Introduction
Triple-negative breast cancers (TNBCs) are defined
as tumors that lack expression of estrogen receptor (ER),
progesterone receptor (PR), and human epidermal growth factor
receptor 2 (HER2) (1). TNBC
patients account for 11–23% of all breast cancers (2–4).
TNBCs follow a more aggressive clinical course than other forms,
such as luminal A and luminal B, and have a poor prognosis
(4). They also have no indications
for hormonal therapy or anti-HER2 therapy. Therefore, treatment of
TNBC patients is restricted to cytotoxic drugs such as
5-fluorouracil (5-FU), vinorelbine (VNB), paclitaxel (PTX),
doxorubicin (DOX), and gemcitabine (GEM) (5). However, TNBCs acquire resistance to
cytotoxic drugs after a series of treatments (6). The development of resistance to
cytotoxic drugs appears to have become a major clinical problem in
the chemotherapy of TNBCs.
Drug efflux mechanisms are the most well-studied
mechanisms of drug resistance. The ABC family proteins, which
include multidrug resistance protein 1 (MDR1) and breast cancer
resistance protein (BCRP), are target molecules to overcome drug
resistance (6–8). However, the combination of these
protein inhibitors and cytotoxic drugs failed to show an improved
outcome over cytotoxic drugs alone (9,10).
Fluoropyrimidine anticancer drugs, as represented by
5-FU and capecitabine, have been used to treat various cancers and
accepted worldwide as first-line anticancer drugs for breast
cancers (11). The mechanisms of
resistance to 5-FU, namely, enhanced activities of thymidylate
synthase (TS) and dihydropyrimidine dehydrogenase (DPYD), are well
known to endow cancer cells with resistance to 5-FU in vitro
and in clinical studies (12–15).
Actually, 5-FU is used in combination with
5-chloro-2,4-dihydroxypyridine, which is a DPYD inhibitor. However,
the effects of this combination are insufficient. The elucidation
of other mechanisms of resistance to 5-FU are thus anticipated.
Investigations of other mechanisms of 5-FU resistance may lead to
the development of novel effective anticancer chemotherapies for
5-FU-resistant patients. To elucidate mechanisms of drug
resistance, the establishment of drug-resistant cancer cell lines
should be one of the most useful approaches for developing model
systems (16–18). However, 5-FU-resistant TNBC cell
lines have not been previously reported, although there have been
some 5-FU-resistant lines of other forms of breast cancer or other
tumors (11).
In this study, we established a 5-FU-resistant cell
line, MDA-MB-231/5-FU, from the human TNBC cancer cell line
MDA-MB-231, by repeated exposure of cells to stepwise increases in
the concentration of 5-FU. Then, we applied a proteomic approach
and the quantification of protein expression to compare proteins
between MDA-MB-231/5-FU and MDA-MB-231 and identify those with
differential expression. MDA-MB-231/5-FU may be a useful tool for
identifying new mechanisms of drug resistance and new drug targets
in TNBCs.
Materials and methods
Chemicals and antibodies
5-FU, DOX and VNB were purchased from Kyowa Hakko
(Tokyo, Japan), CDDP from Pfizer (New York, NY, USA), PTX from
Bristol-Myers (New York, NY, USA), and GEM from Eli Lilly
(Indianapolis, IN, USA). MDR1, p53 and phospho-p53 (Ser15) were
purchased from Cell Signaling Technology (Beverly, MA, USA), DPYD
and TS from GeneTex (San Antonio, TX, USA), BCRP from Abcam
(Cambridge, UK), and β-actin from Sigma (St. Louis, MO, USA).
Horseradish peroxidase (HRP)-conjugated secondary antibodies were
purchased from GE Healthcare (Little Chalfont, Bucks, UK).
Cell lines and culture conditions
The human breast carcinoma cell line MDA-MB-231 was
purchased from the American Type Culture Collection (ATCC,
Rockville, MD, USA). Cells were cultured in Dulbecco’s modified
Eagle’s medium (DMEM; Wako, Osaka, Japan) with 10% fetal bovine
serum (FBS) (Equitech-Bio, Kerrville, TX, USA), 100 U/ml
penicillin, and 100 μg/ml streptomycin (Gibco, Grand Island,
NE, USA) at 37°C under 5% CO2 and 20% O2 in a
humidified chamber.
Establishment of 5-FU-resistant cell
line
5-FU-resistant cells were established from
MDA-MB-231 by exposure to increasing concentrations of 5-FU.
MDA-MB-231 were exposed to an initial 5-FU concentration of 3.84
μmol/l in DMEM plus 10% FBS. The drug concentration was then
increased 1.25 times at each step of resistance, from 3.84
μmol/l up to 23.0 μmol/l. Cells were cultured for at
least four weeks at each step, with medium exchange every three
days. Chemotherapeutic drugs were eliminated from the
5-FU-resistant MDA-MB-231 (MDA-MB-231/5-FU) for 15 days before each
experiment.
Cell proliferation assays
Cell proliferation was examined using a Cell
Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) in
accordance with the manufacturer’s instructions. Briefly, a
suspension of MDA-MB-231 or MDA-MB-231/5-FU (1.5×103
cells/well) in 100 μl of DMEM with 10% FBS was seeded to
96-well plates, and supplemented with 5-FU, DOX, CDDP, VNB, PTX and
GEM. After incubation for 72 h, Cell Counting Kit-8 reagent was
added to each well. After incubation for 90 min, the cell viability
was measured as absorbance at 450 nm using a microplate reader
(Perkin-Elmer, Waltham, MA, USA). Analyses of all samples were
performed in triplicate. The percentage of cell viability was
determined as the ratio of absorbance of the sample versus that
without 5-FU as a control. The IC50 of a
chemotherapeutic drug was determined as the concentration at which
50% inhibition of cell growth was shown compared with the control
cell growth.
Protein extraction
Cells were washed with PBS and lysed in lysis buffer
consisting of 50 mM HEPES, pH 8.0, 150 mM NaCl, 5 mM EDTA, 1%
NP-40, 10% glycerol, 100 mM NaF, 1 mM phenylmethylsulfonyl fluoride
and protease inhibitor cocktail (Nacalai Tesque Inc., Kyoto,
Japan). Lysates were separated by centrifugation, the supernatant
was recovered, and protein concentrations were assayed using the
bicinchoninic acid protein assay reagent (Thermo Fisher Scientific,
Rockford, IL, USA).
iTRAQ sample labeling
TheiTRAQ analysis was performed in a double duplex
manner. Protein lysates (170 μg) from MDA-MB-231 and
MDA-MB-231/5-FU were digested with trypsin and labeled with 114 and
117 iTRAQ reagents according to standard procedures.
Protein identification and relative
quantification
Proteomic analysis was performed on a DiNa-AI Nano
LC System (KYA Technologies, Tokyo, Japan) coupled to a QSTAR Elite
hybrid mass spectrometer (AB Sciex, Framingham, MA, USA) through a
NanoSpray ion source (AB Sciex) as previously described (19). Briefly, mobile phase A was 98%
water [2% acetonitrile (ACN), 0.1% formic acid], and mobile phase B
was 70% ACN (0.1% formic acid, 30% water). The column effluent was
introduced into the spray chamber through a tapered stainless steel
emitter and directly electrosprayed into the QSTAR System ion trap
mass spectrometer in the positive mode for nano-electrospray
ionization-MS/MS analysis. Each sample was run for 150 min. Protein
identification was performed using Analyst QS Software 2.0 (AB
Sciex) in the positive-ion mode. Both sets of data were processed
using ProteinPilot Software 2.0.1 with the Paragon™ search
algorithm (AB Sciex). MS/MS data were searched against the NCBI
database (RefSeq release 54 of July 2012 from the website
ftp://ftp.hgc.jp/pub/mirror/ncbi/refseq/) using a
Homosapiens taxonomy filter. The minimum threshold for protein
identification was set at a protein score of 0.47, corresponding to
a confidence level >66% and 1% false discovery rate.
Annotation analysis
GI accession numbers were uploaded into the DAVID
6.7 (Database for Annotation, Visualization, and Integrated
Discovery) information tool. For Gene Ontology (GO) term analysis,
we studied the ‘Biological Process’ categories using the GO FAT
default settings. For functional annotation searches, we set the
following parameters: ‘Biological Process’, threshold count 3, EASE
0.5; for functional annotation clusters, medium stringency.
Enrichment values (GO terms), enrichment scores (annotation
clusters), and statistical determinants (Fisher’s Exact P-values)
are those calculated using DAVID 6.7 software.
Western blotting
The lysates for western blotting (20 μg of
protein) were separated on sodium dodecyl sulfate-polyacrylamide
gels under reducing conditions, followed by electrophoretic
transfer to polyvinylidene difluoride membranes (Immobilon-P;
Millipore, Billerica, MA, USA). After blocking, the membranes were
probed with the appropriate primary antibodies. Membrane-bound
primary antibodies were detected using secondary antibodies
conjugated with HRP. The chemiluminescence was detected with
LAS-4000 (GE Healthcare) using the enhanced chemiluminescence
technique and quantified using Image Quant TL software (GE
Healthcare).
Results
Establishment of 5-FU-resistant TNBC cell
line
To explore the mechanisms of resistance to 5-FU, we
established a 5-FU-resistant TNBC cell line. To achieve this, a
human TNBC cell line, MDA-MB-231, was treated continuously with
stepwise increases of the concentration of 5-FU every four weeks
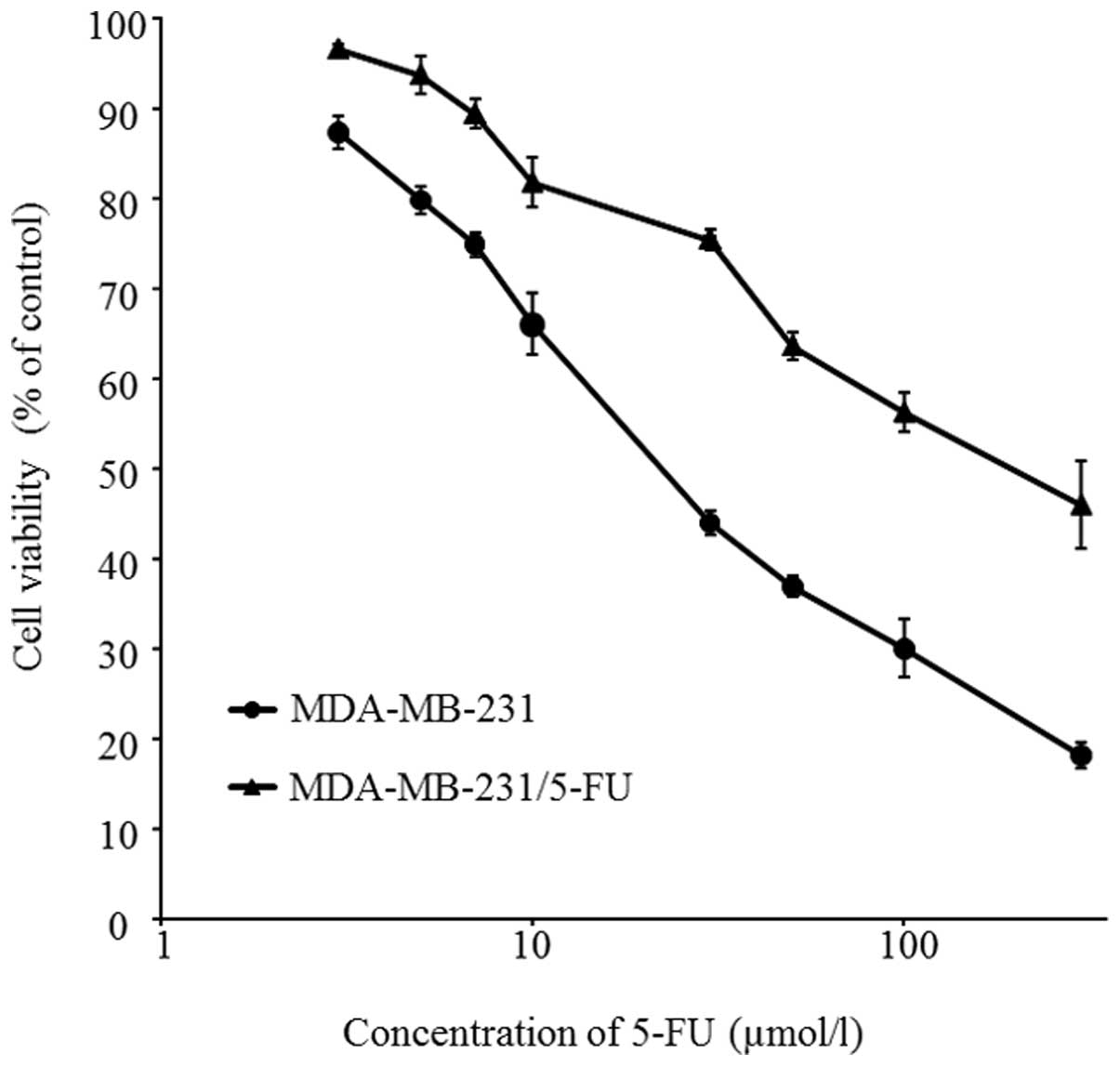
from 3.84 μmol/l to 23.0 μmol/l. Fig. 1 shows cell survival curves of
MDA-MB-231 and 5-FU-resistant cells. The cells were treated with
different concentrations of 5-FU for 72 h. The IC50
values of parent cells and 5-FU-resistant cells to 5-FU were
29.9±2.3 and 165.5±21.8 μmol/l, respectively (P<0.01).
The new cells were thus successfully established as a
5-FU-resistant TNBC cell line: MDA-MB-231/5-FU.
Cross-resistance profiles of
MDA-MB-231/5-FU cells
MDA-MB-231/5-FU acquired resistance to 5-FU; its
resistant index (RI) was 5.5. Generally, multiple drug resistance
involves resistance to one drug accompanied by resistance to
several other anticancer drugs (16). Therefore, we evaluated whether
MDA-MB-231/5-FU acquired cross-resistance to other anticancer drugs
used for TNBCs or with other mechanisms of action. The
IC50 and RI are summarized in Table I. The IC50 values of parent cells
to DOX, CDDP, VNB, PTX and GEM were 38.2±3.3 nmol/l, 2.0±0.3
μmol/l, 2.1±0.8 nmol/l, 1.1±0.7 nmol/l and 33.4±5.7 pmol/l,
respectively. In contrast, the IC50 values of
MDA-MB-231/5-FUto DOX, CDDP, VNB, PTX and GEM were 49.3±1.8 nmol/l,
1.4±0.2 μmol/l, 5.2±0.9 nmol/l, 9.5±2.0 nmol/l and
270.1±15.4 pmol/l, respectively. The RI of DOX, CDDP, VNB, PTX and
GEM were 1.3, 0.7, 2.5, 8.4 and 8.1, respectively. MDA-MB-231/5-FU
acquired cross-resistance to VNB, PTX and GEM. However, these cells
were sensitive to DOX and CDDP.
 | Table I.Cross-resistance of MDA-MB-231/5-FU
cells. |
Table I.
Cross-resistance of MDA-MB-231/5-FU
cells.
| 5-FU | DOX | CDDP | VNB | PTX | GEM |
|---|
|
|
|
|
|
|
|---|
| Cell line | IC50
(μmol/l) | RI | IC50
(nmol/l) | RI | IC50
(μmol/l) | RI | IC50
(nmol/l) | RI | IC50
(nmol/l) | RI | IC50
(pmol/l) | RI |
|---|
| MDA-MB-231 | 29.9 | 5.5a | 38.2 | 1.3 | 2.0 | 0.7 | 2.1 | 2.5b | 1.1 | 8.4a | 33.4 | 8.1a |
|
MDA-MB-231/5-FU | 165.5 | | 49.3 | | 1.4 | | 5.2 | | 9.5 | | 270.1 | |
Western blot analysis of proteins related
to drug resistance
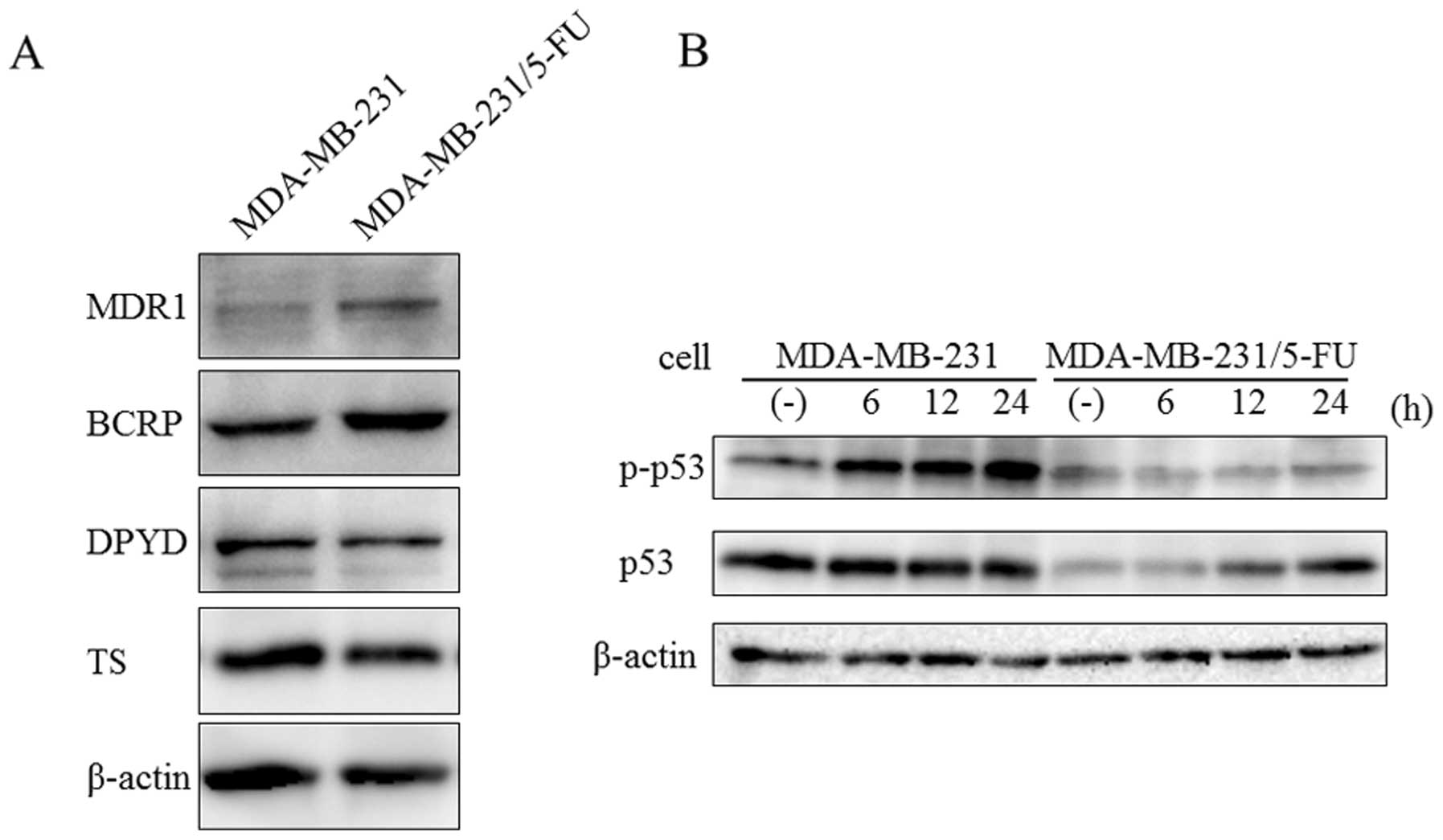
According to previous studies, the mechanisms of
resistance to 5-FU involve increases in 5-FU-degrading enzyme DPYD
and 5-FU-targeting enzyme TS (11–15,20).
On the other hand, ABC family proteins, such as MDR1 and BCRP, are
related to multiple drug resistance in breast cancer (6–8). To
confirm the expression of proteins related to drug resistance, we
examined MDR1, BCRP, DPYD and TS expression by western blot
analysis. MDA-MB-231/5-FU showed increased levels of MDR1 and BCRP1
proteins compared with the parent cells (Fig. 2A). In contrast, there were no
significant differences in DPYD and TS between the parent cells and
MDA-MB-231/5-FU.
p53 plays a major role in cellular responses to DNA
damage and other genomic aberrations (21). Activation of p53 can lead to cell
cycle arrest, DNA repair, or apoptosis. Generally, phosphorylation
of p53 is increased by DNA damage due to 5-FU (22–25).
To evaluate the response to DNA damage, MDA-MB-231 and
MDA-MB-231/5-FU were treated with 30 μM 5-FU for 6, 12 and
24 h. The phosphorylation level of p53 was increased by 5-FU in
MDA-MB-231, but not in its 5-FU-resistant counterpart (Fig. 2B). These results suggested that DNA
damage due to 5-FU was avoided by the over expression of ABC family
proteins.
Quantitative differential proteomics in
MDA-MB-231/5FU cells
To characterize MDA-MB-231/5-FU, we performed
quantitative differential proteomic analysis of MDA-MB-231/5-FU
cells and the parent cells based on the iTRAQ technique. As a
result, 93 proteins with a change of expression of ≥1.2-fold were
considered to be upregulated, whereas 85 proteins with a change
<0.8-fold were downregulated. Table
II shows the proteins for which there was a change of
expression ≥1.2-fold that was significant at the level of
P<0.05. To evaluate the functional differences between parent
cells and MDA-MB-231/5-FU cells, we performed enrichment analysis
(Fig. 3). The upregulated proteins
(≥1.2-fold) were classified into the GO categories of ‘DNA
recombination’, ‘cell cycle’, ‘complex assembly’, ‘cytoskeleton
organization’, ‘transport’, ‘negative regulation of cell death’,
‘chromatin organization’, and ‘cell differentiation’. The
enrichment scores for ‘DNA recombination’, ‘cell cycle’ and
‘complex assembly’ were 1.98, 1.95 and 1.81, respectively.
| Table II.Identification of upregulated
proteins in MDA-MB-231/5FU cells. |
Table II.
Identification of upregulated
proteins in MDA-MB-231/5FU cells.
| Accession no. | Protein name | 117/114 | P-value |
|---|
| gi|4501881 | Actin, α skeletal
muscle | 6.870 | |
| gi|62750354 | Matrin-3 isoform
a | 3.242 | |
| gi|9257257 | WD
repeat-containing protein 1 isoform 1 | 2.333 | 0.007 |
| gi|156523970 | α-2-HS-glycoprotein
preproprotein | 2.216 | 0.000 |
| gi|4506145 | Trypsin-1
preproprotein | 2.193 | 0.000 |
| gi|62414289 | Vimentin | 2.014 | 0.000 |
| gi|4503515 | Eukaryotic
translation initiation factor 3 subunit H | 1.906 | |
| gi|5803013 | Endoplasmic
reticulum resident protein 29 isoform 1 precursor | 1.905 | 0.037 |
| gi|28373194 | Proteasomal
ubiquitin receptor ADRM1 precursor | 1.893 | |
| gi|5031635 | Cofilin-1 | 1.821 | 0.000 |
| gi|4507879 | Voltage-dependent
anion-selective channel protein 1 | 1.781 | 0.013 |
| gi|50053795 | Eukaryotic
translation initiation factor 4B | 1.679 | 0.002 |
| gi|167614506 | Plastin-2 | 1.669 | 0.028 |
| gi|4758516 | Hepatoma-derived
growth factor isoform a | 1.663 | 0.015 |
| gi|4758756 | Nucleosome assembly
protein 1-like 1 | 1.575 | 0.000 |
| gi|112380628 | Lysosome-associated
membrane glycoprotein 1 precursor | 1.567 | |
| gi|4503481 | Elongation factor
1-γ | 1.560 | 0.000 |
| gi|23110935 | Proteasome subunit
α type-1 isoform 1 | 1.493 | 0.024 |
| gi|25777713 | S-phase
kinase-associated protein 1 isoform b | 1.490 | |
| gi|19743823 | Integrin β-1
isoform 1A precursor | 1.488 | 0.001 |
| gi|4506671 | 60S acidic
ribosomal protein P2 | 1.479 | 0.000 |
| gi|5032057 | Protein
S100-A11 | 1.479 | 0.005 |
| gi|4757768 | Rho
GDP-dissociation inhibitor 1 isoform a | 1.454 | 0.003 |
| gi|5901912 | Calmodulin | 1.448 | 0.001 |
| gi|386642862 | Threonine-tRNA
ligase, cytoplasmic isoform 2 | 1.444 | 0.010 |
| gi|4758484 | Glutathione
S-transferase ω-1 isoform 1 | 1.441 | 0.023 |
| gi|4504251 | Histone H2A type
2-A | 1.429 | 0.021 |
| gi|6031192 | Phosphate carrier
protein, mitochondrial isoform a precursor | 1.427 | 0.024 |
| gi|10863927 | Peptidyl-prolyl
cis-trans isomerase A | 1.414 | 0.001 |
| gi|73486658 | Aspartate
aminotransferase, mitochondrial precursor | 1.396 | 0.019 |
| gi|119395750 | Keratin, type II
cytoskeletal 1 | 1.388 | 0.004 |
| gi|385298707 | Hippocalcin-like
protein 1 | 1.370 | 0.005 |
| gi|50592994 | Thioredoxin isoform
1 | 1.356 | 0.045 |
| gi|4503471 | Elongation factor
1-α 1 | 1.305 | 0.002 |
| gi|24307939 | T-complex protein 1
subunit ɛ | 1.297 | 0.003 |
| gi|4758950 | Peptidyl-prolyl
cis-trans isomerase B precursor | 1.289 | 0.009 |
| gi|38327039 | Heat shock 70 kDa
protein 4 | 1.286 | 0.003 |
| gi|42544159 | Heat shock protein
105 kDa | 1.258 | 0.008 |
| gi|98986464 | Transmembrane emp24
domain-containing protein 10 precursor | 1.242 | 0.002 |
| gi|4758012 | Clathrin heavy
chain 1 | 1.221 | 0.011 |
| gi|5453603 | T-complex protein 1
subunit β isoform 1 | 1.215 | 0.022 |
| gi|4506663 | 60S ribosomal
protein L8 | 1.206 | 0.041 |
| gi|5901922 | Hsp90 co-chaperone
Cdc37 | 1.204 | 0.045 |
Discussion
In this study, a 5-FU-resistant TNBC cell line was
established from the TNBC cell line MDA-MB-231 by continuous
exposure to stepwise increases in the concentration of 5-FU. The
IC50 of 5-FU for the 5-FU-resistant MDA-MB-231 was
significantly increased compared with that for MDA-MB-231.
Moreover, MDA-MB-231/5-FU acquired cross-resistance to VNB, PTX and
GEM. To the best of our knowledge, this is the first study on the
establishment of a 5-FU-resistant TNBC cell line. MDA-MB-231/5-FU
should be useful to study the mechanisms underlying the 5-FU
resistance of TNBCs.
Recent studies have reported several determinants of
5-FU resistance mechanisms (15,26,27).
For instance, TS, a 5-FU-targeting enzyme; DPYD, a 5-FU-degrading
enzyme; and OPRT, a 5-FUanabolic enzyme, play key roles in the 5-FU
metabolism pathway. A previous study reported that the expression
of DPYD and TS was enhanced in 5-FU-resistant cell lines (12). However, the expression of DPYD and
TS was not enhanced in MDA-MB-231/5-FU. This suggests that the
mechanisms of 5-FU resistance of MDA-MB-231/5-FU differ from those
generally reported previously. Of note, MDA-MB-231/5-FU showed
cross-resistance to other anticancer drugs, such as PTX, VNB and
GEM. Likewise, it was reported that acquisition of 5-FU resistance
led to the acquisition of cross-resistance to other anticancer
drugs in gastric cancer cells (16). Multiple drug resistance describes a
phenomenon whereby resistance to one drug is accompanied by
resistance to other drugs whose structures and mechanisms of action
may be completely different. Mechanisms of multiple drug resistance
have been associated with increased drug efflux from cells, which
is mediated by an energy-dependent mechanism (8).
The ABC family proteins, which include MDR1 and
BCRP, play key roles in multiple drug resistance in breast cancer
(6–8). Overexpression of MDR1 confers
resistance to a variety of anticancer drugs, which are structurally
and functionally unrelated, including vincristine, VNB, etoposide,
PTX and many others. The expression of MDR1 and BCRP is increased
in MDA-MB-231/5-FU. The overexpression of these proteins may thus
be related to the partial contribution of drug efflux to multiple
drug resistance in these newly established cells. To consider what
kind of protein expression is enhanced other than that of ABC
family proteins, we performed iTRAQ-based quantitative proteomics
on MDA-MB-231/5-FU and the parent cells. The upregulated proteins
were classified into the GO categories of ‘DNA recombination’,
‘cell cycle’, ‘complex assembly’, ‘transport’ and ‘negative
regulation of cell death’. These results suggest that
MDA-NB-231/5-FU cells were resistant to 5-FU by the enhancement of
DNA recombination, regulation of the cell cycle, homologous
recombination and anti-apoptotic functions. These categorized
proteins can be related to mechanisms of drug resistance in
MDA-MB-231/5-FU. S-phase kinase-associated protein 1 (Skp1),
categorized as being involved in ‘DNA recombination’, exhibited a
1.49-fold increase in MDA-MB-231/5-FU compared with that in the
parent cells. Skp1 is composed of the Skp, Cullin and F-box
(SCF)-containing complex, which plays an important role in
regulating the ubiquitination of specific protein substrates and
regulators of cell cycle progression and development. Skp1 binds
directly to F-box motifs found in F-box proteins, such as Skp2,
FBW7 and β-transducin repeat-containing protein (28,29).
SCF protein complex regulates Akt ubiquitination, glycolysis and
tumorigenesis in breast cancer (30). MDA-MB-231/5-FU may thus show
enhanced ubiquitination and cell cycle progression because of the
overexpression of Skp1. Likewise, peptidyl-prolyl cis-trans
isomerase A, originally identified as an intracellular receptor for
cyclosporine A, exhibited a 1.41-fold increase in MDA-MB-231/5-FU
compared with that in the parent cells. The immunosuppressive
activity of cyclosporine A is thought to be mediated by the
engagement of calcineurin by the cyclosporin A-peptidyl-prolyl
cis-trans isomerase A complex, an observation supported by the
finding that peptidylprolyl cis-trans isom-erase A-knockout mice
are resistant to immunosuppression by cyclosporin A (31,32).
Peptidyl-prolyl cis-trans isomerase A was shown to be upregulated
in 5-FU-treated colorectal cancer cells (33). Moreover, the overexpression of
peptidyl-prolyl cistrans isomerase A induced chemoresistance to GEM
(34). In this study,
overexpression of ABC family proteins was observed in
MDA-MB-231/5-FU. However, we maintain that the acquisition of
multidrug resistance was not only due to the increased expression
of ABC family proteins. In accordance with the above findings,
MDA-MB-231/5-FU should be useful to identify factors that
contribute to chemoresistance in TNBCs.
Clinically, TNBC patients are treated with
combination therapy of 5-FU, epirubicin and cyclophosphamide at the
first-line approach. If these drugs have no effect on disease
progression, PTX is applied as a second-line treatment and GEM,
VNB, or other drugs as a third-line treatment. However, our
5-FU-resistant TNBC cell line acquired resistance to 5-FU, VNB, PTX
and GEM. TNBCs are generally more aggressive than the standard
level owing to drug resistance that developed via previous
chemotherapy. This indicates that TNBC patients acquire resistance
to 5-FU via the development of cross-resistance to VNB, PTX and
GEM. Thus, the MDA-MB-231/5-FU established in this study should be
useful for identifying new mechanisms of drug resistance and new
drug targets.
Acknowledgements
The authors also wish to thank members
of the Central Laboratory of Osaka City University Graduate School
of Medicine, for providing technical support. This work was
supported by JSPS KAKENHI Grant number 24650647 (to Katsuyuki
Takahashi).
References
|
1.
|
Foulkes WD, Smith IE and Reis-Filho JS:
Triple-negative breast cancer. N Engl J Med. 363:1938–1948. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Liedtke C, Mazouni C, Hess KR, et al:
Response to neoadjuvant therapy and long-term survival in patients
with triple-negative breast cancer. J Clin Oncol. 26:1275–1281.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Thike AA, Cheok PY, Jara-Lazaro AR, et al:
Triple-negative breast cancer: clinicopathological characteristics
and relationship with basal-like breast cancer. Mod Pathol.
23:123–133. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Dent R, Trudeau M, Pritchard KI, et al:
Triple-negative breast cancer: clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Isakoff SJ: Triple-negative breast cancer:
role of specific chemotherapy agents. Cancer J. 16:53–61. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Marquette C and Nabell L:
Chemotherapy-resistant metastatic breast cancer. Curr Treat Options
Oncol. 13:263–275. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Chen ZS and Tiwari AK: Multidrug
resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic
diseases. FEBS J. 278:3226–3245. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Baguley BC: Multiple drug resistance
mechanisms in cancer. Mol Biotechnol. 46:308–316. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Abraham J, Edgerly M, Wilson R, et al: A
phase I study of the P-glycoprotein antagonist tariquidar in
combination with vinorelbine. Clin Cancer Res. 15:3574–3582. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Ruff P, Vorobiof DA, Jordaan JP, et al: A
randomized, placebo-controlled, double-blind phase 2 study of
docetaxel compared to docetaxel plus zosuquidar (LY335979) in women
with metastatic or locally recurrent breast cancer who have
received one prior chemotherapy regimen. Cancer Chemother
Pharmacol. 64:763–768. 2009. View Article : Google Scholar
|
|
11.
|
Zheng G, Peng F, Ding R, et al:
Identification of proteins responsible for the multiple drug
resistance in 5-fluorouracil-induced breast cancer cell using
proteomics analysis. J Cancer Res Clin Oncol. 136:1477–1488. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Nakamura A, Nakajima G, Okuyama R, et al:
Enhancement of 5-fluorouracil-induced cytotoxicity by leucovorin in
5-fluorouracil-resistant gastric cancer cells with upregulated
expression of thymidylate synthase. Gastric Cancer. Mar
15–2013.(Epub ahead of print).
|
|
13.
|
Kodera Y, Ito S, Fujiwara M, et al: Gene
expression of 5-fluorouracil metabolic enzymes in primary gastric
cancer: correlation with drug sensitivity against 5-fluorouracil.
Cancer Lett. 252:307–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Ichikawa W, Takahashi T, Suto K, et al:
Thymidylate synthase predictive power is overcome by irinotecan
combination therapy with S-1 for gastric cancer. Br J Cancer.
91:1245–1250. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Zhang X, Yashiro M, Qiu H, et al:
Establishment and characterization of multidrug-resistant gastric
cancer cell lines. Anticancer Res. 30:915–921. 2010.PubMed/NCBI
|
|
17.
|
Uchibori K, Kasamatsu A, Sunaga M, et al:
Establishment and characterization of two 5-fluorouracil-resistant
hepatocellular carcinoma cell lines. Int J Oncol. 40:1005–1010.
2012.PubMed/NCBI
|
|
18.
|
Yanagihara K, Takigahira M, Tanaka H, et
al: Establishment and molecular profiling of a novel human
pancreatic cancer panel for 5-FU. Cancer Sci. 99:1859–1864. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Kakehashi A, Ishii N, Shibata T, et al:
Mitochondrial prohibitins and septin 9 are implicated in the onset
of rat hepatocarcinogenesis. Toxicol Sci. 119:61–72. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Park JS, Young Yoon S, Kim JM, et al:
Identification of novel genes associated with the response to 5-FU
treatment in gastric cancer cell lines using a cDNA microarray.
Cancer Lett. 214:19–33. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Shieh SY, Ikeda M, Taya Y, et al: DNA
damage-induced phosphorylation of p53 alleviates inhibition by
MDM2. Cell. 91:325–334. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Tibbetts RS, Brumbaugh KM, Williams JM, et
al: A role for ATR in the DNA damage-induced phosphorylation of
p53. Genes Dev. 13:152–157. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Shin JY, Kim JO, Lee SK, et al: LY294002
may overcome 5-FU resistance via down-regulation of activated p-AKT
in Epstein-Barr virus-positive gastric cancer cells. BMC Cancer.
10:4252010. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Yanamoto S, Iwamoto T, Kawasaki G, et al:
Silencing of the p53R2 gene by RNA interference inhibits growth and
enhances 5-fluorouracil sensitivity of oral cancer cells. Cancer
Lett. 223:67–76. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Chu E, Drake JC, Koeller DM, et al:
Induction of thymidylate synthase associated with multidrug
resistance in human breast and colon cancer cell lines. Mol
Pharmacol. 39:136–143. 1991.PubMed/NCBI
|
|
27.
|
Peters GJ, Backus HH, Freemantle S, et al:
Induction of thymidylate synthase as a 5-fluorouracil resistance
mechanism. Biochim Biophys Acta. 1587:194–205. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Chen J, Shen BY, Deng XX, et al:
SKP1-CULLIN1-F-box (SCF)-mediated DRG2 degradation facilitated
chemotherapeutic drugs induced apoptosis in hepatocellular
carcinoma cells. Biochem Biophys Res Commun. 420:651–655. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Nakayama KI and Nakayama K: Ubiquitin
ligases: cell-cycle control and cancer. Nat Rev Cancer. 6:369–381.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Chan CH, Li CF, Yang WL, et al: The
Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis,
herceptin sensitivity, and tumorigenesis. Cell. 149:1098–1111.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Zhu D, Cardenas ME and Heitman J:
Calcineurin mutants render T lymphocytes resistant to cyclosporin
A. Mol Pharmacol. 50:506–511. 1996.PubMed/NCBI
|
|
32.
|
Colgan J, Asmal M, Yu B, et al:
Cyclophilin A-deficient mice are resistant to immunosuppression by
cyclosporine. J Immunol. 174:6030–6038. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Wong CS, Wong VW, Chan CM, et al:
Identification of 5-fluorouracil response proteins in colorectal
carcinoma cell line SW480 by two-dimensional electrophoresis and
MALDI-TOF mass spectrometry. Oncol Rep. 20:89–98. 2008.PubMed/NCBI
|
|
34.
|
Kuramitsu Y, Taba K, Ryozawa S, et al:
Identification of up- and down-regulated proteins in
gemcitabine-resistant pancreatic cancer cells using two-dimensional
gel electrophoresis and mass spectrometry. Anticancer Res.
30:3367–3372. 2010.PubMed/NCBI
|