Introduction
The tumor suppressor gene p53 plays a pivotal role
in maintenance of genomic stability, possession of anticancer
activity, protection against malignant transformation, induction of
apoptosis when DNA damaged cells are beyond repair and inhibition
of angiogenesis (1). However, more
than 50% of human cancers contain mutated or a deleted p53 gene,
while other tumors have had inactivation of the p53 gene (2). An early study showed restoration of
endogenous p53 function holds considerable promise in the control
of tumor progression (3). In
normal homeostasis of cells, p53 levels are kept low through a
continuous degradation of p53. In response to cell stress, such as
DNA damage induced by ultraviolet irradiation, chemical agents or
oxidative stress, the p53 gene becomes activated and the half-life
of the p53 protein will be increased, leading to p53 accumulation
in stressed cells. Subsequently, the p53 protein will change
conformation to function as a transcription regulator to upregulate
expression of the numerous downstream genes that are associated
with cell cycle arrest, senescence, autophagy and apoptosis
(4–7). In contrast, dysfunctional p53 protein
loses its transcriptional activity and cannot suppress tumor
growth, but rather accelerates tumorigenesis. Thus, investigation
of p53 functions and its degradation could help in gaining
knowledge how to effectively control cancer development and
progression. The p53 protein is regulated by a complex network
(8). Murine double minute 2
(MDM2), one of the important downstream genes of the p53 regulatory
network, can regulate stability and activity of the p53 protein by
binding to the N-terminal part of p53; thus, MDM2 is involved in
cell growth inhibition, apoptosis induction and cell cycle control
(9). MDM2 and p53 interact and
regulate each other through an autoregulatory feedback loop
(10). p53 can transcriptionally
induce MDM2 expression, whereas MDM2 mediates ubiquitylation of
p53, thereby marking p53 for nuclear export and proteasomal
degradation. In contrast, murine double minute X (MDMX), homologous
to MDM2, was indicated to not to be a p53 target gene and MDMX
protein may participate in inhibiting p53 activity by interacting
with p53 Box I, but that it lacks intrinsic ubiquitin-ligase
activity (11); thus, it is also
involved in the autoregulatory feedback loop of MDM2-p53
interaction (12). Therefore, the
functions of the MDM2 and MDMX proteins are interdependent, but are
not interchangeable in the regulation of p53 protein.
To date, studies on the p53 gene as a target of
cancer therapy has made considerable progress, of which restoration
of wild-type p53 function in tumor cells (13) has attracted the greatest attention.
Thus, MDM2, a critical regulator of the p53 protein, is an
attractive target for the development of novel antitumor agents
(14). It has been shown that the
function of the p53 protein can be activated by blocking the
MDM2-p53 interaction, such as through the use of a small-molecule
inhibitor nutlins to block the MDM2-p53 protein interaction
(15). The surprising finding that
decreasing MDM2 does not appear to increase p53 activity can be
explained by recent studies indicating that DNA damage induces MDM2
self-degradation and an MDM2-denpendent degradation of MDMX,
especially in MDMX overexpressed cancer cells (16), suggesting that it is essential for
the p53 response that integrates the distinct and complementary
roles of MDM2 and MDMX in p53 inhibition, and the role of
MDM2-mediated degradation itself and MDMX for p53 activation. MDM2
and MDMX antagonists together lead to more potent activation of p53
in tumor cells for induction of tumor cell cycle arrest and
apoptosis (17,18). Inactivation of p53 occurs in 15–50%
of breast cancer cases (2).
Restoration of p53 activity could lead to tumor regression and is
therefore considered a useful strategy for treatment of breast
cancer. In this study, we synthesized a cell-permeable dual-target
MDM2/MDMX inhibitory protein that contains the transactivator (TAT)
peptide for transduction across membranes and the scaffold protein
(thioredoxin) displaying the MDM2/MDMX inhibitory peptide pDI
(protein disulfide isomerase). This protein can bind to MDM2 and
MDMX to disrupt their interaction with p53. We then investigated
the antitumor activity of this protein in breast cancer cell
lines.
Materials and methods
Cell lines and culture
Human breast adenocarcinoma cell line MCF-7 and
human breast infiltrating duct carcinoma cell line ZR-75-30 (both
with wild-type p53) were obtained from The Medical Research Center
of Xi’an Jiaotong University (Xi’an, China) and cultured in
RPMI-1640 medium containing 10% fetal bovine serum and 1%
benzylpenicillin/streptomycin at 37˚C in a humidified incubator
with 5% CO2. Pancreatin/ethylene diamine tetraacetic
acid (EDTA) (0.05%) was used to detach the cells for
subculture.
Construction of expression vector and
gene expression
We first amplified thioredoxin A (TrxA) to express
the scaffold protein using polymerase chain reaction (PCR) with
p1/p2 primers (Table I) and pET32a
as a template. The 327 bp PCR product was purified and extracted
using the Agarose-Gel extraction kit (Omegas, Norcross, GA). After
DNA-sequence confirmation, the PCR product was inserted into the
pMD18-T vector (Takara, Dalian, China). After amplification and DNA
sequence confirmation, the pMD18-T-TrxA was digested with
NcoI and EcoRV and the released fragment was further
cloned into the prokaryotic expression vector pET40b (Novagen;
Centurion, USA) to yield pET40b-TrxA. TAT sequences were amplified
by using PCR with primers p3/p4 and cloned into pET40b-TrxA at
BamHI and SacI sites to yield pET40b-TAT-TrxA. PDI
was amplified by using PCR with primers p5/p6 and cloned into
pET40b-TAT-TrxA at the RsrII site to yield
pET40b-TAT-TrxA-pDI.
 | Table I.PCR primers used in this study. |
Table I.
PCR primers used in this study.
| p1: | 5′-GATATCGGCCAGGTTAGCGT-3′ |
|
NcoI |
| p2: | 5′-GGTACCCGATGAGCGATAAAATTA-3′ |
|
EcoRV |
| p3: | 5′-GATCCGTATGGCCGCAAGAAGCGTCGCCAGCGCCGTCGCCGGAGCT-3′ |
|
BamHI
SacI |
| p4: | 5′-CCGGCGACGGCGCTGGCGACGCTTCTTGCGGCCATACG-3′ |
| p5: | 5′-GTCCGCCTCTGAGTTTGACGTTTGAGCATTATTGGGCGCAGTTGACGAGCGAAAACG-3′ |
|
RsrII |
| p6: | 5′-GACCGTTTTCGCTCGTCAACTGCGCCCAATAATGCTCAAACGTCAAACTCAGAGGCG-3′ |
|
RsrII |
The pET40b-TAT-TrxA-pDI and the control vector
pET40b-TAT-TrxA were transduced into E. coli BL21 (DE3)
(Novagen) for in vitro expression of the dual-target
MDM2/MDMX inhibitory protein and the control protein, respectively,
according to a previous study (19). Transformed E. coli was
cultured for 8–12 h in Luria-Bertani (LB) medium containing 50
μg/ml kanamycin, seeded into the previous medium at a 1:50
(v/v) ratio, and then further cultured for 3–5 h at 37˚C. Protein
expression was induced by the addition of different concentrations
of Isopropyl β-D-1-Thiogalactopyranoside (IPTG) (0.1, 0.4, 0.7 and
1 mM). The time at which IPTG was added to the cultures was
designated at 1, 3, 5, 7 and 9 h and the temperatures for induction
of protein expression were set to 16, 25 and 37˚C, respectively.
Based on the results, the optimal expression conditions were
determined.
Protein purification
The E. coli culture was centrifuged at 4,000
x g for 20 min at 4˚C; the precipitates were resuspended in a
bacterial lysate buffer containing lysozyme and sonicated on ice
for 30 cycles (9 sec on, 9 sec off). The sonicated bacterial
lysates were centrifuged at 4,000 x g for 10 min at 4˚C and then
briefly washed with Buffer A (2 M urea, 500 mM NaCl, 50 mM PBS, pH
7.5). After centrifuging, the pellets were dissolved in Buffer B (8
M urea, 500 mM NaCl, 50 mM PBS, pH 7.5) containing 10 mM imidazole
at 4˚C overnight. The next day, the mixture was loaded onto a
pre-equilibrated Ni-NTA column for purification of newly
synthesized protein. The column was washed three times with Buffer
B containing 20 mM imidazole and eluted with Buffer B containing
100 mM imidazole.
Protein refolding
To remove the urea and refold the protein, the
purified fractions of protein contents were pooled and gradually
dialyzed in Buffer C (0.1 mM GSH, 0.01 mM GSSG, 500 mM NaCl, 10%
glycerin, 400 mM arginine hydrochloride, 50 mM PBS, pH 7.5) with a
decreasing concentration of urea followed by PBS buffer with 10%
glycerin. Afterwards, the protein concentration was measured
according to the Bradford method (20) using bovine serum albumin (BSA) as
the standard. After filtration and sterilization, the proteins were
stored in 50 mM PBS with 10% glycerin at −80˚C until use (within 6
months).
ELISA
Enzyme-linked immunosorbent assay (ELISA) was
performed as described previously (21) to measure whether the recombinant
dual-target MDM2/MDMX inhibitory protein could inhibit interaction
of MDM2 or MDMX with the p53 protein. Briefly, cell culture plates
were coated with 2 μg/ml of p53 active motif peptide diluted
in PBS overnight at 4˚C. Next, GST-MDM2-1-150 (GST-MDMX-1-200)
(Abnova; Taipei, Taiwan) containing human MDM2 (MDMX), recombinant
protein, control protein but did not contain pDI and dimethyl
sulfoxide (DMSO) were added to the plates, respectively, and
incubated for 1 h. Anti-GST monoclonal antibody (Abcam, Cambridge,
MA) was added into the each well and incubated for 1 h at room
temperature and subsequently washed with PBS thrice. Horseradish
peroxidase (HRP)-conjugated anti-mouse IgG (Abcam) was added to
each well and incubated for 30 min. The optical density (OD) was
measured at 450 nm using an ELISA reader.
Co-immunoprecipitation-western blot
analysis
GST-MDM2-1-150 (GST-MDMX-1-200), anti-His antibody
(Abcam) and recombinant protein or control protein were mixed
together and incubated for 2 h at 4˚C. The binding proteins were
precipitated by using a co-immunoprecipitation kit (Genmed,
Shanghai, China) as described by Momand et al (22). The immunoprecipitated proteins were
identified by using western blot analysis.
Protein extraction and western blot
analysis
To determine the expression of p53, MDM2 and MDMX in
breast cancer cells, the protein samples were fractionated by 10%
sodium dodecyl sulfated-polyacrylamide gel electrophoresis
(SDS-PAGE), and transferred onto a nitrocellulose membrane
(Millipore, Billerica, MA) for western blot analysis. The p53, MDM2
and MDMX proteins were detected using their corresponding primary
antibodies at a concentration of 1 μg/ml followed by
incubation with HRP-conjugated secondary antibodies (Pierce,
Rockford, IL). The protein band was visualized using enhanced
chemiluminescence (ECL, Pierce) and exposed to X-ray film.
Cell viability assay
Cell viability was assessed through a thizolyl blue
(MTT) colorimetric assay. Briefly, breast cancer cells were seeded
onto 96-well culture plates at a density of 2.5×103
cells/well. The recombinant protein, control protein and PBS were
added into each well with different gradient concentrations and the
cells incubated for 48 h. At the end of experiments, 20 μl
of MTT solution (5 mg/ml in 10 mM PBS; Sigma, St. Louis, MO) was
added in each well and the cells incubated for 4 h at 37˚C. The
culture medium was replaced with 200 μl of DMSO to dissolve
the MTT metabolite and the OD measured at 490 nm using a
micro-ELISA reader. Cell viability was calculated as the percentage
absorbance relative to that of the control cultures.
Flow cytometry assay
Flow cytometry was performed to detect cell cycle
distribution and apoptosis. For cell cycle distribution, the cells
were stained with propidium iodide (PI). Breast cancer cells were
treated with different proteins as indicated above. Cells were
collected, washed twice with ice-cold PBS and then fixed in 75%
ice-cold ethanol at 4˚C overnight. Cells were centrifuged at 600 x
g for 5 min and the precipitate incubated with 2 mg/ml RNase A and
then stained with 50 μg/ml PI containing 0.1% Triton X-100
and 0.02 mg/ml EDTA at 37˚C for 30 min. The percentage of cells in
each stage of the cell cycle was determined by a flow cytometer and
calculated by CellQuest software (BD Biosciences, Franklin Lakes,
NJ).
To evaluate apoptosis, the cells were stained with
Annexin V-FITC (fluoresecein isothiocyanate) and PI. Briefly,
breast cancer cells treated with different proteins were collected
and washed with PBS. The cells were resuspended with in binding
buffer at a concentration of 1×106 cells/ml and stained
with 5 μl of Annexin V-FITC and 5 μl of PI and then
subjected to flow cytometric analysis.
Statistical analysis
Statistical analysis was performed by using SPSS
16.0 software (SPSS, Chicago, IL). All data are presented as mean ±
standard deviation. Differences between groups were analyzed using
one-way ANOVA and Student’s t-test was used to evaluate the
statistical significance of the mean. All statistical tests were
two-sided and a p-value ≤0.05 was considered statistically
significant. In each figure error bars represent standard error of
the mean and statistical significance levels are noted as:
*P<0.05, **P<0.01.
Results
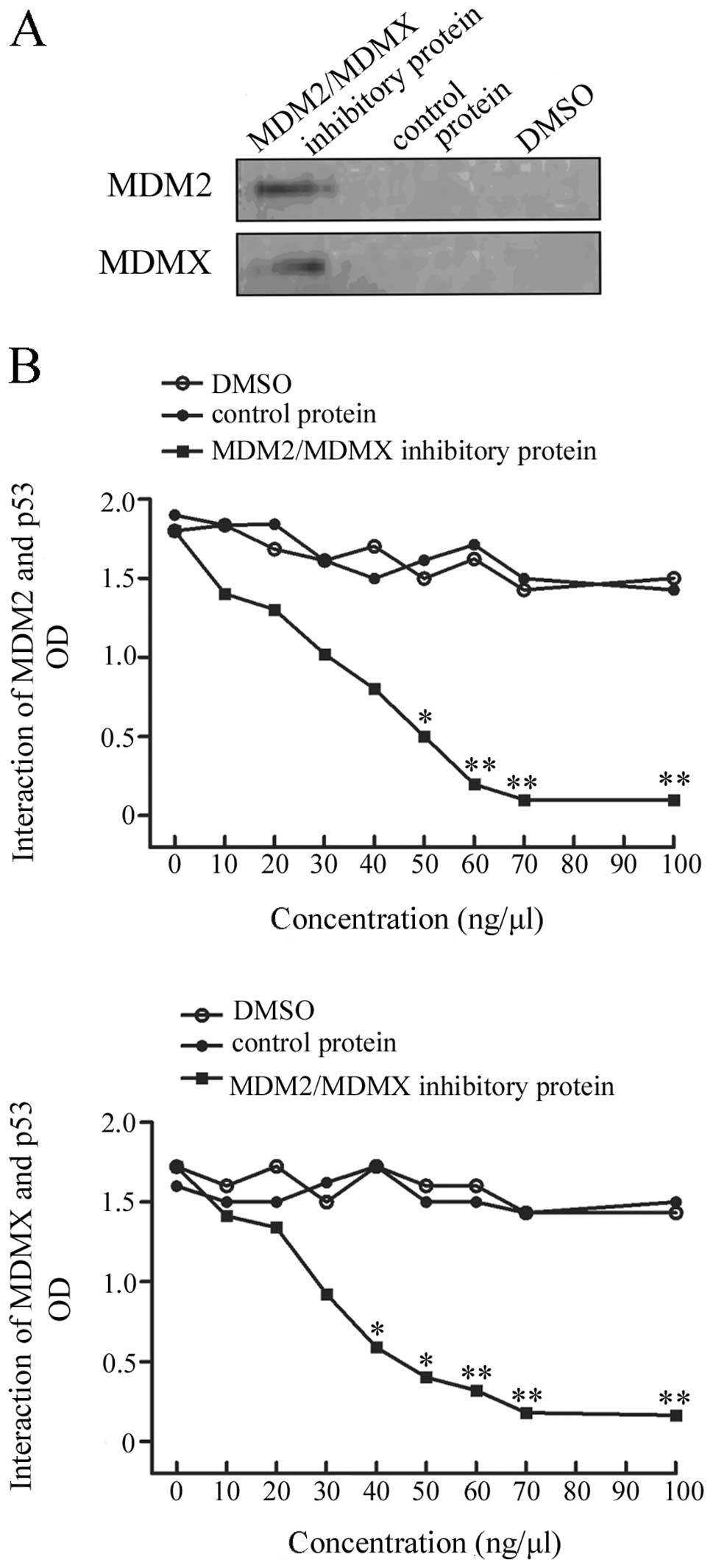
Expression and quality control of the
recombinant dual-target MDM2 and MDMX protein
After in vitro expression and purification
this recombinant protein coupled with MDM2 and MDMX,
co-immunoprecipitation-western blot analysis was performed to check
the quality of the protein. The data showed that this protein was
able to be immunoprecipitated by anti-MDM2 and anti-MDMX
antibodies, indicating that this protein is functional (Fig. 1A).
Inhibition of MDM2-p53 and MDMX-p53
interaction using this recombinant protein
In order to determine whether the recombinant
protein could inhibit MDM2-p53 and MDMX-p53 interaction, an ELISA
was performed. The dual-target MDM2/MDMX inhibitory protein
strongly disrupted both MDM2 and MDMX interaction with p53,
compared to the controls. This protein inhibited interaction of
MDM2/MDMX with p53 in a dose-dependent manner (Fig. 1B).
Inhibition of wild-type p53 breast cancer
cell proliferation by this recombinant protein
Next, the effects of this recombinant protein on
regulation of MCF-7 and ZR-75-30 cell viability were detected by
treating them with different concentrations of the dual-target
MDM2/MDMX inhibitory protein or nutlin-3α. MTT assay data showed
that this recombinant protein inhibited the viability of MCF-7
cells and ZR-75-30 cells in a dose-dependent manner. The
recombinant protein showed a stronger inhibition of cell
proliferation than nutlin-3α, a well characterized MDM2 inhibitor
(Fig. 2).
Induction of breast cancer cell cycle
arrest by the recombinant protein
To assess the cause of the reduced cell viability by
the recombinant protein, we analyzed cell cycle distribution using
a flow cytometric assay. The percentage of the sub-G1 fraction of
breast cancer cell lines MCF-7 and ZR-75-30 treated with this
protein was obviously increased (72.49±0.90% and 76.43±2.07%,
respectively). However, nutlin-3α treatment only induced a slight
increase in the sub-G1 fraction in both MCF-7 cells (51.58±1.58%)
and ZR-75-30 cells (60.53±0.64%) compared to the untreated MCF-7
and ZR-75-30 cells (49.64±1.42% and 48.23±0.64%, respectively)
(P<0.05, Fig. 3A).
Induction of apoptosis in breast cancer
cells by the recombinant protein
Increased sub-G1 phase of the cell cycle may
indicate induced cell apoptosis; thus, we performed a flow
cytometry/Annexin V-PI assay to detect the level of apoptosis.
Treatment of MCF-7 cells with the dual-target MDM2/MDMX inhibitory
protein, nutlin-3α or PBS had differential effects on apoptosis,
resulting in 35.27±0.54, 10.34±1.13 and 7.41±0.83% of apoptotic
cells, respectively. This recombinant protein caused a significant
increase in the percentage of apoptotic cells compared to that of
PBS-control or nutlin-3α-treated cells. The same treatment of
ZR-75-30 cells resulted in 49.69±1.13, 18.92±0.64 and 9.65±0.73% of
apoptotic cells, respectively. The recombinant protein was more
effective than nutlin-3α treatment or PBS (P<0.05, Fig. 3B).
Inhibition of MDM2 and MDMX expression
and activation and stabilization of p53 protein in breast cancer
cells by the recombinant protein
We demonstrated the usefulness of the dual target
MDM2/MDMX inhibitory protein in regulation of breast cancer cell
viability, cell cycle arrest and apoptosis. Next, we determined
whether the dual target MDM2/MDMX inhibitory protein was able to
inhibit MDM2 and MDMX expression and activate and stabilize p53
protein in breast cancer cells. Western blot analysis showed that
the level of p53 expression was dramatically increased in MCF-7
cells treated with the recombinant protein compared to cells
treated with nutlin-3α. ZR-75-30 cells also showed similar results.
However, there was a sharp decrease in MDM2 expression and in MDMX
level in MCF-7 and ZR-75-30 cells cultured with the recombinant
protein compared to that of cells treated with nutlin-3α (Fig. 4A).
Induction of p21, Bax and puma expression
in breast cancer cells by the recombinant protein
The molecular events after p53 gene activation were
assessed by detection of cell apoptosis-regulatory proteins (Bax
and puma) and the cell cycle inhibitory protein p21 using western
blot analysis. In the wild-type p53 breast cancer cell lines, a
marked increase in p21 expression was observed in the cells treated
with the dual-target MDM2/MDMX inhibitory protein. In contrast to
the inhibitor of MDM2 and MDMX, as control, nutlin-3α can also
slightly change p21 expression (Fig.
4B). Breast cancer cells treated with the recombinant protein
resulted in a significant increase in the levels of Bax and puma
proteins, whereas such an effect was not observed in the
nutlin-3α-treated tumor cells.
Discussion
In the current study, we determined the feasibility
of a dual-target MDM2/MDMX inhibitory protein to control breast
cancer by activation and stabilization of p53 protein in
vitro. Our data demonstrated that i) the dual-target MDM2/MDMX
inhibitory protein expressed in E. coli is functional, ii)
treatment with the dual-target MDM2/MDMX inhibitory protein
suppressed the viability of wild-type p53 breast cancer cells and
induced cell cycle arrest and apoptosis of tumor cells, which was
much more effective than that of the MDM2 inhibitor nutlin-3α, iii)
the dual-target MDM2/MDMX inhibitory protein was able to inhibit
MDM2 and MDMX expression and activate and stabilize the p53
protein, and iv) the dual-target MDM2/MDMX inhibitory protein
increased expression of p21, Bax and puma proteins. Thus,
inhibition of MDM2/MDMX proteins could be useful to activate and
stabilize p53 protein for restoration of p53 tumor suppressor
functions in breast cancer or other wild-type p53 silenced
tumors.
Indeed, the ubiquitously expressed tumor suppressor
p53 is a multi-functional protein that regulates cellular stress
responses such as cell cycle arrest, apoptosis and cell senescence
(23). Thus, the aim to restore
p53 activity in human cancer has helped in developing a number of
antitumor therapies in preclinical and clinical trials (24). Early studies utilized a gene
therapy approach to deliver wild-type p53 cDNA into lung and head
and neck cancer patients, which showed p53 antitumor activity
(25–27). However, due to the toxicity and
side-effects of the gene delivery vector or adenovirus, such an
approach was disregarded. However, small molecules have been
engineered to restore expression of wild-type p53 and the
target-specific measure is more desirable (28). Numerous proteins have been
described to regulate p53 pathway. Among these, MDM2 and MDMX stand
out because of their importance. In the early stage, it was found
that the MDM2 or MDMX-deficient mice died in the uterus, but these
deficiencies were viable in a p53-deficient background, which
indicating MDM2 and MDMX are not redundant p53 inhibitors.
In the current study, we regarded MDM2 and MDMX as
the therapeutic targets in order to achieve full activation of p53.
We designed and constructed a prokaryotic expression vector to
carry a dual-target MDM2/MDMX inhibitory protein; the recombinant
protein was expressed in E. coli and was found to be
functional. Our in vitro data showed that this dual-target
MDM2/MDMX inhibitory protein possessed a higher tumor inhibition
rate, caused a larger increase in the sub-G1 fraction of cells, and
promoted more p53 wild-type breast cancer cell apoptosis compared
to nutlin-3α. These data indicate that the dual-target MDM2/MDMX
inhibitory protein can re-activate p53 for its antitumor activity
and is much more effective than the single MDM2 inhibitor nutlin-3α
in antitumor activity. Our results confirmed data from previous
studies that the dual-target MDM2/MDMX inhibitory protein also had
antitumor activity in lung cancer, colon cancer and retinoblastoma
cells (18,29). However, the reason for less
effectiveness of MDM2-p53 inhibitors in nutlin-3α treated tumor
cells may be because they failed to activate p53 especially in
cells overexpressing MDMX (14,30,31),
which further confirmed that MDMX plays a pivotal role inactivation
and stabilization of p53 in cancer cells (32,33).
Furthermore, our current study further investigated
the downstream gene of the p53 protein. Treatment of breast cancer
cells with the dual-target MDM2/MDMX inhibitory protein
significantly induced expression of the p53 protein, but
downregulated MDM2 and MDMX expression compared to nutlin-3α
treatment. Attributed to pDI, this recombinant protein was designed
to downregulate MDM2 and MDMX proteins through binding to these
proteins and disrupting their interaction with p53, which is in
turn to upregulate p53 protein by increasing the half-life of p53
protein due to MDM2, the downstream gene of p53, can promote
degradation of p53 via an autoregulatory feedback loop (34,35).
MDMX as another important downstream gene of the p53 regulatory
network, it shows little ubiquitylation activity towards p53 but
enhanced activity of MDM2 (7) by
an autoregulatory feedback loop of MDM2-p53 interaction (12).
Indeed, the dual-target MDM2/MDMX inhibitory protein
was able to increase levels of p21, Bax and puma proteins,
confirming the activity of p53 protein in the cells. P21 is the
p53-regulated protein to control cell cycle progression and induce
cell cycle arrest when overexpressed, while Bax and puma are the
p53-targeted pro-apoptotic genes involved in cancer cell apoptosis.
Normally, p53 regulates p21 expression to coordinate cell cycle
G0/G1 phase checkpoint. High expression of both p21 and p53 is one
of the reasons why the cell cycle arrests in G0/G1 phase in breast
cancer cells treated with inhibitor of MDM2 and MDMX. Thus,
expression of p21, puma and Bax protein was the effect of p53
activation and the latter was through suppression of MDM2 and MDMX
by the dual-target MDM2/MDMX inhibitory protein in wild-type p53
breast cancer cells.
Further studies will evaluate the feasibility of
this approach in a clinical trial. The recent studies estimated
that the MDM2 and MDMX antagonists could be used in the treatment
of 2–3 millions patients diagnosed with cancer per year, which
could provide a strong incentive for the search for p53-based
anticancer strategies, but the potential toxicity of such
inhibitory proteins to normal cells remains to be determined.
Moreover, potential addiction of tumor cells to overexpressed MDM2
or MDMX may make them uniquely vulnerable to p53 activating agents.
In addition, combination with a low and non-toxic concentration of
standard cytotoxic chemotherapeutic agents will produce favorable
therapeutic indices. Clearly, the ability to obtain diverse
p53-activating strategies opens up exciting opportunities for
development and implementation of new therapeutic strategies for
the large population of cancer patients. However, this study is a
proof-of-principle for dual targeting of MDM2/MDMX to activate p53
protein antitumor activity. To overcome the bottleneck of this
recombinant protein, a better strategy or approach is needed to
generate a peptide with more stability, longer half-life and no
immunogenicity to be used in patients. Thus, there is a long way
ahead before the current knowledge can be applied for breast cancer
therapeutics in the clinic.
Acknowledgements
This study was supported by ‘the
Fundamental Research Funds for Central University’.
References
|
1.
|
Harris SL and Levine AJ: The p53 pathway:
positive and negative feedback loops. Oncogene. 24:2899–2908. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Hollstein M, Sidransky D, Vogelstein B and
Harris CC: p53 mutations in human cancers. Science. 253:49–53.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Ventura A, Kirsch DG, McLaughlin ME, et
al: Restoration of p53 function leads to tumour regression in vivo.
Nature. 445:661–665. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Rother K, Kirschner R, Sanger K, Bohlig L,
Mossner J and Engeland K: p53 downregulates expression of the G1/S
cell cycle phosphatase Cdc25A. Oncogene. 26:1949–1953. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Yu J and Zhang L: The transcriptional
targets of p53 in apoptosis control. Biochem Biophys Res Commun.
331:851–858. 2005. View Article : Google Scholar
|
|
6.
|
Xue W, Zender L, Miething C, et al:
Senescence and tumour clearance is triggered by p53 restoration in
murine liver carcinomas. Nature. 445:656–660. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Crighton D, Wilkinson S, O’Prey J, et al:
DRAM, a p53-induced modulator of autophagy, is critical for
apoptosis. Cell. 126:121–134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Boehme KA and Blattner C: Regulation of
p53 - insights into a complex process. Crit Rev Biochem Mol Biol.
44:367–392. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Piette J, Neel H and Marechal V: Mdm2:
keeping p53 under control. Oncogene. 15:1001–1010. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Wu X, Bayle JH, Olson D and Levine AJ: The
p53-mdm-2 autoregulatory feedback loop. Genes Dev. 7:1126–1132.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Hock A and Vousden KH: Regulation of the
p53 pathway by ubiquitin and related proteins. Int J Biochem Cell
Biol. 42:1618–1621. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Marine JC and Jochemsen AG: Mdmx as an
essential regulator of p53 activity. Biochem Biophys Res Commun.
331:750–760. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Wiman KG: Strategies for therapeutic
targeting of the p53 pathway in cancer. Cell Death Differ.
13:921–926. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Bond GL, Hu W and Levine AJ: MDM2 is a
central node in the p53 pathway: 12 years and counting. Curr Cancer
Drug Targets. 5:3–8. 2005.PubMed/NCBI
|
|
15.
|
Vassilev LT, Vu BT, Graves B, et al: In
vivo activation of the p53 pathway by small-molecule antagonists of
MDM2. Science. 303:844–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Hu B, Gilkes DM, Farooqi B, Sebti SM and
Chen J: MDMX overexpression prevents p53 activation by the MDM2
inhibitor Nutlin. J Biol Chem. 281:33030–33035. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Toledo F and Wahl GM: MDM2 and MDM4: p53
regulators as targets in anticancer therapy. Int J Biochem Cell
Biol. 39:1476–1482. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Hu B, Gilkes DM and Chen J: Efficient p53
activation and apoptosis by simultaneous disruption of binding to
MDM2 and MDMX. Cancer Res. 67:8810–8817. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Wu SP, Fu AL, Wang YX, et al: A novel
therapeutic approach to 6-OHDA-induced Parkinson’s disease in rats
via supplementation of PTD-conjugated tyrosine hydroxylase. Biochem
Biophys Res Commun. 346:1–6. 2006.
|
|
20.
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Bottger A, Bottger V, Garcia-Echeverria C,
et al: Molecular characterization of the hdm2-p53 interaction. J
Mol Biol. 269:744–756. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Momand J, Zambetti GP, Olson DC, George D
and Levine AJ: The mdm-2 oncogene product forms a complex with the
p53 protein and inhibits p53-mediated transactivation. Cell.
69:1237–1245. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Zifou JT and Lowe SW: Tumor suppressive
function of p53. Cold Spring Harb Perspect Biol.
1:a0018832009.PubMed/NCBI
|
|
24.
|
Fischer U and Schulze-Osthoff K: New
approaches and therapeutics targeting apoptosis in disease.
Pharmacol Rev. 57:187–215. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Roth JA, Nguyen D, Lawrence DD, et al:
Retrovirus-mediated wild-type p53 gene transfer to tumors of
patients with lung cancer. Nat Med. 2:985–991. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Clayman GL, el-Naggar AK, Lippman SM, et
al: Adenovirus-mediated p53 gene transfer in patients with advanced
recurrent head and neck squamous cell carcinoma. J Clin Oncol.
16:2221–2232. 1998.PubMed/NCBI
|
|
27.
|
Swisher SG, Roth JA, Nemunaitis J, et al:
Adenovirus-mediated p53 gene transfer in advanced non-small-cell
lung cancer. J Natl Cancer Inst. 91:763–771. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Ritter T, Lehmann M and Volk HD:
Improvements in gene therapy: averting the immune response to
adenoviral vectors. BioDrugs. 16:3–10. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
McEvoy J, Ulyanov A, Brennan R, et al:
Analysis of MDM2 and MDM4 single nucleotide polymorphisms, mRNA
splicing and protein expression in retinoblastoma. PLoS One.
7:e427392012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Patton JT, Mayo LD, Singhi AD, Gudkov AV,
Stark GR and Jackson MW: Levels of HdmX expression dictate the
sensitivity of normal and transformed cells to Nutlin-3. Cancer
Res. 66:3169–3176. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Wade M, Wong ET, Tang M, Stommel JM and
Wahl GM: Hdmx modulates the outcome of p53 activation in human
tumor cells. J Biol Chem. 281:33036–33044. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Francoz S, Froment P, Bogaerts S, et al:
Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating
and quiescent cells in vivo. Proc Natl Acad Sci USA. 103:3232–3237.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Toledo F, Krummel KA, Lee CJ, et al: A
mouse p53 mutant lacking the proline-rich domain rescues Mdm4
deficiency and provides insight into the Mdm2-Mdm4-p53 regulatory
network. Cancer Cell. 9:273–285. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Stommel JM and Wahl GM: A new twist in the
feedback loop: stress-activated MDM2 destabilization is required
for p53 activation. Cell Cycle. 4:411–417. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Cai X and Yuan ZM: Stochastic modeling and
simulation of the p53-MDM2/MDMX loop. J Comput Biol. 16:917–933.
2009. View Article : Google Scholar : PubMed/NCBI
|