Introduction
Reactive oxygen species (ROS) are mutagenic and
hence may promote cancer (1). ROS
modulate growth signals and regulate gene expression, leading to
the sustained proliferation of cancer cells (2). Oxidative stress caused by metabolic,
dietary, and environmental factors leads to excessive production of
ROS, which can then induce genetic alterations, including
frameshift mutations in tumor suppressor genes (3,4) such
as the type II transforming growth factor-β (TGF-β) receptor
(5). These mutations may allow
colon epithelial cells to escape growth restriction mediated by
ligation of TGF-β to its type II receptor.
In addition to genetic alterations, epigenetic
changes also result in the loss of or aberrant expression of genes
associated with carcinogenesis (6). ROS trigger DNA methylation of tumor
suppressor gene promoters; Lim et al have suggested a
functional pathway model of ROS-induced epigenetic changes in which
persistently elevated levels of ROS induce CpG methylation in the
promoter region of the gene encoding the tumor suppressor
E-cadherin, via specific recognition of an E-box motif by Snail
(7). In addition, the tumor
suppressor Runt-related transcription factor 3 (RUNX3) is silenced
by ROS-induced epigenetic regulation in colorectal cancer cells
(8).
Akt signaling upregulates the Wnt signaling pathway
via inhibitory phosphorylation of glycogen synthase kinase-3β
(GSK-3β), which results in stabilization of β-catenin and its
relocation from the cell membrane to the nucleus, where it is
recruited into TCF/LEF transcriptional regulatory complexes
(9–11). TCF/LEF complexes bind to enhancer
regions of target genes involved in proliferation, invasion, and
inhibition of apoptosis, including c-Myc and cyclin D1. These
effects contribute directly to colon cancer development.
RUNX3 is a tumor suppressor that is involved in
various cancers, including gastric cancer (12,13).
RUNX3 knockout mice exhibit gastric epithelial hyperplasia, reduced
levels of apoptosis, and reduced sensitivity to TGF-β (14). Approximately 45–60% of human
gastric cancers display a loss of RUNX3 expression due to
hemizygous deletion and promoter hypermethylation (14). By interacting with Smad2/3, Smad4,
p300, and FoxO3a to regulate the transcription of target genes,
RUNX3 is involved in the TGF-β-mediated signaling pathway (15). RUNX3 suppresses gastric
tumorigenesis by upregulating p21 (16), Bim (17), and Claudin-1 (18). The gastrointestinal tract,
particularly the colon and rectum, is constantly exposed to ROS
originating from endogenous and exogenous sources (19). The involvement of ROS in colorectal
cancer remains speculative; however, numerous epidemiological
studies suggest that oxidative stress is an important factor in
cancer initiation and progression (13,20,21).
We recently reported that oxidative stress
downregulates RUNX3 expression by upregulating DNA
methyltransferase 1 and histone deacetylase, resulting in
hypermethylation of the RUNX3 promoter (8). In this study, we investigated whether
oxidative stress is able to regulate the Akt signaling pathway by
reducing the expression of the RUNX3 tumor suppressor in colorectal
cancer cells.
Materials and methods
Cell culture
The human colorectal cancer cell line SNU-407
(Korean Cell Line Bank, Seoul, Republic of Korea) was maintained at
37°C in a humidified atmosphere of 5% CO2. The cells
were cultured in RPMI-1640 medium containing 10% fetal calf serum,
streptomycin (100 μg/ml), and penicillin (100 U/ml).
Western blot analysis
Nuclear extracts were prepared using a Nuclear
Protein Extraction kit (Cayman Chemical, Ann Arbor, MI, USA). The
nuclear extracts were lysed on ice for 4 min in 1 ml of lysis
buffer comprising 10 mM Tris-HCl (pH 7.9), 10 mM NaCl, 3 mM
MgCl2, and 1% NP-40. After centrifugation for 10 min at
3000 × g, the pellets were re-suspended in 50 μl of
extraction buffer (20 mM HEPES, pH 7.9, 20% glycerol, 1.5 mM
MgCl2, 0.2 mM EDTA, 1 mM DTT, and 1 mM PMSF), incubated
on ice for 30 min, and then centrifuged at 13000 × g for 5 min.
After determination of the protein concentration, the supernatants
were stored at −70°C. Aliquots of the lysates (40 μg of
protein) were boiled for 5 min and then electrophoresed on a 10%
SDS-polyacrylamide gel. Blots of the gels were transferred to
nitrocellulose membranes, which were subsequently incubated with
primary antibodies and then with secondary immunoglobulin-G
horseradish peroxidase conjugates (Pierce, Rockford, IL, USA). The
protein bands were detected using an enhanced chemiluminescence
western blotting detection kit (Amersham, Little Chalfont,
Buckinghamshire, UK) and were visualized using a luminescent image
analyzer.
Immunocytochemistry
Cells plated on coverslips were fixed in 4%
paraformaldehyde for 30 min and then permeabilized with PBS
containing 0.1% Triton X-100 for 2.5 min. Cells were treated with
blocking medium (PBS containing 3% bovine serum albumin) for 1 h
and incubated with an anti-RUNX3 antibody diluted in blocking
medium for 2 h. The primary anti-RUNX3 antibody was detected by
incubation with a 1:500 dilution of the FITC-conjugated secondary
antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h.
After washing with PBS, the stained cells were mounted onto
microscope slides in mounting medium containing DAPI (Vector,
Burlingame, CA, USA). Images were collected using the LSM 510
program on a Zeiss confocal microscope.
Trypan blue staining
Trypan blue solution (0.4%) was added to the cell
suspension at a 1:5 ratio and the suspension was incubated at room
temperature for 5 min. Viable cells that did not show any staining
were counted under a microscope using a hemocytometer.
RNA isolation and reverse
transcription-PCR (RT-PCR)
Total RNA was isolated from cells using Trizol
reagent (GibcoBRL, Grand Island, NY, USA) and the cDNA was
amplified using 1 μl of reverse transcription reaction
buffer, primers, dNTPs, and 0.5 U of Taq DNA polymerase in a final
volume of 25 μl, as described previously (22). The PCR conditions were as follows:
initial denaturation at 94°C for 5 min, followed by 35 cycles of
94°C for 1 min, 55°C for 1 min, and 72°C for 1 min, and then a
final elongation step at 72°C for 7 min. The following primers were
used to amplify human RUNX3 and Akt: RUNX3 sense,
5′-GGCAATGACGAGAACTAC-3′ (located in exon 2); RUNX3 antisense,
5′-GGAGAATGGGTTCAGTTC-3′ (located in exon 5); Akt sense,
5′-GCAGCACGTGTACGAGAAGA-3′; and Akt antisense,
5′-GGTGTCAGTCTCCGACGTG-3′. The amplified products were resolved by
1% agarose gel electrophoresis, stained with ethidium bromide, and
then photographed under UV light using Image Quant™ TL analysis
software (Amersham Bioscience, Sweden).
Transient transfection of small
interfering RNA (siRNA)
Cells were seeded into 24-well plates at a density
of 1.5×105 cells/well and allowed to reach approximately
50% confluency. Cells were then transfected with 10–50 nM of a
mismatched siRNA control (siControl; Santa Cruz Biotechnology) or
an siRNA targeted against RUNX3 (Santa Cruz Biotechnology) using
Lipofectamine RNAiMax reagent (Invitrogen, Carlsbad, CA, USA),
according to the manufacturer’s instructions. Twenty-four hours
after transfection, the cells were treated with 5-fluorouracil for
a further 48 h and then examined using an MTT assay.
Chromatin immunoprecipitation (ChIP)
assay
The ChIP assay was performed using the SimpleChIP™
enzymatic chromatin IP kit (Cell Signaling Technology, Danvers, MA,
USA) according to the manufacturer’s protocol with slight
modifications. Briefly, cells were pre-treated with 1 mM N-acetyl
cysteine for 1 h, treated with H2O2 for 48 h,
and then cross-linked by the addition of 1% formaldehyde. Chromatin
was prepared and digested with nuclease for 12 min at 37°C. The
ChIP assay was performed using a DNMT1 antibody (Abcam, Cambridge,
MA, USA) and normal rabbit IgG. The antibodies were added to the
chromatin digests and the mixtures were incubated overnight at 4°C
with constant rotation. ChIP-grade protein G magnetic beads were
added to capture the immunoprecipitated complexes. The beads were
washed and the immunoprecipitates were eluted with ChIP elution
buffer. The cross-links were reversed by incubation at 65°C for 30
min and then proteinase K was added and the samples were incubated
for a further 2 h at 65°C. The immunoprecipitated DNA fragments
were purified using spin columns and the DNA recovered from the
immunoprecipitated complexes was subjected to 35 cycles of PCR
amplification. The primers used to amplify the DNA fragment were as
follows (23): RUNX3 binding site
1 (RBS1) sense, 5′-TTTCCATCCTGCTAAGTACTT-3′; RBS1 antisense,
5′-GATCCCAACATGGGTCTTTCC-3′; RBS2 sense,
5′-ACTTGTCTGAACCTCTCTTTG-3′; RBS2 antisense,
5′-AAAGCAAAGAAATTCAAACAT-3′; catenin binding site sense,
5′-GAGCGCATGCTAAGCTGAAA-3′; and catenin binding site antisense,
5′-GGACAGACGGCCAAAGAATC-3′. The PCR products were separated on 2%
agarose gels and the DNA bands were visualized using the Image
program (NIH, Bethesda, MD, USA).
Statistical analysis
All measurements were performed in triplicate and
all data are represented as the mean ± SEM. The results were
subjected to an analysis of variance using Tukey’s test. P<0.05
was considered statistically significant.
Results
ROS downregulate RUNX3 expression and
upregulate Akt expression in SNU-407 cells
In a previous study, we demonstrated that oxidative
stress attenuates RUNX3 expression in human colorectal cancer
SNU-407 cells (8). To confirm this
finding, SNU-407 cells were treated with 100 μM
H2O2 for 48 h and a significant decrease in
nuclear RUNX3 protein levels were detected by immunoblot and
confocal imaging analyses (Figs.
1A and 1B). In addition,
growth of the SNU-407 cells was significantly increased by the
H2O2 treatment (Fig. 1C). These results suggest that RUNX3
has a negative influence on the proliferation of SNU-407 cells.
RUNX3 represses Akt1 expression through transcriptional inhibition
and the loss of RUNX3 promotes tumorigenesis via activation of the
Akt1/β-catenin/cyclin D1 signaling pathway (23). In agreement with these findings,
immunoblot analyses revealed that treatment of SNU-407 cells with
H2O2 resulted in substantial increases in
total Akt and phosphorylated Akt protein levels (Fig. 1D).
ROS reverse RUNX3-mediated repression of
Akt transcription
Next, to investigate the inverse correlation between
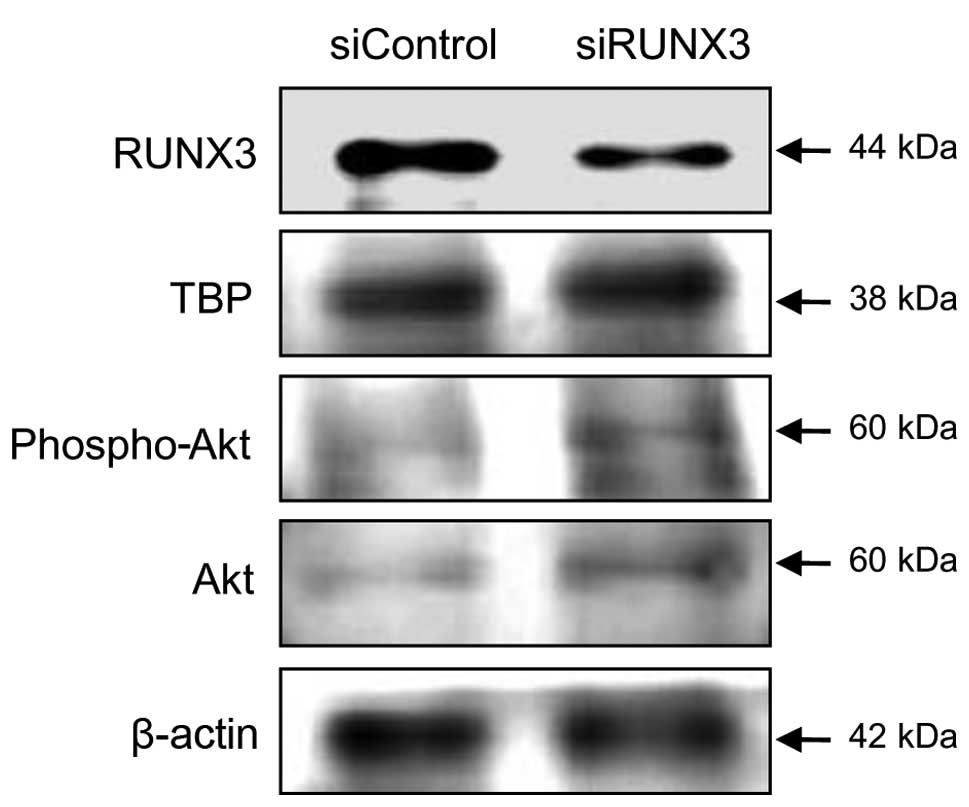
RUNX3 and Akt expression, a siRNA was used to knockdown endogenous
RUNX3 in SNU-407 cells, which express high levels of the tumor
suppressor. Total and phosphorylated Akt protein levels were higher
in cells treated with a RUNX3-specific siRNA than in cells treated
with a control siRNA (Fig. 2). The
inverse correlation between RUNX3 and Akt expression was further
confirmed by RT-PCR. Cells treated with H2O2
for up to 48 h showed a time-dependent decrease in the level of
RUNX3 mRNA and a parallel increase in the level of Akt mRNA
(Fig. 3A). Two RUNX3-binding sites
(RBS1 and RBS2) with the consensus sequence ‘TGTGGT’ are present in
the Akt1 promoter region. A ChIP assay revealed that, compared with
control cells, those treated with H2O2
displayed decreased binding of RUNX3 to the RBS1 and RBS2 sites
(Figs. 3B and 3C).
ROS regulate the Akt1/β-catenin signaling
pathway
There are many reports that ROS regulate cell
survival by modulating the Akt signaling pathway (24,25).
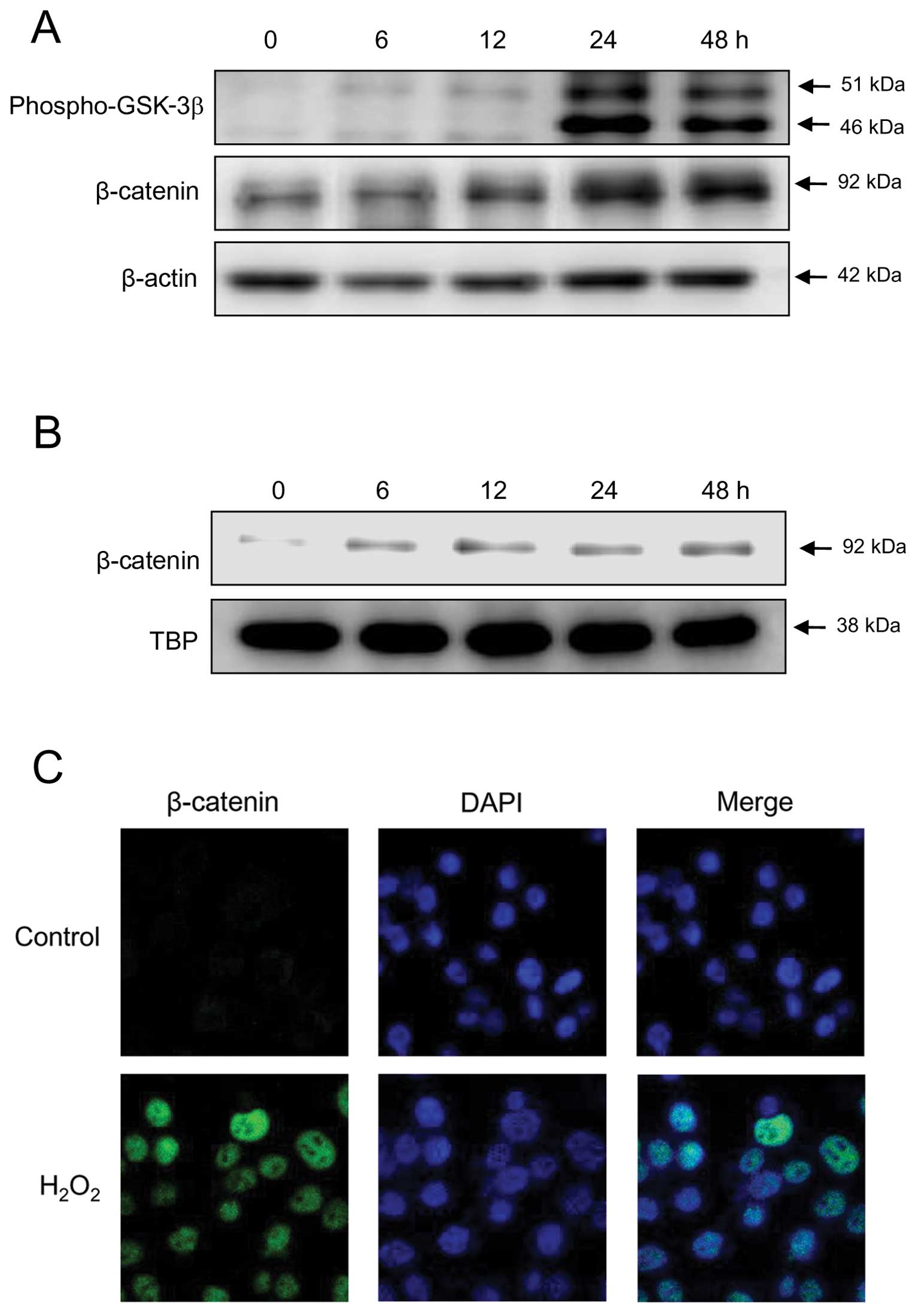
Treatment of SNU-407 cells with H2O2 induced
β-catenin expression and GSK-3β phosphorylation (Fig. 4A). A previous study demonstrated
that RUNX3 reduces the nuclear localization, transactivation, and
stability of β-catenin protein (23); therefore, we examined the effect of
ROS on the expression and localization of endogenous β-catenin in
SNU-407 cells using immunoblot and immunocytochemical analyses.
Levels of β-catenin in the nucleus were dramatically higher in
H2O2-treated cells than in control cells
(Fig. 4B and 4C), indicating that ROS induce the
expression and nuclear localization of β-catenin in RUNX3-positive
cells.
ROS regulate the β-catenin-mediated
induction of cyclin D1
Cyclin D1 is a downstream effector of the
Akt1/β-catenin signaling pathway that preferentially binds to and
activates CDK4 and CDK6 at G1 phase to initiate cell cycle
progression (26,27). Therefore, the effects of ROS on the
expression of cyclin D1 and its associated signaling molecules were
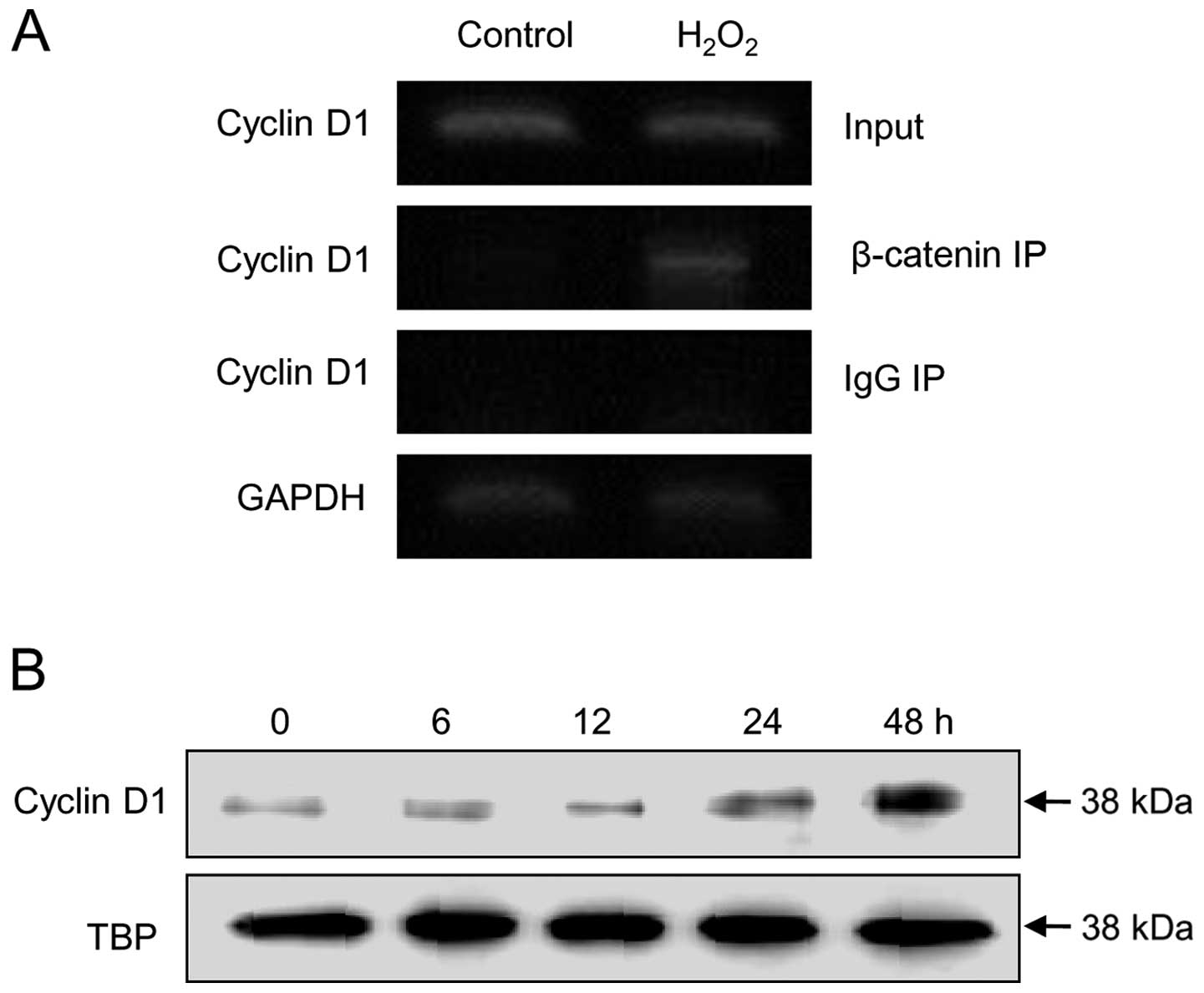
examined. The cyclin D1 promoter contains adjacent TCF/LEF (at −81
bp) and CREB (at −58 bp) binding sites (28). To determine whether ROS induce
cyclin D1 via their effects on β-catenin, ChIP assays were used to
investigate the interaction of β-catenin with the cyclin D1
promoter. Exposure of SNU-407 cells to H2O2
for 48 h increased the amount of cyclin D1 promoter DNA that
co-immunoprecipitated with β-catenin (Fig. 5A), indicating that ROS initiate the
recruitment of β-catenin to the cyclin D1 promoter. Exposure of the
cells to H2O2 also increased
β-catenin-mediated cyclin D1 protein expression in a time-dependent
manner (Fig. 5B). Taken together,
these results indicate that ROS silence RUNX3 expression and
activate the Akt1/β-catenin/cyclin D1 signaling pathway, which
promotes tumorigenesis in colorectal cancer.
Discussion
An imbalance in the production or removal of ROS can
be either directly or indirectly involved in the initiation,
promotion, and progression phases of carcinogenesis (29). The generation of ROS is involved in
the Akt signaling pathway (30,31)
and Akt may foster tumorigenesis in multiple ways (32,33).
For example, Akt stabilizes Myc and cyclin D1 and induces
degradation of the CDK inhibitor p27, all of which promote cell
cycle progression (34).
This study demonstrates that ROS repress RUNX3 and
induce Akt expression and the subsequent GSK-3β/β-catenin/cyclin D1
cascade. Knockdown of endogenous RUNX3 in SNU-407 cells increased
the expression and phosphorylation of Akt, as well as cell
proliferation. The ROS-induced upregulation of Akt and β-catenin
led to the induction of cyclin D1 expression. Therefore, we
conclude that downregulation of RUNX3 by ROS leads to activation of
the Akt signaling pathway in colorectal cancer.
Over-activation of Akt in cancer cells often occurs
as the result of chromosome deletion, genetic mutation, or
amplification of the genes that can regulate the activity of Akt.
In fact, deletion of the PTEN gene (35) and mutation of the gene encoding
phosphoinositide-3-kinase, catalytic, alpha polypeptide have been
associated with over-activation of Akt (36). However, the occurrence of genetic
alterations of Akt itself is relatively low in gastric cancer
(37). Recent studies demonstrate
that β-catenin/TCF/LEF, STAT3, and CREB regulate the activity of
the Akt 1 promoter (38,39). In addition, binding sites for AP1,
Sp1, and NF-κB in the Akt 1 promoter have been predicted by
software screening (39).
The tumor suppressor RUNX3 is frequently inactivated
in gastric cancer tissues (14)
and its aberrant activity is closely related to metastatic outcome
(40). It has been postulated that
the mechanism for RUNX3 inactivation in cancer cells and tissues
involves hypermethylation of its promoter region (14,41).
The RUNX3 gene is located on human chromosome 1p36 (14), a region containing many genes that
play roles in the maintenance of chromosome stability, suppression
of tumorigenesis, control of apoptosis, and DNA methylation
(42). Deletions in the 1p36
region are common in colorectal cancers (42–45),
suggesting that gene loss in this region affects chromosome
stability (43). RUNX3 interacts
with mSin3 and Groucho/TLE, which associate with histone
deacetylases and SUV39H1, a histone methyltransferase, to inhibit
or silence gene transcription (46). The two RUNX3-binding sites in the
Akt promoter are adjacent to STAT3-binding motifs and
β-catenin/TCF-binding elements. It is possible that binding of
RUNX3 to this region blocks the ability of other transcriptional
activators to bind to the Akt promoter. RUNX3 can interact with the
β-catenin/TCF complex and this interaction reduces its ability to
promote the transcription of target genes in colorectal cancer
(47). In this study, we
demonstrated that β-catenin is activated by ROS; however, we were
not able to investigate the possibility that RUNX3 interacts with
the β-catenin/TCF complex before binding to the Akt promoter,
leading to the inhibition of Akt transcription. Further studies are
required to investigate the association between RUNX3 and β-catenin
during tumorigenesis of colorectal cancer.
Taken together, the data presented here suggest that
oxidative stress may play an important role in inhibiting the
activation of the tumor suppressor RUNX3 and the subsequent
regulation of the Akt/β-catenin/cyclin D1 cascade in human
colorectal cancer cells. This effect of ROS may be associated with
the progression of colorectal cancer.
Acknowledgements
This study was supported by a grant
from the National R&D Program for Cancer Control, Ministry for
Health and Welfare, Republic of Korea (1120340).
References
|
1.
|
Shibutani S, Takeshita M and Grollman AP:
Insertion of specific bases during DNA synthesis past the
oxidation-damaged base 8-oxodG. Nature. 349:431–434. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Hussain SP, Hofseth LJ and Harris CC:
Radical causes of cancer. Nat Rev Cancer. 3:276–285. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Gasche C, Chang CL, Rhees J, Goel A, et
al: Oxidative stress increases frameshift mutations in human
colorectal cancer cells. Cancer Res. 61:7444–7448. 2001.PubMed/NCBI
|
|
4.
|
Zienolddiny S, Ryberg D and Haugen A:
Induction of microsatellite mutations by oxidative agents in human
lung cancer cell lines. Carcinogenesis. 21:1521–1526. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Markowitz S, Wang J, Myeroff L, et al:
Inactivation of the type II TGF-beta receptor in colon cancer cells
with microsatellite instability. Science. 268:1336–1338. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Sugimura T and Ushijima T: Genetic and
epigenetic alterations in carcinogenesis. Mutat Res. 462:235–246.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Lim SO, Gu JM, Kim MS, et al: Epigenetic
changes induced by reactive oxygen species in hepatocellular
carcinoma: methylation of the E-cadherin promoter.
Gastroenterology. 135:2128–2140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Kang KA, Zhang R, Kim GY, Bae SC, et al:
Epigenetic changes induced by oxidative stress in colorectal cancer
cells: methylation of tumor suppressor RUNX3. Tumour Biol.
33:403–412. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Lovatt M and Bijlmakers MJ: Stabilisation
of beta-catenin downstream of T cell receptor signalling. PLoS One.
5:e127942010. View Article : Google Scholar
|
|
10.
|
McManus EJ, Sakamoto K, Armit LJ, et al:
Role that phosphorylation of GSK3 plays in insulin and Wnt
signalling defined by knockin analysis. EMBO J. 24:1571–1583. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Fuchs SY, Ougolkov AV, Spiegelman VS and
Minamoto T: Oncogenic beta-catenin signaling networks in colorectal
cancer. Cell Cycle. 4:1522–1539. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Bae SC and Choi JK: Tumor suppressor
activity of RUNX3. Oncogene. 23:4336–4340. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Subramaniam MM, Chan JY, Yeoh GK, et al:
Molecular pathology of RUNX3 in human carcinogenesis. Biochim
Biophys Acta. 1796:315–331. 2009.PubMed/NCBI
|
|
14.
|
Li QL, Ito K, Sakakura C, et al: Causal
relationship between the loss of RUNX3 expression and gastric
cancer. Cell. 109:113–124. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Chuang LS and Ito Y: RUNX3 is
multifunctional in carcinogenesis of multiple solid tumors.
Oncogene. 29:2605–2615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Chi XZ, Yang JO, Lee KY, et al: RUNX3
suppresses gastric epithelial cell growth by inducing
p21(WAF1/Cip1) expression. Mol Cell Biol. 25:8097–8107. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Yano T, Ito K, Fukamachi H, et al: The
RUNX3 tumor suppressor upregulates Bim in gastric epithelial cells
undergoing transforming growth factor beta-induced apoptosis. Mol
Cell Biol. 26:4474–4488. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Chang TL, Ito K, Ko TK, et al: Claudin-1
has tumor suppressive activity and is a direct target of RUNX3 in
gastric epithelial cells. Gastroenterology. 138:255–265. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Blau S, Rubistein A, Bass P, et al:
Differences in the reducing power along the rat GI tract: lower
antioxidant capacity of the colon. Mol Cell Biochem. 194:185–191.
1999. View Article : Google Scholar
|
|
20.
|
Benhar M, Engelberg D and Levitzki A: ROS,
stress-activated kinases and stress-signaling in cancer. EMBO Rep.
3:420–425. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Skrzydlewska E, Sulkowski S, Koda M, et
al: Lipid peroxidation and antioxidant status in colorectal cancer.
World J Gastroenterol. 11:403–406. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Chen JC, Huang KC and Lin WW: HMG-CoA
reductase inhibitors upregulate heme oxygenase-1 expression in
murine RAW264.7 macrophages via ERK, p38 MAPK and protein kinase G
pathways. Cell Signal. 18:32–39. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Lin FC, Liu YP, Lai CH, et al:
RUNX3-mediated transcriptional inhibition of Akt suppresses
tumorigenesis of human gastric cancer cells. Oncogene.
31:4302–4316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Lee YJ, Jeong HY, Kim YB, et al: Reactive
oxygen species and PI3K/Akt signaling play key roles in the
induction of Nrf2-driven heme oxygenase-1 expression in
sulforaphane-treated human mesothelioma MSTO-211H cells. Food Chem
Toxicol. 50:116–123. 2012. View Article : Google Scholar
|
|
25.
|
Irani K: Oxidant signaling in vascular
cell growth, death, and survival: a review of the roles of reactive
oxygen species in smooth muscle and endothelial cell mitogenic and
apoptotic signaling. Circ Res. 87:179–183. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Osaki M, Oshimura M and Ito H: PI3K-Akt
pathway: its functions and alterations in human cancer. Apoptosis.
9:667–676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: a changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Pradeep A, Sharma C, Sathyanarayana P, et
al: Gastrin-mediated activation of cyclin D1 transcription involves
beta-catenin and CREB pathways in gastric cancer cells. Oncogene.
23:3689–3699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Kang DH: Oxidative stress, DNA damage, and
breast cancer. AACN Clinical Issues. 13:540–549. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Wang X, McCullough KD, Franke TF and
Holbrook NJ: Epidermal growth factor receptor-dependent Akt
activation by oxidative stress enhances cell survival. J Biol Chem.
275:14624–14631. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Huang C, Li J, Ding M, et al: UV Induces
phosphorylation of protein kinase B (Akt) at Ser-473 and Thr-308 in
mouse epidermal Cl 41 cells through hydrogen peroxide. J Biol Chem.
276:40234–40240. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Manning BD and Cantley LC: AKT/PKB
signaling: navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Plas DR and Thompson CB: Akt-dependent
transformation: there is more to growth than just surviving.
Oncogene. 24:7435–7442. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Los M, Maddika S, Erb B and
Schulze-Osthoff K: Switching Akt: from survival signaling to deadly
response. Bioessays. 31:492–495. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Oki E, Baba H, Tokunaga E, et al: Akt
phosphorylation associates with LOH of PTEN and leads to
chemoresistance for gastric cancer. Int J Cancer. 117:376–380.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Li VS, Wong CW, Chan TL, et al: Mutations
of PIK3CA in gastric adenocarcinoma. BMC Cancer. 5:292005.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Soung YH, Lee JW, Nam SW, et al:
Mutational analysis of AKT1, AKT2 and AKT3 genes in common human
carcinomas. Oncology. 70:285–289. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Dihlmann S, Kloor M, Fallsehr C and von
Knebel Doeberitz M: Regulation of AKT1 expression by
beta-catenin/Tcf/Lef signaling in colorectal cancer cells.
Carcinogenesis. 26:1503–1512. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Misra UK and Pizzo SV: Upregulation of
AKT1 protein expression in forskolin-stimulated macrophage:
evidence from ChIP analysis that CREB binds to and activates the
AKT1 promoter. J Cell Biochem. 100:1022–1033. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Fahrner JA, Eguchi S, Herman JG and Baylin
SB: Dependence of histone modifications and gene expression on DNA
hypermethylation in cancer. Cancer Res. 62:7213–7218.
2002.PubMed/NCBI
|
|
41.
|
Li QL, Kim HR, Kim WJ, et al:
Transcriptional silencing of the RUNX3 gene by CpG hypermethylation
is associated with lung cancer. Biochem Biophys Res Commun.
314:223–228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Di Vinci A, Infusini E, Nigro S, et al:
Intratumor distribution of 1p deletions in human colorectal
adenocarcinoma is commonly homogeneous: indirect evidence of early
involvement in colorectal tumorigenesis. Cancer. 83:415–422.
1998.
|
|
43.
|
Di Vinci A, Infusini E, Peveri C, et al:
Correlation between 1p deletions and aneusomy in human colorectal
adenomas. Int J Cancer. 75:45–50. 1998.PubMed/NCBI
|
|
44.
|
Tanaka K, Yanoshita R, Konishi M, et al:
Suppression of tumourigenicity in human colon carcinoma cells by
introduction of normal chromosome 1p36 region. Oncogene.
8:2253–2258. 1993.
|
|
45.
|
Praml C, Finke LH, Herfarth C, et al:
Deletion mapping defines different regions in 1p34.2-pter that may
harbor genetic information related to human colorectal cancer.
Oncogene. 11:1357–1362. 1995.
|
|
46.
|
Durst KL and Hiebert SW: Role of RUNX
family members in transcriptional repression and gene silencing.
Oncogene. 23:4220–4224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Ito Y: RUNX genes in development and
cancer: regulation of viral gene expression and the discovery of
RUNX family genes. Adv Cancer Res. 99:33–76. 2008. View Article : Google Scholar : PubMed/NCBI
|