Introduction
Colorectal cancer (CRC) is one of the most common
human malignant cancers with over one million new cases and more
than a half million deaths around the world each year (1). To date, chemotherapy remains one of
the main therapeutic approaches for advanced CRC; and
5-fluorouracil (5-FU)-based regimens are considered as standard
chemotherapeutics (2,3). However, due to drug resistance and
toxicity against normal tissues, systemic chemotherapy using
5-FU-based regimens produces objective response rates of <10–20%
(2,4–6).
These problems limit the effectiveness of current CRC chemotherapy,
highlighting the urgent need for the development of novel antitumor
agents.
Angiogenesis, a process involving the growth of new
blood vessels from the pre-existing vasculature (7,8), is
essential in various human biological processes including wound
healing, reproduction and embryonic development (9–11).
However, dyregulation of angiogenesis also plays a critical role in
the development of solid tumors, supplying nutrients and oxygen to
support continuous growth of tumor as well as providing an avenue
for hematogenous metastasis (11–13).
Tumor angiogenesis is highly regulated by multiple intracellular
signaling transduction cascades such as Hedgehog, signal transducer
and activator of transcription 3 (STAT3), Akt and p70S6 kinase
(p70S6K) pathways that are known to malfunction in many types of
cancer. The Hedgehog (HH) signaling pathway is important for
embryonic development (14); its
aberrant activation has been associated with many human cancers
including CRC (15–19). Mammals have three Hedgehog
homologues (Sonic Hedgehog, Indian Hedgehog and Desert Hedgehog),
of which Sonic Hedgehog (SHH) is the best studied. Activation of HH
signaling is initiated by binding of Hh to the trans-membrane
receptor Patched (Ptch). This results in the release of
Ptch-mediated suppression of Smoothened (Smo). Smo subsequently
activates the Gli family of transcription factors that regulate the
expression of various angiogenic mediators promoting angiogenesis
(20–23). STAT3 plays an essential role in
cell survival, proliferation and angiogenesis (24). After activation via
phosphorylation, STAT3 proteins in the cytoplasm dimerize and
translocate to the nucleus where they regulate the expression of
critical genes involved in cancer progression. Constitutive
activation of STAT3 is strongly associated with cancer development
and commonly suggests a poor prognosis (25,26).
PI3K-dependent Akt pathway is essential for cell proliferation and
survival and has been shown to be activated in several cancer types
(27–29). After activation by extracellular
stimuli, PI3K is able to phosphorylate PI(4)P and PI(4,5) P2
to generate PI(3,4)P2 and PI(3,4,5)P3,
respectively. These lipids serve as plasma membrane docking sites
for proteins containing pleckstrin-homology (PH) domains, such as
Akt and its upstream activator 3-phosphoinositide-dependent protein
kinase-1 (PDK1). The colocalization of PDK1 and Akt in plasma
membrane results in the phosphorylation of Akt, which in turn
activates mTOR (mammalian target of rapamycin) leading to the
phosphorylation/activation of p70S6K. The Akt-mTOR-p70S6K signaling
pathway is considered as a central regulatory pathway involved in
the regulation of cell proliferation, differentiation, survival and
angiogenesis (30–33). Therefore, inhibition of
angiogenesis via modulation of these signalings has become a major
focus for anticancer drug development.
Due to drug resistance and cytotoxicity of
currently-used chemotherapies, natural products, including
traditional Chinese medicine (TCM), have received great interest
since they have relatively few side-effects as compared to modern
chemotherapeutics and have been used for thousands of years as
important alternative remedies for various diseases including
cancer (34,35). Thus, identifying naturally
occurring agents is a promising approach for anticancer treatment.
Ursolic acid (UA), a pentacyclic triterpene acid, is a biologically
active compound present in many traditional Chinese medicinal
herbs, such as Hedyotic diffusa, Spica prunellae, Patrinia
scabiosaefolia and Scutellaria barbata that have long
been used in China for the clinical treatment of CRC (36–39).
Previous studies report that UA exhibits a broad range of
pharmacological properties such as anti-inflammatory, antiviral,
antioxidant, hepatoprotective, cytotoxic, antitumor,
anti-angiogenesis, and anti-metastatic activities (40). Recent studies have shown that UA
inhibits the proliferation and induces apoptosis and/inhibits
proliferation of colon carcinoma cells (13,41–43).
In order to further elucidate the mechanism of tumorcidal activity
of UA, in the present study we evaluated its effect on CRC growth
and angiogenesis in vivo and in vitro, and
investigated the underlying molecular mechanisms of its action.
Materials and methods
Materials and reagents
Usolic acid (UA) was purchased from Sigma-Aldrich
Chemical (St. Louis, MO, USA). Matrigel was provided by
Becton-Dickinson (San Jose, CA, USA). Roswell Park Memorial
Institute medium-1640 (RPMI-1640), Dulbecco’s modified Eagle’s
medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin,
trypsin-EDTA and TRIzol reagent were purchased from Invitrogen
(Carlsbad, CA, USA). SuperScript II reverse transcriptase was
obtained from Promega (Madison, WI, USA). The In Vitro
Angiogenesis Assay kit was purchased from Millipore (Billerica, MA,
USA). Human VEGF-A and bFGF ELISA kits were obtained from Shanghai
Xitang Biological Technology Ltd. (Shanghai, China). All antibodies
were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
BCA Protein assay kit was purchased from Tiangen Biotech (Beijing)
Co., Ltd. Bio-Plex phosphoprotein assay kits were purchased from
Bio-Rad (Hercules, CA, USA). All other chemicals, unless otherwise
stated, were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
Human colon carcinoma HT-29 cells were obtained from
the Cell Bank of Chinese Academy of Sciences (Shanghai, China).
Human umbilical vein endothelial cells (HUVECs) were purchased from
Xiangya Cell Center, University of Zhongnan (Hunan, China). HT-29
cells and HUVECs were grown in DMEM and RPMI-1640, respectively.
Both DMEM and RPMI-1640 were supplemented with 10% (v/v) FBS, 100
U/ml penicillin, and 100 μg/ml streptomycin. All cell lines
were cultured at 37°C, 5% CO2 and in a humidified
environment.
Animals
Male BALB/c athymic (nude) mice (with an initial
body weight of 20–22 g) were obtained from Shanghai SLAC Laboratory
Animal Co., Ltd. (Shanghai, China) and housed under pathogen-free
conditions with controlled temperature (22°C), humidity, and a 12-h
light/dark cycle. Food and water were given ad libitum
throughout the experiment. All animal treatments were performed
strictly in accordance with international ethical guidelines and
the National Institutes of Health Guide concerning the Care and Use
of Laboratory Animals. The experiments were approved by the
Institutional Animal Care and Use Committee of Fujian University of
Traditional Chinese Medicine.
In vivo nude mouse xenograft study
CRC xenograft mice were produced with HT-29 cells.
The cells were grown in culture and then detached by
trypsinization, washed, and resuspended in serum-free DMEM.
Resuspended cells (1.5×106) mixed with Matrigel (1:1)
were subcutaneously injected into the right flank of mice to
initiate tumor growth. At 5 days following xenograft implantation
(tumor size ∼3 mm in diameter), mice were randomized into two
groups (n=10) and treated with UA (dissolved in PBS) 12.5 mg/kg or
saline by daily intraperitoneal injection, 6 days a week for 16
days. Body weight and tumor size were measured. Tumor size was
determined by measuring the major (L) and minor (W) diameter with a
caliper. The tumor volume was calculated according to the following
formula: tumor volume = π/6 × L × W2. At the end of the
experiment, the animals were anaesthetized with pelltobarbitalum
natricum, and tumors were excised. A portion of each tumor was
fixed in 10% buffered formalin and the remaining tissue snap-frozen
in liquid nitrogen and stored at −80°C.
Immunohistochemistical analysis of CRC
tumor tissues
Six tumors were randomly selected from UA-treatment
or control groups. Tumor tissues were fixed in 10% formaldehyde for
12 h, paraffin-embedded, sectioned and placed on slides. The slides
were subjected to antigen retrieval and endogenous peroxidase
activity was quenched with hydrogen peroxide. Non-specific binding
was blocked with normal serum in PBS (0.1% Tween-20). Rabbit
polyclonal antibodies against CD31, SHH, Gli-1, VEGF-A and bFGF
(all in 1:200 dilution, Santa Cruz Biotechnology) were used to
detect the relevant proteins. The binding of the primary antibody
was demonstrated with a biotinylated secondary antibody,
horseradish peroxidase (HRP)-conjugated streptavidin (Dako) and
diaminobenzidine (DAB) as the chromogen. The tissues were
counterstained with diluted Harris hematoxylin. After staining,
five high-power fields (at magnification of ×400) were randomly
selected in each slide. The proportion of positive cells in each
field was determined using the true color multi-functional cell
image analysis management system (Image-Pro Plus, Media
Cybernetics, USA). To control for non-specific staining, PBS was
used to replace the primary antibody as a negative control.
Chick chorioallantoic membrane (CAM)
assay
A CAM assay was performed to determine the in
vivo anti-angiogenic activity of UA. Briefly, 10 μl of
UA (25 μg/μl) was loaded onto a 0.5-cm diameter
Whatman filter paper. The filter was then applied to the CAM of a
7-day embryo. After incubation for 72 h at 37°C, the region
surrounding the filter was photographed with a digital camera. The
number of blood vessels was quantified manually in a circular
perimeter surrounding the implants, at a distance of 0.25 cm from
the edge of the filter. Assays were performed twice with a final
total of 10 eggs for each data point.
Cell viability evaluation by MTT
assay
UA was dissolved in DMSO and diluted to working
concentrations with culture medium. The final concentration of DMSO
in the medium for all cell-based experiments was 0.1% HUVECs were
seeded into 96-well plates at a density of 1.0×104
cells/well in 0.1 ml medium and incubated at 37°C under a 5%
CO2 for 24 h. The cells were treated with various
concentrations of UA for 24 h or with 40 μM of UA for
different periods of time. Treatment with 0.1% DMSO was included as
the vehicle control. At the end of the treatment, 10 μl MTT
[5 mg/ml in phosphate buffered saline (PBS)] were added to each
well, and the samples were incubated for an additional 4 h at 37°C.
The purple-blue MTT formazan precipitate was dissolved in 100
μl DMSO. Absorbance was measured at 570 nm using an ELISA
reader (BioTek, Model EXL800, USA).
Migration assay of HUVECs
Migration of HUVECs was performed by the wound
healing method. HUVECs were seeded into 12-well plates at a density
of 2×105 cells/well in 1 ml medium. After 24 h of
incubation, cells were scraped away vertically in each well using a
P100 pipette tip. Three randomly selected views along the scraped
line were photographed in each well using phase-contrast inverted
microscopy at a magnification of ×100. Cells were treated with
various concentrations of UA for 24 h, and a second set of images
were taken using the same method. A reduction in the size of the
scraped region is indicative of cell migration.
Tube formation assay of HUVECs
HUVEC tube formation was examined using the ECMatrix
assay kit (Millipore) following the manufacturer’s instructions.
Briefly, confluent HUVECs were harvested and diluted
(1×104 cells) in 50 μl of medium containing
various concentrations of UA. The harvested cells were seeded with
ECMatrix gel (1:1 v/v) into 96-well plates and incubated for 9 h at
37°C. The cells were photographed using phase-contrast inverted
microscopy at a magnification of ×100. The level of HUVEC tube
formation was quantified by calculating the length of the tubes in
three randomly chosen fields from each well.
RT-PCR
Total RNA was isolated from tumor tissues (three
tumors were randomly selected from UA-treatment or control groups)
or HT-29 cells with TRIzol reagent. Oligo(dT)-primed RNA (1
μg, isolated from tumor tissues or cells) was
reverse-transcribed with SuperScript II reverse transcriptase
(Promega) according to the manufacturer’s instructions. The
obtained cDNA was used to determine the level of VEGF-A, bFGF, Shh
and Gli-1 mRNA by PCR with Taq DNA polymerase (Fermentas). GAPDH
was used as an internal control. Samples were analyzed by gel
electrophoresis (1.5% agarose). The DNA bands were examined using a
gel documentation system (Model Gel Doc 2000, Bio-Rad).
Western blot analysis
HT-29 cells (2.5×105) in 5 ml medium were
seeded into 25 cm2 flasks and treated with the indicated
concentrations of UA for 24 h. Treated cells were lysed in
mammalian cell lysis buffer (M-PER, Thermo Scientific, Rockford,
IL, USA) containing protease (EMD Biosciences) and phosphatase
inhibitor (Sigma-Aldrich) cocktails and centrifuged at 14,000 × g
for 15 min. Protein concentrations in cell lysate supernatants were
determined by BCA protein assay. Equal amounts of protein from each
tumor or cell lysate were resolved on 12% Tris-glycine gels and
transferred onto PVDF membranes. The membranes were blocked for 2 h
with 5% non-fat dry milk and incubated with the desired primary
antibody directed against Shh, Gli-1, or β-actin (all in 1:1,000
dilutions) overnight at 4°C. Appropriate HRP-conjugated secondary
antibodies with chemiluminescence detection were used to image the
antibody-detected proteins.
Bio-Plex Phosphoprotein assay
Eight tumors were randomly selected from
UA-treatment or control groups and homogenized. For analysis of
phosphorylation of Akt and p70S6K in vitro, HT-29 cells
(2.5×105) were seeded into 25-cm2 flasks in 5
ml medium and treated with 40 μM of UA for 24 h. To detect
STAT3 phosphorylation in vitro, HT-29 cells were first grown
in complete DMEM (10% FBS) until ∼70% confluency and subsequently
cultured in FBS-free medium overnight. The medium was replaced with
DMEM with 10% FBS and cells were pre-treated with UA (40 μM)
for 1 h followed by stimulation with 10 ng/ml of IL-6 for 15 min.
Tumor tissues and treated cells were lysed using a commercially
available lysis kit (Bio-Rad Laboratories) and centrifuged at
14,000 × g for 15 min. Protein concentrations of the clarified
supernatants were determined by BCA protein assay. The presence of
p-STAT3, p-Akt and p-p70S6K was detected using a bead-based
multiplex assay for phosphoproteins (Bio-Plex Phosphoprotein assay,
Bio-Rad Laboratories) according to the manufacturer’s protocol.
Data were collected and analyzed using the Bio-Plex 200 suspension
array system (Bio-Rad).
Measurement of VEGF-A and bFGF protein
expression in HT-29 cells by ELISA
HT-29 cells were seeded into 6-well plates at a
density of 2×105 cells/well in 2 ml medium and treated
with the indicated concentrations of UA for 24 h. The medium and
cells from each well were collected and stored at −80°C until
analyzed. The level of VEGF-A or bFGF in the media was measured
using an ELISA kit (Xitang Biological Technology Ltd.) according to
the manufacturer’s instructions. The concentrations of VEGF-A and
bFGF were determined by comparison to serial dilutions of VEGF-A
and bFGF purified standards.
Statistical analysis
Data are presented as the mean ± SD for the
indicated number of independently performed experiments. The data
were analyzed using the SPSS package for Windows (Version 17.0).
Statistical analysis was carried out with Student’s t-test and
ANOVA. Differences with P<0.05 were considered to be
statistically significant.
Results
UA inhibits tumor growth in colorectal
cancer (CRC) xenograft mice
The in vivo efficacy of UA against tumor
growth was evaluated by measuring tumor volume in CRC xenograft
mice. As shown in Fig. 1,
administration of UA significantly inhibited tumor growth
throughout the study, as compared with the control group
(P<0.05), whereas the body weight gain in experimental animals
was not affected by UA treatment, suggesting that UA is potent in
suppressing colon tumor growth in vivo, without apparent
adverse effects.
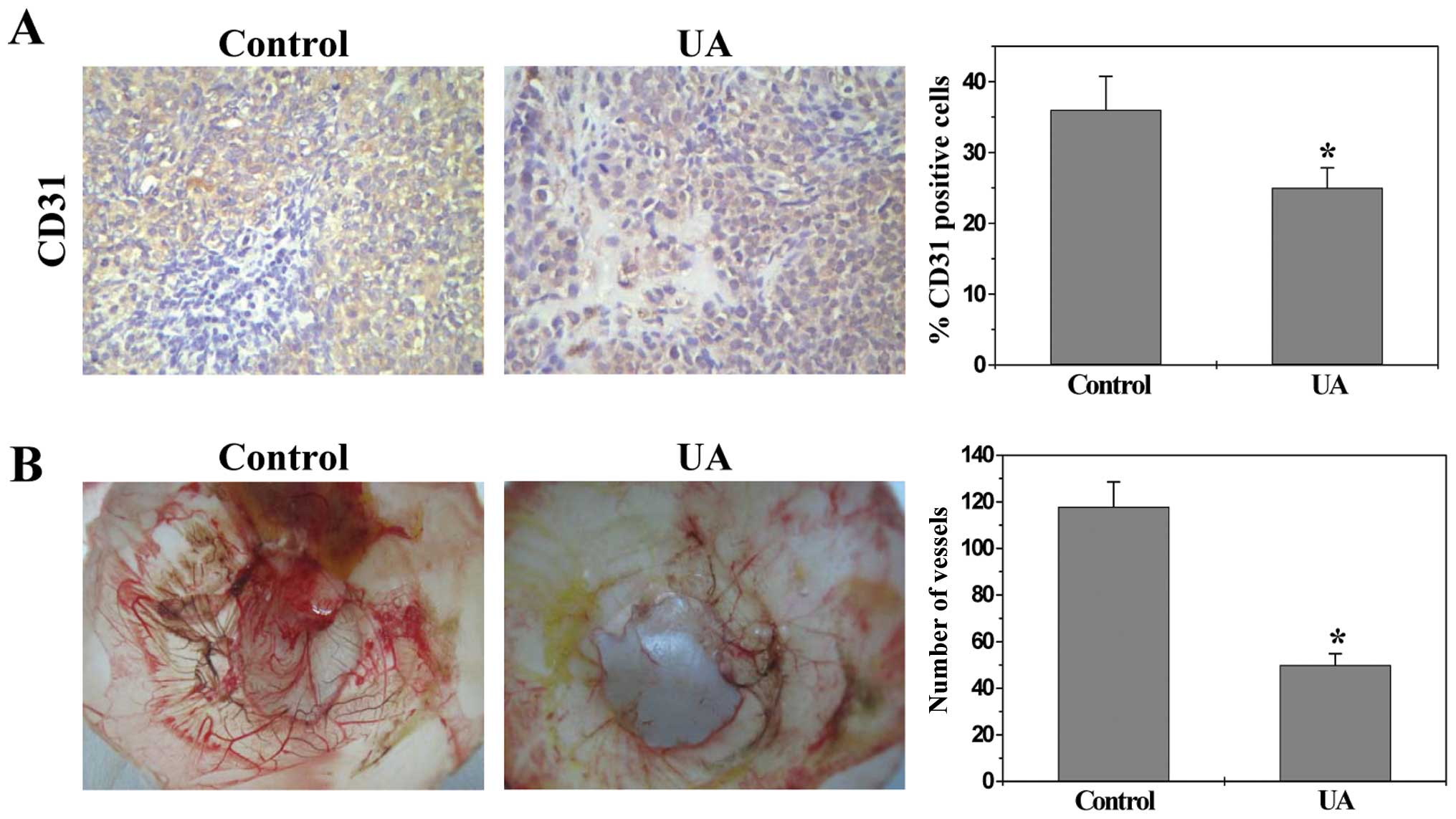
UA inhibits angiogenesis in vivo and in
vitro
To determine the effect of UA on tumor angiogenesis,
we examined the intratumoral microvessel density (MVD) in CRC mice
by evaluating expression of the endothelial cell-specific marker
CD31. Data from immunohistochemical staining (IHC) assay showed
that the percentage of CD31-positive cells in control or UA-treated
mice was 36.0±4.8 or 25.0±2.8%, respectively (Fig. 2A, P<0.01), demonstrating UA’s
inhibitory activity on tumor angiogenesis. The in vivo
anti-angiogenic activity of UA was further confirmed using a chick
chorioallantoic membrane (CAM) model. As shown in Fig. 2B, UA treatment significantly
reduced the total number of blood vessels in chicken embryos as
compared to untreated controls (P<0.01).
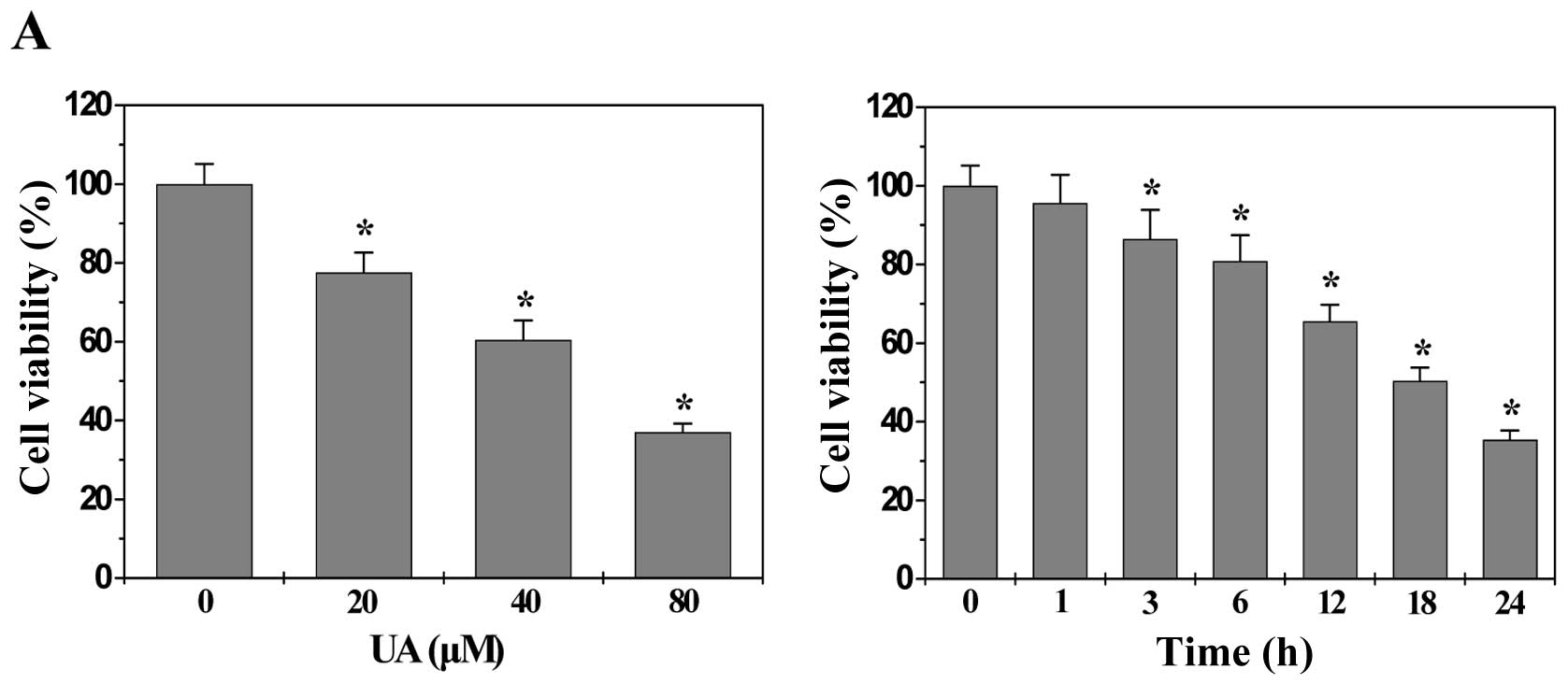
The processes of angiogenesis include endothelial
cell (EC) proliferation, migration, and alignment into tubular
structures. To evaluate the anti-angiogenic effect of UA we modeled
each of these processes with HUVECs in vitro. As shown in
Fig. 3A, UA treatment dose- and
time-dependently decreased the proliferation (viability) of HUVECs
compared to untreated control cells (P<0.01). In addition, UA
treatment inhibited HUVEC migration after monolayer wounding
(Fig. 3B). Moreover, we examined
the effect of UA on capillary tube formation of HUVECs using an
extracellular matrix, in which cultured ECs rapidly align and form
hollow tube-like structures. As shown in Fig. 3C, untreated HUVECs formed elongated
tube-like structures, whereas UA treatment resulted in a
significant decrease in capillary tube formation (P<0.01). Taken
together, these data suggested that UA-caused inhibition of colon
tumor growth is accompanied by its anti-angiogenic activity.
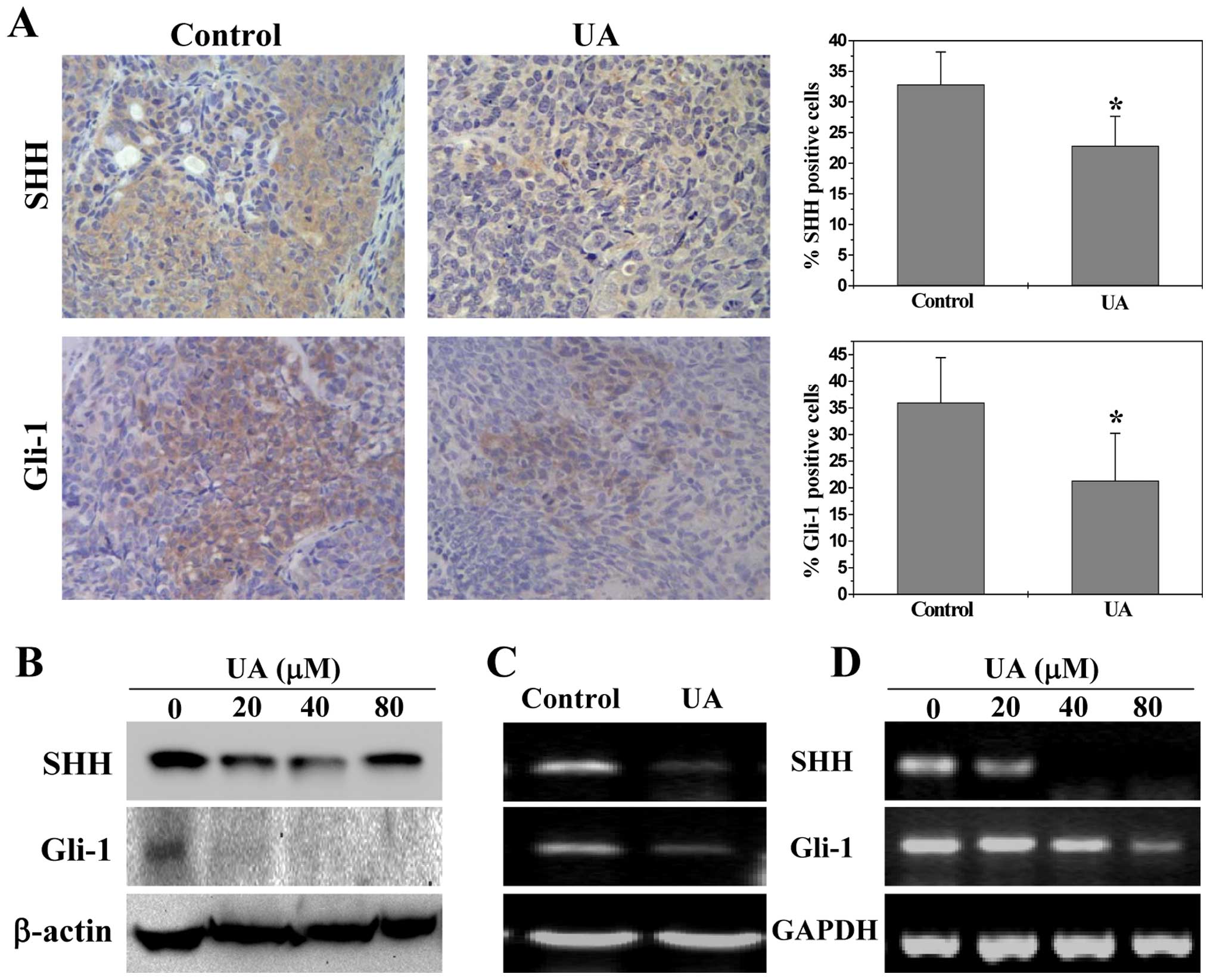
UA suppresses multiple signaling pathways
in vivo and in vitro
To explore the underlying mechanisms of
anti-angiogenic activities of UA, we determined its effect on the
activation of several CRC-related signal transduction cascades.
Activation of STAT3, Akt and p70S6K is mediated by its
phosphorylation, we therefore investigated the effect of UA on
STAT3, Akt and p70S6K activation in CRC xenograft tumor tissues and
HT-29 cells by Bio-Plex Phosphoprotein assay. We found after UA
treatment the phosphorylation level of STAT3, Akt and p70S6K in
both tumors (Fig. 4A) and HT-29
cells (Fig. 4B) was decreased as
compared to controls (P<0.01). The activation of SHH pathway was
evaluated by examining the expression of the key mediators of SHH
pathway in CRC xenograft tumors and HT-29 cells. As shown in
Fig. 5A, the percentage of cells
in the CRC xenograft tumors expressing SHH and Gli-1 in the control
group was 32.8±5.3 and 36.0±8.4%, respectively, whereas the levels
in UA-treated mice were 22.8±4.8 and 21.3±8.9%, respectively
(P<0.01). Similarly, UA treatment significantly reduced the
protein expression of SHH and Gli-1 in HT-29 cells (Fig. 5B). Data from RT-PCR showed that the
pattern of mRNA expression was similar to their respective protein
levels (Fig. 5C and D).
Collectively, these data suggest that UA significantly suppresses
the activation of multiple signaling pathways mediating tumor
angiogenesis.
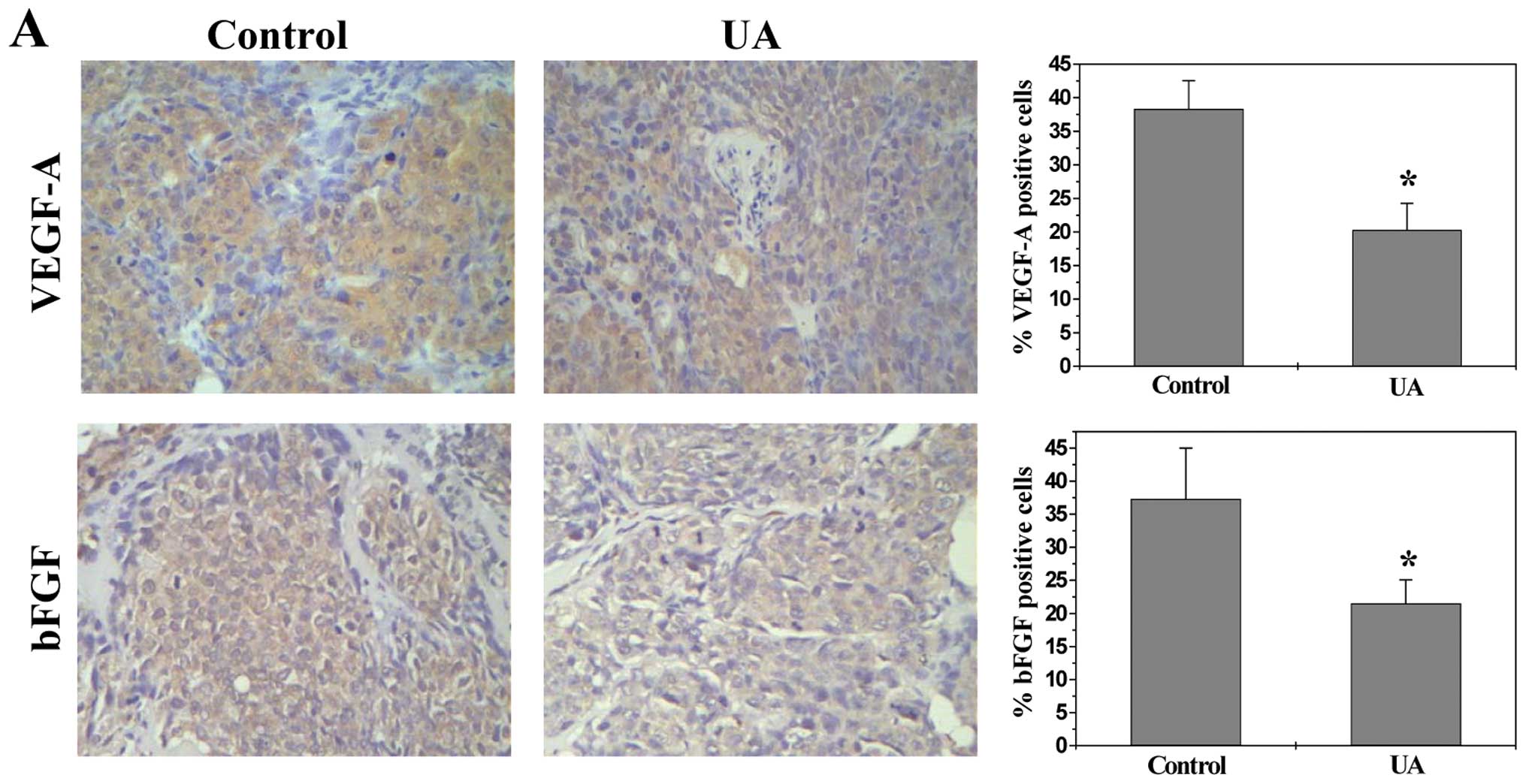
UA inhibits the expression of VEGF-A and
bFGF
VEGF-A and bFGF are critical target gene of the
above-mentioned pathways (30,31,43,44).
As most potent angiogenic stimulators, VEGF-A and bFGF are commonly
overexpressed in many kinds of human cancer correlating with tumor
progression and poorer prognosis (45–48).
To further investigate the mechanism whereby UA inhibited
angiogenesis, we determined its effect on VEGF-A and bFGF
expression. Data from RT-PCR, IHC and ELISA analyses indicated that
UA treatment profoundly decreased mRNA and protein levels of VEGF-A
and bFGF in both CRC xenograft tumor tissues and HT-29 cells
(Fig. 6).
Discussion
Due to its essential role in the growth, progression
and metastasis of solid tumors, angiogenesis has become an
attractive target for anticancer chemotherapy. A variety of
anti-angiogenic agents is currently in preclinical development,
with some of them now entering clinical trials. However, the
administration of angiogenesis inhibitors may cause cardiovascular
complications, including impaired wound healing, bleeding,
hypertension, proteinuria and thrombosis (45–48),
due to their intrinsic cytotoxicity against non-tumor associated
endothelial cells. In addition, since multiple signaling pathways
are involved in the process of tumor angiogenesis, most
currently-used angiogenic inhibitors, which typically are designed
to affect a single target, may be insufficient and probably lead to
resistance (49). These problems
highlight the urgent need for the development of multi-target
agents with minimal side effects and toxicity. Natural products
have received great interest since they have relatively fewer side
effects as compared to modern chemotherapeutics and have been shown
to display multiple therapeutic effects for various diseases
including cancer. Ursolic acid (UA), a major active compound of
many traditional Chinese medicinal herbs, has been shown to possess
anticancer activity. However, the precise mechanism of its
potential tumoricidal activity remains largely unclear. Therefore,
before UA can be further developed as an anticancer agent, the mode
of action for its antitumor effects should be fully elucidated.
In the present study, using a CRC mouse xenograft
model we demonstrated that UA could inhibit cancer growth in
vivo, without apparent sign of toxicity. In addition, we found
that UA significantly reduced the intratumoral microvessel density
(MVD) in CRC xenograft mice and the total number of blood vessels
in chick chorioallantoic membrane, suggesting that that UA-caused
inhibition of colon tumor growth may be associated with its
anti-angiogenic activity. Moreover, using human umbilical vein
endothelial cells (HUVEC), we found that UA dose- and/or
time-dependently inhibited several typical features of angiogenic
process, i.e. suppressing endothelial cell proliferation,
inhibiting migration and capillary tube formation of endothelial
cells, further demonstrating the anti-angiogenic activity of
UA.
Tumor angiogenesis is tightly regulated by multiple
signal transduction cascades including SHH, STAT3 and Akt pathways.
Activation of these signals upregulates the expression of various
angiogenic factors including VEGF-A and bFGF which exert
pro-angiogenic function via binding to their specific receptors
located on vascular endothelial cells (36,50,51),
eventually promoting angiogenesis. In this study we found that UC
treatment inhibited the activation of STAT3, Akt and SHH pathways
both in vivo in CRC tumors and in vitro in human
colon carcinoma HT-29 cells since UA significantly suppressed the
phosphorylation of STAT3, Akt and p70S6K, as well as the mRNA and
protein expression of the key mediators of SHH signaling.
Consistently, UA treatment profoundly downregulated the expression
of VEGF-A and bFGF in both CRC tumors and HT-29 cells.
In conclusion, here we proposed that inhibition of
tumor angiogenesis via suppression of multiple signaling pathways
might be one of the mechanisms whereby UA can be effective in
cancer treatment.
Abbreviations:
|
UA
|
ursolic acid;
|
|
CRC
|
colorectal cancer;
|
|
CAM
|
chorioallantoic membrane;
|
|
HUVEC
|
human umbilical vein endothelial
cell;
|
|
DMSO
|
dimethyl sulfoxide;
|
|
MTT
|
3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide;
|
|
IHC
|
immunohistochemical staining;
|
|
VEGF
|
vascular endothelial growth
factor;
|
|
bFGF
|
basic fibroblast growth factor;
|
|
MVD
|
microvessel density;
|
|
SHH
|
sonic hedgehog signal pathway;
|
|
STAT3
|
signal transducer and activator of
transcription 3;
|
|
Akt
|
AKT8 in rodent T-cell lymphoma;
|
|
p70S6K
|
p70S6 kinase
|
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (no. 81073097).
References
|
1.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2.
|
Gustin DM and Brenner DE: Chemoprevention
of colon cancer:current status and future prospects. Cancer
Metastasis Rev. 21:323–348. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Lin JM, Chen YQ, Wei LH, Chen XZ, Xu W,
Hong ZF, Sferra TJ and Peng J: Hedyotis Diffusa Willd
extract induces apoptosis via activation of the
mitochondrion-dependent pathway in human colon carcinoma cells. Int
J Oncol. 37:1331–1338. 2010.
|
|
4.
|
Van Cutsem E and Costa F: Progress in the
adjuvant treatment of colon cancer: has it influenced clinical
practice? JAMA. 294:2758–2760. 2005.PubMed/NCBI
|
|
5.
|
Longley DB, Allen WL and Johnston PG: Drug
resistance, predictive markers and pharmacogenomics in colorectal
cancer. Biochim Biophys Acta. 1766:184–196. 2006.PubMed/NCBI
|
|
6.
|
Lippman SM: The dilemma and promise of
cancer chemoprevention. Nat Clin Pract Oncol. 10:5232006.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Folkman J: Anti-angiogenesis: new concept
for therapy of solid tumors. Ann Surg. 75:409–416. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Holash J, Wiegand SJ and Yancopoulos GD:
New model of tumor angiogenesis: dynamic balance between vessel
regression and growth mediated by angiopoietins and VEGF. Oncogene.
18:5356–5362. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Folkman J: Seminars in Medicine of the
Beth Israel Hospital, Boston. Clinical applications of research on
angiogenesis. N Engl J Med. 333:1757–1763. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Folkman J: Angiogenesis: an organizing
principle for drug discovery? Nat Rev Drug Discov. 6:273–286. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Folkman J: Angiogenesis. Annu Rev Med.
57:1–18. 2006. View Article : Google Scholar
|
|
12.
|
Folkman J: Tumor angiogenesis: therapeutic
implications. N Engl J Med. 285:1182–1186. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Cook KM and Figg WD: Angiogenesis
inhibitors: current strategies and future prospects. CA Cancer J
Clin. 60:222–243. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Ingham PW, Nakano Y and Seger C:
Mechanisms and functions of Hedgehog signalling across the metazoa.
Nat Rev Genet. 12:393–406. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Theunissen JW and de Sauvage FJ: Paracrine
Hedgehog signaling in cancer. Cancer Res. 69:6007–6010. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Das S, Tucker JA, Khullar S, Samant RS and
Shevde LA: Hedgehog signaling in tumor cells facilitates
osteoblast-enhanced osteolytic metastases. PLoS One. 7:e343742012.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Sahebjam S, Siu LL and Razak AA: The
utility of hedgehog signaling pathway inhibition for cancer.
Oncologist. 17:1090–1099. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Yoshikawa K, Shimada M, Miyamoto H,
Higashijima J, Miyatani T, Nishioka M, Kurita N, Iwata T and Uehara
H: Sonic hedgehog relates to colorectal carcinogenesis. J
Gastroenterol. 44:1113–1117. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Varnat F, Duquet A, Malerba M, Zbinden M,
Mas C, Gervaz P and Ruiz i Altaba A: Human colon cancer epithelial
cells harbour active HEDGEHOG-GLI signalling that is essential for
tumour growth, recurrence, metastasis and stem cell survival and
expansion. EMBO Mol Med. 1:338–351. 2009. View Article : Google Scholar
|
|
20.
|
Mazumdar T, DeVecchio J, Shi T, Jones J,
Agyeman A and Houghton JA: Hedgehog signaling drives cellular
survival in human colon carcinoma cells. Cancer Res. 71:1092–1102.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Lum L and Beachy PA: The Hedgehog response
network: sensors, switches, and routers. Science. 304:1755–1759.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Varjosalo M and Taipale J: Hedgehog:
functions and mechanisms. Genes Dev. 22:2454–2472. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Lin JM, Wei LH, Shen AL, Cai QY, Xu W, Li
H, Zhan YZ, Hong ZF and Peng J: Hedyotis diffusa Willd
extract suppresses Sonic hedgehog signaling leading to the
inhibition of colorectal cancer angiogenesis. Int J Oncol.
42:651–656. 2013. View Article : Google Scholar
|
|
24.
|
Auzenne EJ, Klostergaard J, Mandal PK,
Liao WS, Lu Z, Gao F, Bast RC Jr, Robertson FM and McMurray JS: A
phosphopeptide mimetic prodrug targeting the SH2 domain of Stat3
inhibits tumor growth and angiogenesis. J Exp Ther Oncol.
10:155–162. 2012.PubMed/NCBI
|
|
25.
|
Bromberg J and Wang TC: Inflammation and
cancer: IL-6 and STAT3 complete the link. Cancer Cell. 15:79–80.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Kusaba T, Nakayama T, Yamazumi K, Yakata
Y, Yoshizaki A, Inoue K, Nagayasu T and Sekine I: Activation of
STAT3 is a marker of poor prognosis in human colorectal cancer.
Oncol Rep. 15:1445–1451. 2006.PubMed/NCBI
|
|
27.
|
Franke TF, Kaplan DR, Cantley LC and Toker
A: Direct regulation of the Akt proto-oncogene product by
phosphatidylinositol-3, 4-bisphosphate. Science. 275:665–668. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Clarke RB: p27KIP1
phosphorylation by PKB/Akt leads to poor breast cancer prognosis.
Breast Cancer Res. 5:162–163. 2003. View
Article : Google Scholar
|
|
29.
|
Chang F, Lee JT, Navolanic PM, Steelman
LS, Shelton JG, Blalock WL, Franklin RA and McCubrey JA:
Involvement of PI3K/Akt pathway in cell cycle progression,
apoptosis, and neoplastic transformation: a target for cancer
chemotherapy. Leukemia. 17:590–603. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Sun D, Liu Y, Yu Q, Zhou Y, Zhang R, Chen
X, Hong A and Liu J: The effects of luminescent ruthenium(II)
polypyridyl functionalized selenium nanoparticles on bFGF-induced
angiogenesis and AKT/ERK signaling. Biomaterials. 34:171–180. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Al-Ansari MM, Hendrayani SF, Tulbah A,
Al-Tweigeri T, Shehata AI and Aboussekhra A: p16INK4A
represses breast stromal fibroblasts migration/invasion and their
VEGF-A-dependent promotion of angiogenesis through Akt inhibition.
Neoplasia. 14:1269–1277. 2012.
|
|
32.
|
Pratheeshkumar P, Budhraja A, Son YO, Wang
X, Zhang Z, Ding S, Wang L, Hitron A, Lee JC, Xu M, Chen G, Luo J
and Shi X: Quercetin inhibits angiogenesis mediated human prostate
tumor growth by targeting VEGFR-2 regulated AKT/mTOR/P70S6K
signaling pathways. PLoS One. 7:e475162012. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Li W, Tan D, Zhang Z, Liang JJ and Brown
RE: Activation of Akt-mTOR-p70S6K pathway in angiogenesis in
hepatocellular carcinoma. Oncol Rep. 20:713–719. 2008.PubMed/NCBI
|
|
34.
|
Gordaliza M: Natural products as leads to
anticancer drugs. Clin Transl Oncol. 9:767–776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Ji HF, Li XJ and Zhang HY: Natural
products and drug discovery. EMBO Rep. 10:194–200. 2009.PubMed/NCBI
|
|
36.
|
Lin JM, Wei LH, Xu W, Hong ZF, Liu XX and
Peng J: Effect of Hedyotis Diffusa Willd extract on tumor
angiogenesis. Mol Med Rep. 4:1283–1288. 2011.PubMed/NCBI
|
|
37.
|
Peng J, Chen YQ, Lin JM, Zhuang QC, Xu W,
Hong ZF and Sferra TJ: Patrinia Scabiosaefolia extract
suppresses proliferation and promotes apoptosis by inhibiting STAT3
pathway in human multiple myeloma cells. Mol Med Rep. 4:313–318.
2011.
|
|
38.
|
Wei LH, Chen YQ, Lin JM, Zhao JY, Chen XZ,
Xu W, Liu XX, Sferra TJ and Peng J: Scutellaria Barbata D.
Don induces apoptosis of human colon carcinoma cell via activation
of the mitochondrion-dependent pathway. J Med Plants Res.
5:1962–1970. 2011.
|
|
39.
|
Zheng LP, Chen YQ, Lin W, Zhuang QC, Chen
XZ, Xu W, Liu XX, Peng J and Sferra TJ: Spica Prunellae
extract promotes mitochondrion-dependent apoptosis in a human colon
carcinoma cell line. Afr J Pharm Pharmacol. 5:327–335. 2011.
View Article : Google Scholar
|
|
40.
|
Ikeda Y, Murakami A and Ohigashi H:
Ursolic acid: an anti- and pro-inflammatory triterpenoid. Mol Nutr
Food Res. 52:26–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Andersson D, Liu JJ, Nilsson A and Duan
RD: Ursolic acid inhibits proliferation and stimulates apoptosis in
HT29 cells following activation of alkaline sphingomyelinase.
Anticancer Res. 23:3317–3322. 2003.PubMed/NCBI
|
|
42.
|
Prasad S, Yadav VR, Sung B, Reuter S,
Kannappan R, Deorukhkar A, Diagaradjane P, Wei C,
Baladandayuthapani V, Krishnan S, Guha S and Aggarwal BB: Ursolic
acid inhibits growth and metastasis of human colorectal cancer in
an orthotopic nude mouse model by targeting multiple cell signaling
pathways: chemosensitization with capecitabine. Clin Cancer Res.
18:4942–4953. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Pola R, Ling LE, Silver M, Corbley MJ,
Kearney M, Pepinsky RB, Shapiro R, Taylor FR, Baker DP and Asahara
T: The morphogen Sonic hedgehog is an indirect angiogenic agent
upregulating two families of angiogenic growth factors. Nat Med.
7:706–711. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Kujawski M, Kortylewski M, Lee H, Herrmann
A, Kay H and Yu H: Stat3 mediates myeloid cell-dependent tumor
angiogenesis in mice. J Clin Invest. 118:3367–3677. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Muñoz-Chápuli R, Quesada AR and Angel
Medina M: Angiogenesis and signal transduction in endothelial
cells. Cell Mol Life Sci. 61:2224–2243. 2004.
|
|
46.
|
Chen HX and Cleck JN: Adverse effects of
anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol.
6:465–477. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Zangari M, Fink LM, Elice F, Zhan F,
Adcock DM and Tricot GJ: Thrombotic events in patients with cancer
receiving antiangiogenesis agents. J Clin Oncol. 27:4865–4873.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Higa GM and Abraham J: Biological
mechanisms of bevacizumab-associated adverse events. Expert Rev
Anticancer Ther. 9:999–1007. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Eikesdal HP and Kalluri R: Drug resistance
associated with antiangiogenesis therapy. Semin Cancer Biol.
19:310–317. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Rak J and Kerbel RS: bFGF and tumor
angiogenesis - Back in the limelight? Nat Med. 3:1083–1084. 1997.
View Article : Google Scholar : PubMed/NCBI
|