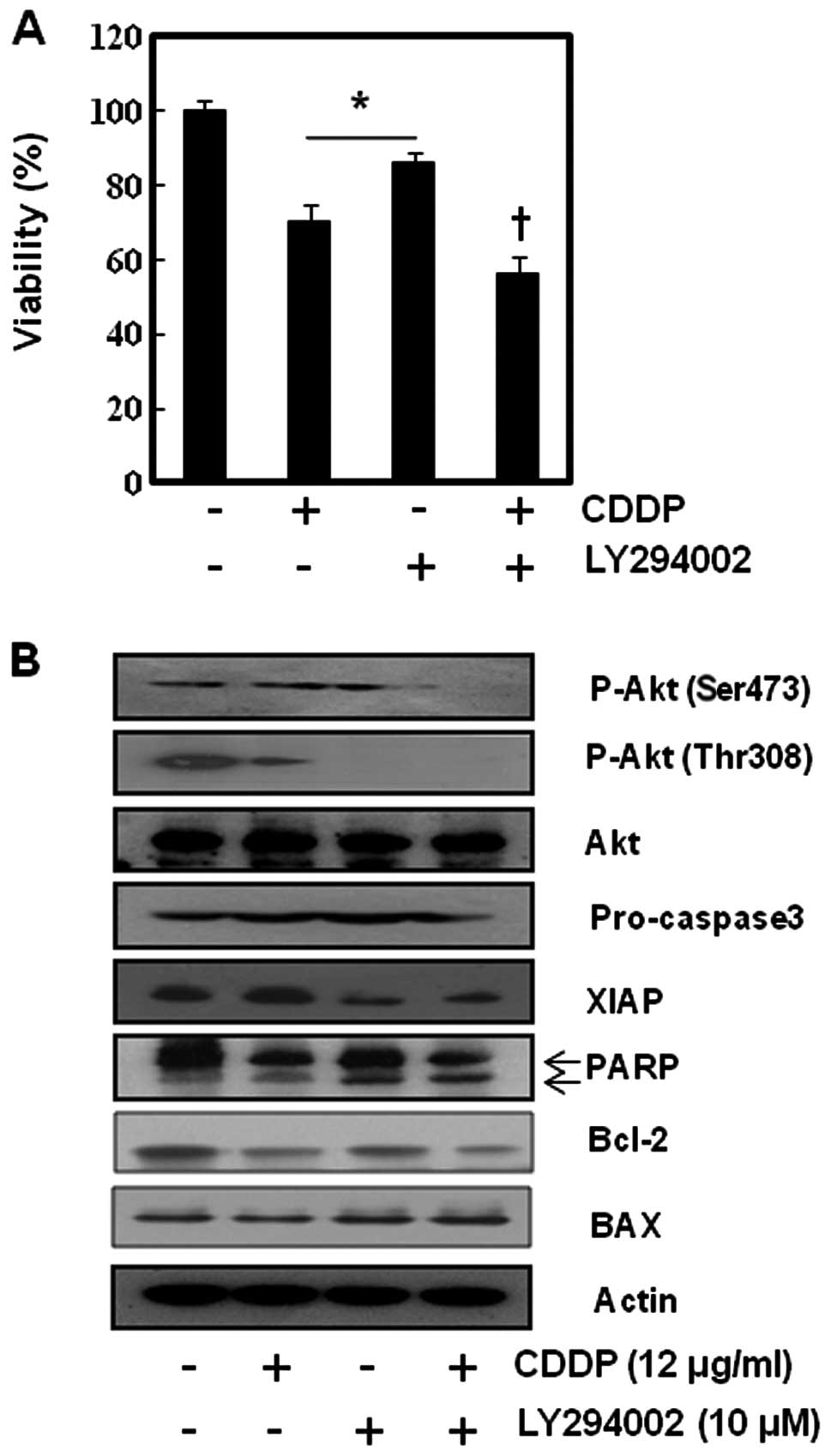
cells we inhibited the Akt activity of SNU-16 cells using LY294002,
a representative PI3k/Akt inhibitor. As shown in Fig. 6, LY294002 slightly accentuated the
cytotoxicity of CDDP in MMT assay and no synergism was observed
between LY294002 (PI3k/Akt inhibitor) and CDDP in SNU-16 cells. To
confirm this finding at the molecular level, we performed western
blot analysis for apoptosis-related factors and p-Akt. The
suppression of Akt phosphorylation led to inhibition of XIAP and
activation of apoptosis-related enzyme (PARP and caspase 3). We
found that LY294002 enhanced the cytotoxicity of CDDP by
suppressing XIAP. This result suggests that inhibition of Akt may
not significantly enhance the sensitivity of CDDP in SNU-16 cells
even though CDDP induces Akt activation.
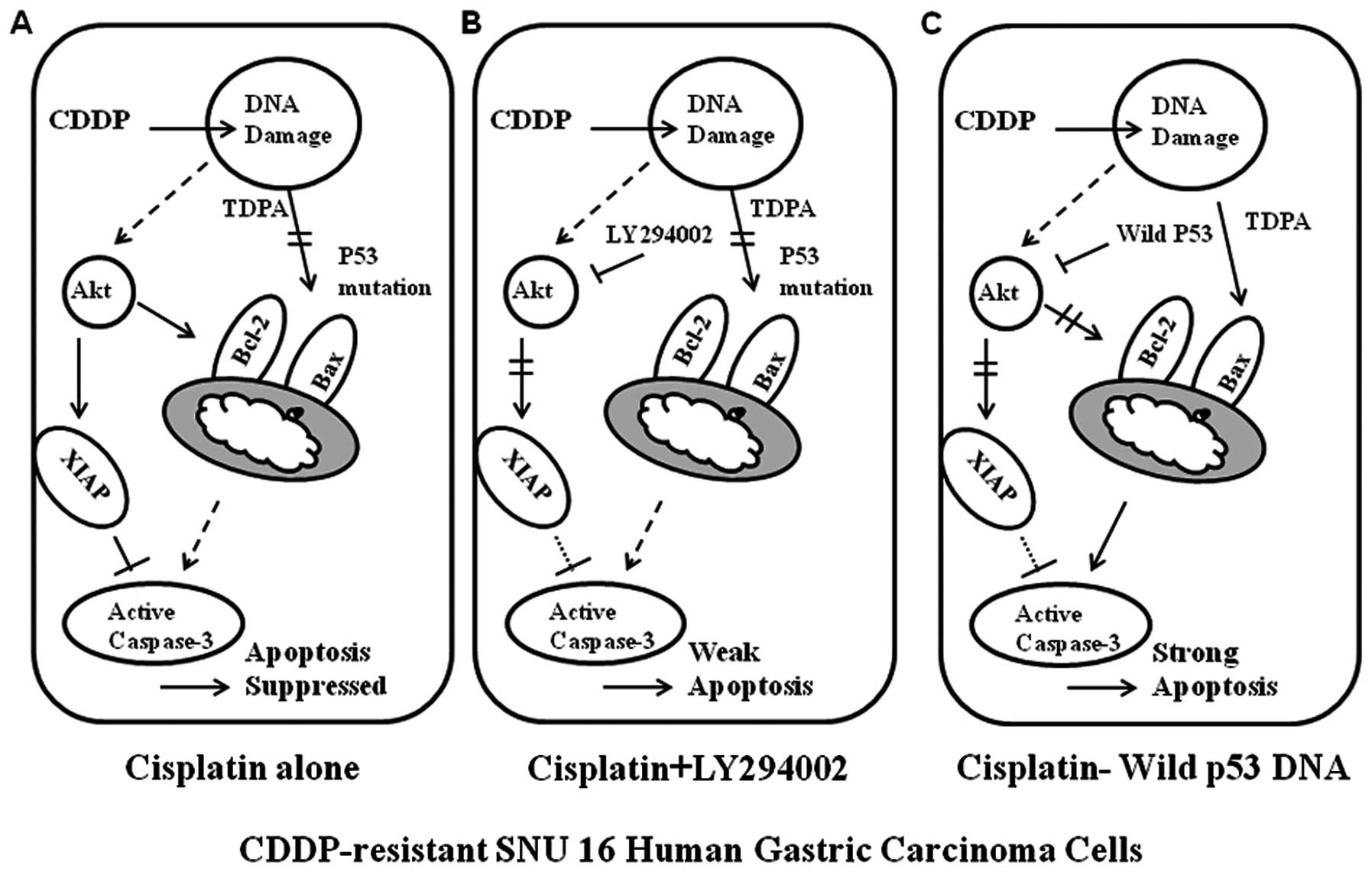
The resistance to CDDP of SNU-16 cells
can be overcome by p53 augmentation through inhibition of Akt as
well as induction of Bax
We assumed that the reason why PI3k/Akt inhibitor
did not induce synergism with CDDP is the lack of the proapoptotic
drive due to loss of p53 function. Therefore, we transfected SNU-16
cells with wild-type p53, and then tested the sensitivity to CDDP.
MTT assay revealed that in the SNU-16 cells transfected with
wild-type p53, the sensitivity to CDDP was significantly enhanced
(Fig. 6A). To confirm this finding
at the molecular level, we performed western blot analysis for p53
and downstream molecules of PI3k/Akt pathway. Successful
transfection with wild-type p53 was confirmed with GFP.
Interestingly, the transfection of wild-type p53 alone did not
significantly influence phosphorylation of p70S6K, or of Akt as
well as Bax induction. However, addition of CDDP on the
p53-trasfected cells not only augmented Bax induction but also
suppressed Bcl-2 through inhibition of Akt phosphorylation. These
results suggest that restoration of p53 function can overcome the
resistance to CDDP by augmenting the proapoptotic drive through
p53-mediated transcriptional activation and by inhibiting the
anti-apoptotic drive through inhibition of Akt activity (Figs. 7 and 8).
Discussion
Our study was designed to demonstrate the difference
in apoptotic processes in CDDP-induced apoptosis between
constitutively CDDP-sensitive and CDDP-resistant gastric cancer
cells in vitro, and to find a method to overcome the
resistance to CDDP. SNU-16 cells are the most resistant to CDDP and
SNU-1 the most sensitive among the 3 cell lines (SNU-1, SNU-5 and
SNU-16). The major contributor to the big difference in
CDDP-induced cell death between SNU-1 cells and SNU-16 cells was
loss of MMP (ΔΨm). The loss of MMP
(ΔΨm) is one of the main events of the apoptotic
process induced by chemotherapeutic drugs (12,13),
and this results in either caspase-dependent or independent
apoptosis (14,15). In this study, significant loss of
MMP (ΔΨm) by CDDP treatment was observed in SNU-1
cells, but not in SNU-16 cells. This finding indicates that lack of
the proapoptotic drive or a surplus of anti-apoptotic drive induced
the failure of CDDP in inducing loss of MMP (ΔΨm)
in SNU-16 cells. Our data indicated that SNU-16 cells were
p53-non-functioning cells. Actually, SNU-16 cells have a missense
mutation of codon 205, TAT to TTT, Tyr to Phe (16). This area belongs to the p53 DNA
binding domain (17). This is
consistent with our findings. Here, we also tested whether an Akt
inhibitor can enhance the CDDP sensitivity in p53 mutation cancer
cells by suppressing the anti-apoptotic proteins because a previous
study suggested that the mutation status of p53 might not predict
the chemo-response, and that Akt activation might be involved in
CDDP resistance. Unlike the previous report, inhibition of PI3K/Akt
pathway was not adequate to overcome CDDP resistance in SNU-16
cells. We also tested whether the anthocyanins enhanced CDDP
sensitivity, because it has been reported that anthocyanins
isolated from Vitis coignetiae Pulliat can enhance apoptosis
by suppressing anti-apoptotic proteins such as Bcl-2, and XIAP
through the inhibition of Akt and NF-κB that are involved in drug
resistance (18,19). Similar to the results of LY294002,
the anthocyanins also slightly enhanced the CDDP sensitivity of SNU
16 cells, but they did not show a clear synergism (data not
shown).
Here, we demonstrated that the restoration of p53
functions in SNU-16 cells enhancing CDDP-induced apoptosis not only
by inducing apoptotic factors through p53-mediated transcriptional
activation but also by inhibiting anti-apoptotic proteins through
inhibition of Akt activity. This finding was also observed in
wild-type SNU-1 cells (Fig. 4);
CDDP not only augmented Bax expression but also suppressed Bcl-2
expression in SNU-1 cells. This can be explained by previous
studies that suggested that DNA damage can activate pTEN through
p53 activation followed by inhibition of Akt (20,21).
Our data encourage the use of gene therapy with wild-type p53 in
cancer treatment. This result is supported by the successful
results of p53 gene therapy in combination with CDDP in in
vitro and xenograft cancer models, and in the patients with
small cell lung cancer (22,23).
However, there is still controversy surrounding p53 gene therapy
because there are also negative results showing no additional
benefit with combination therapy (24).
The limitation of this study is that we only
compared three gastric cell lines and validated the role of p53
restoration only in SNU-16 cells. In addition, there may be many
other ways to enhance CDDP sensitivity. Therefore, these issues
will require investigation. To reveal the clinical significance, an
in vivo study followed by a clinical trial is warranted. In
conclusion, this study suggests that the primary contributor to
resistance to CDDP in SNU-16 cells may well be a failure of
induction of apoptosis due to lack of induction of proapoptotic
activities rather than an increase in anti-apoptotic activity, and
that restoration of p53 function can overcome the resistance to
CDDP not only by augmenting the proapoptotic drive through
p53-mediated transcriptional activation but also by inhibiting the
anti-apoptotic drive through inhibition of Akt activity. This study
supports that the restoration of p53 is still important in
CDDP-induced apoptosis in p53 mutant SNU-16 human gastric cancer
cells.
Acknowledgements
This study was supported by a grant
from the National R&D Program for Cancer Control, Ministry for
Health, Welfare and Family Affairs, Republic of Korea (0820050),
and from Priority Research Center Program through the National
Research Foundation of Korea (NRF) funded by the Ministry of
Education, Science and Technology (2010-0029621).
References
|
1.
|
Li J, Feng Q, Kim JM, Schneiderman D,
Liston P, Li M, Vanderhyden B, Faught W, Fung MF, Senterman M,
Korneluk RG and Tsang BK: Human ovarian cancer and cisplatin
resistance: possible role of inhibitor of apoptosis proteins.
Endocrinology. 142:370–380. 2001.PubMed/NCBI
|
|
2.
|
Zwelling LA, Anderson T and Kohn KW:
DNA-protein and DNA interstrand cross-linking by cis- and
trans-platinum(II) diamminedichloride in L1210 mouse leukemia cells
and relation to cytotoxicity. Cancer Res. 39:365–369.
1979.PubMed/NCBI
|
|
3.
|
Fichtinger-Schepman AM, van der Veer JL,
den Hartog JH, Lohman PH and Reedijk J: Adducts of the antitumor
drug cis-diamminedichloroplatinum(II) with DNA: formation,
identification, and quantitation. Biochemistry. 24:707–713. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Bottone MG, Soldani C, Veneroni P, Avella
D, Pisu M and Bernocchi G: Cell proliferation, apoptosis and
mitochondrial damage in rat B50 neuronal cells after cisplatin
treatment. Cell Prolif. 41:506–520. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Melendez-Zajgla J, Cruz E, Maldonado V and
Espinoza AM: Mitochondrial changes during the apoptotic process of
HeLa cells exposed to cisplatin. Biochem Mol Biol Int. 47:765–771.
1999.PubMed/NCBI
|
|
6.
|
Henkels KM and Turchi JJ:
Cisplatin-induced apoptosis proceeds by caspase-3-dependent and
-independent pathways in cisplatin-resistant and -sensitive human
ovarian cancer cell lines. Cancer Res. 59:3077–3083.
1999.PubMed/NCBI
|
|
7.
|
Miyajima A, Nakashima J, Yoshioka K,
Tachibana M, Tazaki H and Murai M: Role of reactive oxygen species
in cis-dichlorodiammineplatinum-induced cytotoxicity on bladder
cancer cells. Br J Cancer. 76:206–210. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Pugazhenthi S, Nesterova A, Sable C,
Heidenreich KA, Boxer LM, Heasley LE and Reusch JE: Akt/protein
kinase B up-regulates Bcl-2 expression through cAMP-response
element-binding protein. J Biol Chem. 275:10761–10766. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Mitsiades CS, Mitsiades N, Poulaki V,
Schlossman R, Akiyama M, Chauhan D, Hideshima T, Treon SP, Munshi
NC, Richardson PG and Anderson KC: Activation of NF-kappaB and
upregulation of intracellular anti-apoptotic proteins via the
IGF-1/Akt signaling in human multiple myeloma cells: therapeutic
implications. Oncogene. 21:5673–5683. 2002. View Article : Google Scholar
|
|
10.
|
Fraser M, Bai T and Tsang BK: Akt promotes
cisplatin resistance in human ovarian cancer cells through
inhibition of p53 phosphorylation and nuclear function. Int J
Cancer. 122:534–546. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Chipuk JE and Green DR: Dissecting
p53-dependent apoptosis. Cell Death Differ. 13:994–1002. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Crompton M: The mitochondrial permeability
transition pore and its role in cell death. Biochem J. 341:233–249.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Zamzami N and Kroemer G: The mitochondrion
in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol.
2:67–71. 2001.PubMed/NCBI
|
|
16.
|
Ku JL and Park JG: Biology of SNU cell
lines. Cancer Res Treat. 37:1–19. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Somasundaram K: Tumor suppressor p53:
regulation and function. Front Biosci. 5:D424–D437. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Shin DY, Lee WS, Lu JN, Kang MH, Ryu CH,
Kim GY, Kang HS, Shin SC and Choi YH: Induction of apoptosis in
human colon cancer HCT-116 cells by anthocyanins through
suppression of Akt and activation of p38-MAPK. Int J Oncol.
35:1499–1504. 2009.PubMed/NCBI
|
|
19.
|
Yun JW, Lee WS, Kim MJ, Lu JN, Kang MH,
Kim HG, Kim DC, Choi EJ, Choi JY, Lee YK, Ryu CH, Kim G, Choi YH,
Park OJ and Shin SC: Characterization of a profile of the
anthocyanins isolated from Vitis coignetiae Pulliat and
their anti-invasive activity on HT-29 human colon cancer cells.
Food Chem Toxicol. 48:903–909. 2010.PubMed/NCBI
|
|
20.
|
Stambolic V, MacPherson D, Sas D, Lin Y,
Snow B, Jang Y, Benchimol S and Mak TW: Regulation of PTEN
transcription by p53. Mol Cell. 8:317–325. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Feng Z: p53 regulation of the
IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring
Harb Perspect Biol. 2:a0010572010. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Wang WD, Li R, Chen ZT, Li DZ, Duan YZ and
Cao ZH: Cisplatin-controlled p53 gene therapy for human non-small
cell lung cancer xenografts in athymic nude mice via the CArG
elements. Cancer Sci. 96:706–712. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Antonia SJ, Mirza N, Fricke I, Chiappori
A, Thompson P, Williams N, Bepler G, Simon G, Janssen W, Lee JH,
Menander K, Chada S and Gabrilovich DI: Combination of p53 cancer
vaccine with chemotherapy in patients with extensive stage small
cell lung cancer. Clin Cancer Res. 12:878–887. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Guan YS, Liu Y, Zou Q, He Q, La Z, Yang L
and Hu Y: Adenovirus-mediated wild-type p53 gene transfer in
combination with bronchial arterial infusion for treatment of
advanced non-small-cell lung cancer, one year follow-up. J Zhejiang
Univ Sci B. 10:331–340. 2009.PubMed/NCBI
|