Introduction
Histone deacetylase inhibitors (HDACIs) and
proteasome inhibitors (PIs) have recently emerged as new groups of
therapeutic agents that are effective in treating a variety of
malignancies (1). HDACIs are
anticancer drugs that have moved rapidly through clinical
development, and in 2006, vorinostat (SAHA, Zolinza) was approved
by the FDA for the treatment of cutaneous T cell lymphoma (2). The targets of these compounds include
class I, II and IV HDACs, which function in deacetylating histone
proteins to induce chromatin remodeling and altered gene
transcription. Numerous non-histone proteins can also be modified
by acetylation, and thus, the inhibition of HDAC activity can
affect various molecular processes. This broad effect on protein
function may account for the pleiotropic responses generated by
HDACIs, including the induction of tumor cell apoptosis, cell cycle
arrest, antitumor activity in vivo, cell differentiation,
morphological changes in oncogene-transformed cells, invasion and
angiogenesis (3).
The ability of HDACIs to selectively induce tumor
cells to undergo apoptosis has been key to their therapeutic
efficacy in pre-clinical models. Moreover, HDACIs can augment the
apoptotic effects of other anticancer agents that have diverse
molecular targets. Although HDACIs are promising anti-cancer drugs,
particularly given their ability to be combined with other agents,
identifying the key molecular events that determine the biological
response of cells to HDACI treatment remains a challenge (3).
APC, an HDACI isolated from Fusarium sp., was
first reported to be a reversible inhibitor of the in vitro
development of apicomplexan parasites. APC acts by inhibiting the
HDAC enzyme of the parasite, and it was later shown to promote the
anti-proliferative activity and differentiation of mammalian cells
(4). APC has been shown to exhibit
antitumor activities in several human cancers, including leukemia,
cervical cancer, gastric cancer and breast cancer (5). In addition, APC has been found to
reverse the transformation of the human cervical cancer cells, HeLa
and H-ras-transformed human breast cancer cells (6). Studies have demonstrated that APC
induces apoptosis through the selective induction of Fas/Fas
ligand, resulting in the release of cytochrome c from the
mitochondria and the subsequent activation of caspase-9 and
caspase-3 (7). Very recently, APC
has been shown to induce apoptosis by endoplasmic stress and
mitochondrial dysfunction via PLCγ1 activation, Ca2+
release, and reactive oxygen species generation (8).
Accumulating evidence has suggested that
transcriptional activation by HDACIs requires a mechanism other
than chromatin remodeling, such as through histone
hyperacetylation, which is associated with protein kinase signaling
pathways (9), or acetylation of
non-histone proteins, including p53 or NF-κB (10). The NF-κB signaling pathway is
appreciated as one of the pivotal modulators of specific gene
expression and differential cellular responses by HDACIs.
NF-κB is a well-known transcription factor that
regulates the expression of a large number of genes in response to
a variety of cellular conditions. Recent studies have demonstrated
that NF-κB provides an anti-apoptotic signal in many different
cancer cells (11). NF-κB is
thought to reside in the cytoplasm in an inactive form, bound by
the IκB family of inhibitory proteins (12). However, it may shuttle between the
nucleus and cytoplasm in unstimulated cells (13). Stimulation of cells with specific
inducers activates the IκB kinase (IKK) complex, leading to the
phosphorylation of serines 32 and 36 of IκBα or serines 19 and 23
of IκBβ (14). This
phosphorylation event triggers rapid ubiquitination and subsequent
degradation of IκB proteins through the 26S proteasome complex,
allowing the free NF-κB protein to translocate into the nucleus and
activate the transcription of its target genes (15). The interaction of NF-κB with
histone acetyltransferase (HAT)-containing coactivators, including
p300/CBP, the steroid receptor-coactivator-1, and the
p300/CBS-associated factor, has been shown to activate
transcription. Previously, it has been reported that the
acetylation of NF-κB is responsible for sustaining NF-κB-dependent
transcription (16), and this
process is regulated by the HDAC family of proteins, including
HDAC-1, -2 and -3 (17).
The proteasome is a proteolytic complex that is
responsible for the intracellular degradation of numerous
ubiquitinated proteins that are involved in apoptosis and cell
cycle regulation. Bortezomib (Velcad; Millennium Pharmaceuticals,
Boston, MA, USA), a first-class proteasome inhibitor that is
approved by the FDA for the treatment of multiple myeloma and
mantel cell lymphoma, acts by targeting the catalytic 20S core of
the proteasome and inducing apoptosis in cancer cells (18). Among other mechanisms, proteasome
inhibitors lead to the cytoplasmic accumulation of the IκB protein,
resulting in reduced NF-κB activity. The purpose of the current
study was to determine whether the small-molecule proteasome
inhibitors, MG132, P1-1 and EPM, could sensitize colorectal cancer
cells to APC-mediated apoptosis. We also assessed whether combined
treatments involving APC and the MG132, PI-1, and EPM PIs could
sensitize colorectal cancer cells to conventional chemotherapeutic
drugs and induce them to undergo apoptosis. Finally, the potential
underlying molecular mechanisms that control the cell cycle,
apoptosis, survival, and stress-regulatory pathways were
investigated.
Materials and methods
Cell culture
Human colorectal cancer cell lines (SW1116 and
SW837) and normal human fibroblasts (CRL1554) were obtained from
the ATCC (American Type Culture Collection, VA, USA). The SW1116
and SW837 cell lines were cultivated in 90% Leibovitz’s L15 medium
containing 10% fetal bovine serum. The L15 medium was formulated
for use in a free gas exchange with atmospheric air. The CRL1554
cells were cultivated in Dulbecco’s modified Eagle’s medium (90%)
containing fetal bovine serum (10%).
Chemicals
Apicidin
[cyclo(N-O-methyl-L-tryptophanyl-L-isoleucinyl-D-pipecolinyl-L-2-amino-8-oxodecanoyl)]
was purchased from Sigma (St. Louis, MO, USA). The following
proteasome inhibitors: MG132, proteasome inhibitor 1 (PI-1) and
epoxomicin (EPM) were obtained from Biomol International, Enzo Life
Sciences International, Inc. (Plymouth Meeting, PA, USA). Falcon
plastic ware was purchased from BD Biosciences (San Jose, CA, USA).
Trypsin, Leibovitz’s L-15 and EMEM media, fetal bovine serum (FBS),
and penicillin/ streptomycin (200X solutions) were obtained from
Mediatech, Inc. (Herndon, VA, USA). An Annexin V-FITC apoptosis
detection kit was obtained from BD Hoffmann-La Roche, Inc. (Nutley,
NJ, USA). A DNA-prep kit was obtained from Beckman Coulter (Miami,
FL, USA). Primers, Taqman probes and all other reagents for RT-PCR
and real-time qPCR were obtained from Applied Biosystems (Foster
City, CA, USA). Cayman’s NF-κB (p65) transcription factor assay kit
was obtained from Cayman Chemical (Ann Arbor, MI, USA). HDAC
activity assay and nuclear/cytosol fractionation kits were obtained
from BioVision, Inc. (Milpitas, CA, USA). The 20S proteasome assay
kit for drug discovery was purchased from Biomol International,
Enzo Life Sciences International, Inc. PhosphoDetect™ JNK,
PhosphoDetect ERK1/2 and PhosphoDetect Akt ELISA kits were obtained
from Calbiochem-Novabiochem (Beeston, Nottingham, UK). All other
chemicals were purchased from Sigma Chemicals (St. Louis, MO,
USA).
Cell proliferation
The effects of the proteasome inhibitors on the
antimitogenic activity of APC on colorectal cancer cells was
evaluated by the 3-(4, 5-dimethylthiazol-2-yl)-2,
5-diphenyltetrazolium bromide (MTT) assay as previously described
(19). Cancer cells were incubated
with various concentrations of APC (0.06-1.0 μM) for 24 h
followed by incubation with MG132 (0.15, 0.3 μM), PI-1 (7.8,
15.6 nM) or EPM (2.8, 5.6 nM) for 72 h. The culture media were then
discarded, and 100 μl of MTT (5 mg/ml in culture medium,
sterile-filtered) was added to each well. The plate was then
incubated for 4 h at 37°C. The MTT solution was aspirated, and the
resulting formazan crystals were dissolved by incubation for 20 min
in 200 μl/well of DMSO: ethanol (1:1 v/v) at ambient
temperature. Changes in absorbance were measured at λ540 and 650
nm. Control cells were incubated in media supplemented with DMSO at
a final concentration of 0.2%; the growth and survival of cells are
not affected at this concentration of DMSO.
Colony formation assay
The effect of APC, proteasome inhibitors (MG132,
PI-1 or EPM) and APC/PI combinations on the colony formation of
colorectal cancer cells was determined by a colony formation assay
as previously described (19).
SW1116 and SW837 cells were plated (2.5×105 cells/ml) in
24-well plates and incubated in a non-CO2 incubator for
18 h followed by further incubation for 24 h with APC (3.4
μM), proteasome inhibitors [MG132 (1.5 μM), PI-1 (36
nM) or EPM (26 nM)] and APC/PI combinations. The cells were then
trypsinized, counted, plated at 500 cells/ml in a 6-well plate, and
incubated in a non-CO2 incubator for 10–14 days. The
treated cells were fixed in 100% methanol for 30 min at room
temperature and stained with 0.1% crystal violet for 1 h, and the
stained colonies were counted and compared with a control.
The effect of APC, proteasome inhibitors (MG132,
PI-1 and EPM) and APC/PI combinations on normal human fibro-blast
cells (CRL1554) was also monitored as described above using an
inverted microscope and an MTT assay.
HDAC activity
The colorimetric HDAC activity assay kit (BioVision,
Inc.) was used to monitor histone deacetylase activity in cancer
cell nuclear extracts according to the manufacturer’s instructions.
The SW1116 and SW837 cancer cell lines (2.5×105
cells/ml) were plated in 24-well plates and incubated in a
non-CO2 incubator for 18 h followed by treatment with
APC (3.4 μM) for 24 h. The nuclear extracts of both
untreated and APC-treated cells (50 μl) were diluted to 85
μl (final volume) with ddH2O and plated in a
96-well plate. For background reading, only 85 μl of
ddH2O was added. A HeLa cell nuclear extract (10
μl) was diluted with 75 μl of ddH2O and
used as a positive control. For the negative control, the tested
nuclear extract was diluted into 83 μl, and then 2 μl
of TSA (HDACI, 1 mM) was added; alternatively, a known sample
containing no HDAC activity was used. Ten microliters of the 10X
HDAC assay buffer and 5 μl of the HDAC colorimetric
substrate [Boc-Lys (Ac)-pNA, 10 mM] was added, the solution was
mixed thoroughly, and the plates were incubated at 37°C for 1 h.
The reaction was stopped by adding 10 μl of lysine
developer, and the plates were incubated at 37°C for 30 min. The
plates were then read in an ELISA plate reader at 400 or 405
nm.
Proteasome activity
Proteasome activity in the cancer cell extracts was
monitored using the 20S proteasome assay kit for drug discovery
according to the manufacturer’s instructions. SW1116 and SW837
cells (2.5×105 cells/ml) were plated in 24-well plates
and incubated in a non-CO2 incubator for 18 h followed
by treatment with proteasome inhibitors [MG132 (1.5 μM),
PI-1 (36 nM) or EPM (26 nM)] for 24 h. Cell extracts of untreated
and proteasome inhibitor-treated cancer cells were prepared using a
nuclear/cytosolic fractionation kit (BioVision, Inc.). The
cytosolic extracts (0.5 μg) and positive and negative
controls were then incubated with 75 μM proteasome substrate
(Suc-LLVY-AMC) in 100 μl of assay buffer (20 mM Tris-HCl, pH
8.0) for 90 min at 37°C. A VersaFluor™ fluorometer with excitation
at λ360 nm and emission at λ460 nm (Bio-Rad) was used to monitor
fluorescence release from AMC (7-amido-4-methyl-coumarin).
NF-κB activity
NF-κB (p65) activity was determined by Cayman’s
NF-κB (p65) transcription factor assay according to the
manufacturer’s instructions. SW1116 and SW837 cells
(2.5×105 cells/ml) were plated in 24-well plates and
incubated in a non-CO2 incubator for 18 h. The cells
were then incubated with APC (3.4 μM), proteasome inhibitors
[MG132 (1.5 μM), PI-1 (36 nM) or EPM (26 nM)] and APC/PI
combinations for 24 h, and nuclear extracts were purified using a
nuclear/cytosol fractionation kit. A specific double-stranded DNA
sequence containing the NF-κB response element was immobilized onto
the wells of a 96-well plate as part of the assay. A specific
primary antibody directed against NF-κB (p65) was used for
detection in the nuclear extracts or in a positive control, and a
second antibody conjugated to HRP was used for the readout at λ450
nm.
Reactive oxygen species (ROS) assay
ROS were analyzed as previously described (8) using 2′, 7′-dichlorofluorescein
diacetate (DCFH-DA) (Sigma Chemicals). DCFH-DA is a stable,
non-fluorescent cell-permeable compound that becomes fluorescent
(dichlorofluorescein) in the presence of active radicals and emits
green fluorescence upon excitation at 485 nm. The extent of ROS
generation was measured by quantifying fluorescence intensity.
SW1116 and SW837 cells (2.5×105 cells/ml) were plated in
24-well plates and incubated in a non-CO2 incubator for
18 h before treatment with APC (3.4 μM), proteasome
inhibitors [MG132 (1.5 μM), PI-1 (36 nM) or EPM (26 nM)] and
APC/PI combinations (or solvent alone) for 48 h. The cells were
subsequently washed and incubated with 10 μM DCFH-DA in
phosphate buffered saline at 37°C for 30 min. Fluorescence was
analyzed on a microtiter plate reader at 485-nm excitation and
535-nm emission. Production of ROS was determined by comparing the
intensity of fluorescence between treated and untreated cells. The
functional role of ROS generation on cell death was assessed using
the free radical scavenger, L-N-acetylcysteine (L-NAC) (Sigma
Chemicals). Cells were preincubated with 15 mM L-NAC for 3 h,
treated with APC, PIs and APC/PI combinations for 48 h and assessed
for cell death as described above.
Cell cycle analysis
The distribution of cells in cell cycle phases
(G0/G1, S, and G2/M) was
determined by flow cytometry by measuring the DNA content of nuclei
labeled with propidium iodide as previously described (20). SW1116 cancer cells
(2.5×105 cells/ml) were plated in 24-well plates and
incubated at 37°C in a non-CO2 incubator. Eighteen hours
after seeding the cells in culture, the cells were treated with APC
(3.4 μM), proteasome inhibitors [MG132 (1.5 μM), PI-1
(36 nM) or EPM (26 nM)] and APC/PI combinations for 24 h. Untreated
and drug-treated cells were collected by trypsinization, washed
with cold phosphate buffered saline (PBS) and counted. A DNA-prep
kit (Beckman Coulter) and a DNA-prep EPICS workstation (Beckman
Coulter) were used to process the cells as follows: the cells were
treated with a cell-membrane permeabilizing agent followed by
propidium iodide and RNase and incubated at room temperature for 15
min before analysis by flow cytometry (Beckman Coulter, Nyon,
Switzerland). The percentage of cells in various cell cycle phases
was calculated using the Phoenix statistical software package with
advanced DNA cell cycle software (Phoenix Flow System, San Diego,
CA, USA).
Assessment of apoptosis
Induction of apoptosis in colorectal cancer cells
treated with APC, PIs and APC/PI combinations was determined using
an Annexin V-FITC apoptosis detection kit according to the
manufacturer’s instructions. SW1116 cancer cells
(2.5×105 cells/ml) were plated in 24-well plates and
incubated at 37°C in a non-CO2 incubator. The cells were
then treated with APC (3.4 μM), proteasome inhibitors [MG132
(1.5 μM), PI-1 (36 nM) or EPM (26 nM)] and APC/PI
combinations for 24 h. Control and treated cells were resuspended
in a 100 μl staining solution containing Annexin V-FITC and
propidium iodide in HEPES buffer, incubated at room temperature for
15 min, and analyzed by flow cytometry. Annexin V binds to cells
expressing phosphatidylserine on the outer layer of the cell
membrane, whereas propidium iodide stains the cellular DNA of cells
with a compromised cell membrane. This allows the discrimination of
live cells (unstained with either fluorochrome) from apoptotic
cells (stained only with Annexin V) and necrotic cells (stained
with both Annexin V and propidium iodide).
mRNA expression of genes associated with
apoptosis and cell cycle regulation
Expression of cell cycle- and apoptosis-associated
genes in control and drug-treated cells was determined by real-time
polymerase chain reaction (qRT-PCR) using an ABI 7000 SDS system
(Applied Biosystems) and the comparative ΔΔCt method (19). Ready-made assays-on-demand that
target specific genes with probes and primers were obtained from
Applied Biosystems. The targets and their Applied Biosystems assay
numbers for the cell cycle regulatory genes were: cdk1
(Hs00364293_m1), cdk2 (Hs00608082_m1), cdk4
(Hs00364847_m1), cdk6 (Hs00608037_m1.), cdc25A
(Hs00153168_m1), p15 (Hs00394703_m1), p19
(Hs00176481_m1), p21 (Hs00355782_m1), and p27
(Hs00197366_m1).
The targets and their Applied Biosystems assay
numbers for the pro-apoptotic, anti-apoptotic and caspase genes
were: Bax (Hs00180269_m1), Bim (Hs00375807_m1),
Apaf1 (Hs00559441_m1), cIAP-1 (Hs0023691_m1),
c-IAP-2 (Hs00985029_m1), Bcl2 (Hs00608023_m1),
x-IAP (Hs00236913_m1), casp2 (Hs00154242_m1),
casp3 (Hs00234387_m1), casp6 (Hs00154250_m1),
casp7 (Hs00169152_m1), and casp9 (Hs00154260_m1).
GAPDH was used as an endogenous control to normalize the expression
values for each sample. For the comparative Ct method, we performed
a two-step RT-PCR to obtain cDNA and carried out real-time
quantitation using the target gene expression assays and Taqman
Universal Master mix (Applied Biosystems).
SW1116 cancer cells (2.5×105 cells/ml)
were plated in 24-well plates and incubated in a non-CO2
incubator for 18 h. The cells were then treated with APC (3.4
μM), proteasome inhibitors [MG132 (1.5 μM), PI-1 (36
nM) or EPM (26 nM)] and APC/PI combinations for 24 h. mRNA was
extracted using the nucleospin RNAII ready-to-use system
(Macherey-Nagel), and 200 ng/μl of mRNA was used in the RT
reaction. Contaminating DNA was eliminated with DNase-I treatment
for 20 min at 25°C, followed by heat inactivation for 10 min at
65°C prior to cDNA synthesis using the high capacity cDNA reverse
transcription kit (Applied Biosystem) according to the
manufacturer’s instructions. For each sample, 2.5 μl of cDNA
and 12.5 μl of Taqman Universal Master mix (2X) were used,
and the final volume was adjusted to 25 μl with
nuclease-free water on an optical 96-well reaction plate (Applied
Biosystems). Real-time PCR was performed on an ABI 7000 SDS system
using ABI Prism’s SDS collection software version 1.1 (Applied
Biosystems). Real-time PCR conditions followed the protocol given
by the manufacturer of the Taqman Universal Master mix: step 1,
95°C for 10 min; step 2, 94°C for 15 sec; and step 3, 60°C for 1
min. The samples were analyzed using SDS collection software
version 1.1 by setting the baseline between 3 and 15 and the
threshold at 0.2. The amount of target normalized to an endogenous
reference and relative to a calibrator (untreated) was determined
by 2−ΔΔCt, and the log comparative Ct is presented
graphically.
Expression of phospho-JNK1/2,
phospho-ERK1/2 and phospho-Akt
The phosphorylated forms of JNK1 and JNK2 at
Thr183 and Tyr185, the phosphorylated forms
of ERK1 at Thr202 and Tyr204 and ERK2 at
Thr185 and Tyr187 and the phosphorylated Akt
at Thr308 were determined in both treated and untreated
cancer cell extracts using the following PhosphoDetect ELISA kits:
JNK1/2 (pThr183/pTyr185)
(Calbiochem-Novabiochem: CBA007), ERK1/2
(pThr185/pTyr187) (Calbiochem-Novabiochem:
CBA006) and Akt (pThr308) (Calbiochem-Novabiochem:
CBA004). SW1116 and SW837 cancer cells (2.5×105
cells/ml) were plated in 24-well plates and incubated in a
non-CO2 incubator for 18 h. The cells were then treated
with APC (3.4 μM), proteasome inhibitors [MG132 (1.5
μM), PI-1 (36 nM) or EPM (26 nM)] and APC/PI combinations
for 48 h. The phosphoprotein assays were performed according to the
manufacturer’s instructions.
Chemosensitization potential of the APC
and proteasome inhibitor combinations
The potential of the combined treatments with APC
and proteasome inhibitors (MG132, PI-1 or EPM) to sensitize human
colorectal cancer cells to standard chemotherapeutic drugs was
investigated as previously described (20) with some modifications. Briefly,
SW1116 and SW837 cells were plated (27×103 cells/well)
in 96-well plates at 37°C in a non-CO2 incubator.
Eighteen hours after starting the culture, the cells were treated
for 24 h with various concentrations of the following:
camptothecin, CPT (64×10−10 – 1×10−4 M), 5FU
(41.6×10−9 – 0.65×10−3 M), oxaliplatin, OXP
(4×10−10 – 0.06×10−4 M), doxorubicin, DOX
(55×10−11 – 0.85×10−5 M), carboplatin, CAP
(43.5×10−10 – 0.86×10−4 M), cisplatin, CIP
(26.88×10−9 – 0.42×10−3 M), taxol, TAX
(93.44×10−10 – 1.4×10−4 M), cyclophosphamide,
CPA (14×10−9 – 0.22×10−3 M), vincristine, VCR
(16×10−11 – 2.5×10−5 M), etoposide, ETP
(25.6×10−10 – 0.4×10−4 M), ellipticine, ELP
(12.8×10−10 – 0.2×10−4 M), amsacrine, AMS
(80×10−11 – 1.25×10−5 M), homo-harringtonine,
HHG (12.8×10−11 – 0.2×10−5 M), or
aphidicolin, APD (17.28×10−11 – 0.27×10−5 M).
The drug was then removed, and the cells were washed with HBSS and
treated with combinations of APC (62 nM)/MG132 (0.25 μM),
APC (240 nM)/MG132 (0.16 μM), APC (96 nM)/PI-1 (10 nM), APC
(48 nM)/ PI-1 (7.8 nM), APC (250 nM)/EPM (1.4 nM) and APC (125
nM)/EPM (2.8 nM) for 72 h. Cell growth was monitored by MTT
assay.
Interaction of APC/proteasome inhibitors
and standard chemotherapeutic drugs in colorectal cancer cells
To evaluate the type of interaction between the
combined treatment with APC/MG132, PI-1 or EPM and standard
chemotherapeutic drugs in human colorectal cancer cells, the cells
were treated as described above with APC/MG132, PI-1 or EPM and
standard chemotherapeutic drugs individually and in combination.
The type of drug interaction was determined as previously described
(21) using the following
formulae: SFA + B > (SFA) ×
(SFB) = antagonistic, SFA + B =
(SFA) × (SFB) = additive and SFA +
B < (SFA) × (SFB) = synergistic,
where SF is the surviving fraction, A and B indicate the agent used
alone, and (A + B) refers to the agents used in combination.
Statistical analyses
Statistical analyses were performed with SPSS-19
software. The results are expressed as the mean ± SEM. The
statistical significance of the differences between control and
treated groups were determined by one-way ANOVA. P-values <0.05
were considered statistically significant.
Results
Augmentation of HDACI (APC)-mediated
lethality by its combination with proteasome inhibitors
To determine the effect that the combined exposure
to APC and PIs would have on human colorectal cancer cells and
whether proteasome inhibitors could increase APC lethality, the
cancer cells were treated with various concentrations of APC
(0.06-1.0 μM) in the presence and absence of the following
tested proteasome inhibitors: MG132 (0.16, 0.3 μM), PI-1
(7.8, 15.6 nm), and EPM (1.4, 2.8 nM). These concentrations were
determined by a dose response study (data not shown). The combined
treatment of APC (0.06–1.0 μM) with MG132 (0.16 μM)
produced a growth inhibition of IC50= 0.34 μM
(P≤0.0.882) in SW1116 cells (Fig.
1Aa) and IC50=0.35 μM (P≤0.649) in SW837
cells (Fig. 1Ad). These values
were similar to those produced by a single treatment with APC.
MG132 (0.16 μM) did not improve the sensitivity of SW1116
(sensitization ratio, SR= 0.95) or SW837 (SR= 0.92) cells to APC.
However, the combined treatment of APC and MG132 (0.3 μM)
produced a much higher and more significant growth inhibition of
SW1116 cells (IC90=0.29 μM, P≤0.0001) (Fig. 1Aa) and a more significant growth
inhibition of SW837 cells (IC90= 0.37 μM,
P≤0.0001) (Fig. 1Ad) when compared
with the single treatment with APC (IC90=0.77 μM
for SW1116 and IC90=0.94 μM for SW837 cells).
MG132 (0.3 μM) increased the sensitivity of both SW1116
(SR=2.64) and SW837 (SR=2.5) cells to APC.
The combined treatment of APC (0.06–1.0 μM)
with PI-1 (7.8 nM) produced a much higher and more significant
growth inhibition of SW1116 (IC90=0.28 μM,
P≤0.0001) (Fig. 1Ab) and SW837
(IC90=0.37 μM, P≤0.0001) (Fig. 1Ae) cells than that produced by the
single treatment with APC (IC90=0.78 μM for
SW1116 and IC90=0.8 μM for SW837 cells) (Fig. 1Ab, Ae). PI-1 (7.8 nM) increased the
sensitivity of both SW1116 (SR=2.8) and SW837 (SR=2.15) cells to
APC. The combined treatment of APC and PI-1 (15.6 nM) showed a much
higher and more significant growth inhibition of SW1116
(IC90=0.16 μM, P≤0.0001) (Fig. 1Ab) and SW837 (IC90= 0.24
μM, P≤0.0001) (Fig. 1Ae)
cells when compared with the single treatment with APC. PI-1 (15.6
nM) greatly increased the sensitivity of both SW1116 (SR=4.67) and
SW837 (SR=3.3) cells to APC.
The combined treatment of APC (0.06–1.0 μM)
with EPM (2.8 nM) demonstrated a slightly higher growth inhibition
of SW1116 (IC50=0.31 μM, P≤0.441) cells than that
demonstrated by the single treatment with APC (IC50=
0.34 μM) (Fig. 1Ac). On the
other hand, the combined treatment with APC and EPM (2.8 nM) showed
a slightly higher but insignificant growth inhibition of SW837
(IC50=0.34 μM, P≤0.447) (Fig. 1Af) cells when compared with the
single treatment with APC (IC50= 0.39 μM). EPM
(2.8 nM) did not improve the sensitivity of SW1116 (SR=1.11) and
SW837 (SR=1.10) cells to APC. The combined treatment of APC and EPM
(5.6 nM) produced a higher but insignificant growth inhibition of
SW1116 (IC50=0.18 μM, P≤0.494) (Fig. 1Ac) cells, whereas this combination
markedly inhibited the growth of SW837 (IC50=0.11
μM, P≤0.0001) (Fig. 1Af)
cells, when compared with the single treatment with APC. Finally,
EPM (5.6 nM) increased the sensitivity of both SW1116 (SR=1.91) and
SW837 (SR=3.29) cells to APC.
Colony formation assays were performed to confirm
the results of the inhibition studies. The treatment of SW1116
cells with a combination of APC and MG132 significantly inhibited
the colony formation of the cells (mean number of colonies = 60,
P≤0.0001) when compared with the untreated SW1116 cells (mean
number of colonies = 209). Additionally, this treatment
significantly inhibited the colony formation of SW1116 cells
(P≤0.0001) when compared with single treatments with either APC
(mean number of colonies = 99) or MG132 (mean number of colonies =
108) (Fig. 1Ba).
The treatment of SW1116 cells with a combination of
APC and PI-1 significantly inhibited the colony formation of SW1116
cells (mean number of colonies = 84, P≤0.0001) when compared with
the untreated cells (mean number of colonies = 209). Additionally,
this treatment significantly inhibited the colony formation of
SW1116 cells (P≤0.0001) when compared with the single treatment
with PI-1 (mean number of colonies = 130). However, the difference
when this combination was compared with the colonies formed upon
APC treatment alone was statistically insignificant (P≤0.061)
(Fig. 1Bb).
The treatment of SW1116 cells with the combination
of APC and EPM significantly inhibited the colony formation of
SW1116 cells (mean number of colonies = 58, P≤0.0001) when compared
with the untreated SW1116 cells (mean number of colonies = 209).
Similarly, this combination significantly inhibited the colony
formation of SW1116 cells (P≤0.0001) when compared with single
treatments with either APC (mean number of colonies = 99) or EPM
(mean number of colonies = 141) (Fig.
1Bc).
Inhibition of HDACs, the 26S proteasome
and NF-κB binding to DNA and generation of ROS in cancer cells
HDAC activity was measured in the nuclear fractions
of untreated and APC-treated colorectal cancer cells to determine
whether the antimitogenic activity of APC was associated with the
inhibition of intra-nuclear HDAC activity. SW1116 cancer cells
treated with APC showed a significant inhibition of HDAC activity
(P≤0.0001) when compared with untreated cancer cells (Fig. 2A). To determine whether APC
activated the DNA binding activity of NF-κB and whether the tested
proteasome inhibitors could interrupt this activity, the cancer
cells were treated with APC, tested proteasome inhibitors and their
combinations. The DNA binding activities were monitored in
untreated and treated cell extracts.
APC significantly increased the DNA binding activity
of NF-κB when compared with untreated cancer cells (P≤0.022). In
contrast, this activity was decreased after treatment with MG132
(P≤0.87), PI-1 (P≤0.419) and EPM (P≤0.352) when compared with the
untreated cancer cells; however, this decrease was insignificant. A
significant decrease in the DNA binding activity of NF-κB was
observed after the combined treatment with APC/MG132 (P≤0.008),
APC/PI-1 (P≤0.01), and APC/EPM (P≤0.001) when compared with
untreated cancer cells. These findings raise the possibility that
the interaction between APC and PIs reflects the inactivation of
the cytoprotective NF-κB signaling pathway (Fig. 2B).
26S proteasome activity was also determined in
untreated and proteasome inhibitor-treated cancer cell extracts to
establish whether the antimitogenic activities of the tested
proteasome inhibitors were linked to the inhibition of
intracellular proteasome activity. The MG132 (P≤0.001), PI-1
(P≤0.001) and EPM (P≤0.0001) proteasome inhibitors exhibited
significant inhibition of 26S proteasome activity in SW1116
colorectal cancer cells when compared with untreated cancer cells
(Fig. 2C). Additionally, treatment
with APC (P≤0.001) and the combinations of APC/MG132, APC/PI-1 or
APC/EPM significantly inhibited the proteasome activity of
colorectal cancer cells (P≤0.0001) when compared with untreated
cancer cells (Fig. 2C).
Reactive oxygen species (ROS) have been implicated
in HDAC and proteasome inhibitor-induced cytotoxicity in a number
of malignancies. Thus, the effects of APC, the tested proteasome
inhibitors, and APC/PI combinations on ROS generation were examined
in SW1116 cells. ROS levels were markedly increased (P≤0.0001)
following the combined treatment with APC/MG132, APC/EPM and
APC/PI-1 when compared with untreated cells and single treatments
with APC and the tested proteasome inhibitors (Fig. 2D). Additionally, single treatments
with MG132 (P≤0.0001), EPM (P≤0.0001) or PI-1 (P≤0.004) induced
significant ROS when compared with untreated cancer cells (Fig. 2D). The induction of ROS was
abrogated by pretreatment with L-NAC (data not shown).
Cell cycle perturbation and induction of
apoptosis in cancer cells treated with APC, proteasome inhibitors
and APC/PI combinations
To study the effects of APC, the tested PIs, and
APC/PI combinations on cell cycle perturbation, the distribution of
cancer cells in the various cell cycle phases
(G0/G1, S, and G2/M) was examined
by flow cytometry. Treatment of SW1116 cells with APC resulted in
the accumulation of cancer cells in S-phase (57.3 vs. 41.5% for UT)
and a strong decrease in cells in the G1/G0
phase (33.2 vs. 40.3% for UT) and G2/M phase (9.3 vs.
18% for UT) (Fig. 3A).
Single treatment with the tested proteasome
inhibitors (MG132, PI-1 and EPM) gave results consistent with those
recently reported by Abaza et al (22). Treatment of SW1116 cells with MG132
resulted in a marked accumulation of the cancer cells in S-phase
(57 vs. 25.5% for UT) and a decrease in cells in the
G1/G0 phase (25.5 vs. 40.3% for UT) and G2/M
phase (17.4 vs. 18% for UT) (22).
The combined treatment of APC with G132 arrested the cancer cells
in the G1/G0 phase (51.4 vs. 40.3% for UT)
and S-phase (44.9 vs. 41.5% for UT), and there was a decrease in
the G2/M phase (3.5% vs. 18% for UT). The combined
treatment of APC and MG132 markedly induced apoptosis as evident
from the percentage of cells in SubG1 (21.2%) when
compared with UT (SubG1=0) and single treatments with APC
(SubG1=4.8%) or MG132 (SubG1=1.1%) (Fig. 3A).
The treatment of SW1116 cells with PI-1 resulted in
the accumulation of cancer cells in the G1-G0
phase (49.3 vs. 40.5% for UT) and a decrease in cells in the
S-phase (33.2 vs. 41.5% for UT) and G2-M phase (17.3 vs.
18% for UT) (22). The treatment
with a combination of APC and PI-1 resulted in the accumulation of
SW1116 cells in the S-phase (52.5 vs. 41.5% for UT) and
G2/M phase (23.4 vs. 18% for UT) and a decrease in cells
in the G1/G0-phase (24 vs. 40.3% for UT). The
combined treatment of APC and PI-1 induced apoptosis
(SubG1=8%) when compared with the single treatment with
PI-1 (SubG0=0.2%) or APC (SubG1=4.8%)
(Fig. 3A).
Treatment of SW1116 cancer cells with EPM resulted
in the accumulation of cancer cells in the S-phase (51.2 vs. 41.5%
for UT) and a decrease in cells in the
G1/G0-phase (31.3 vs. 40.3% for UT) and
G2/M-phase (17.3 vs. 18% for UT) (22). The combined treatment of APC and
EPM also resulted in the accumulation of cancer cells in the
S-phase (55.7 vs. 41.5% for UT) and a decrease in cells in the
G1/G0-phase (33.9 vs. 40.3% for UT) and the
G2/M-phase (10.3 vs. 18% for UT). The combined treatment
increased apoptosis (SubG1= 6%) when compared with the
single treatment with APC (4.8%) (Fig.
3A). In a parallel study, the cell cycle phase distribution of
SW837 cells treated with APC, the tested proteasome inhibitors, and
APC/PI combinations gave similar results to those obtained with
SW1116 cells (data not shown).
The increase in the percentage of SW1116 cells in
sub-G1 after treatment with combinations of APC and
proteasome inhibitors compared with untreated SW1116 cells or
SW1116 cells treated with either APC or proteasome inhibitor alone
indicates an increase in the percentage of apoptotic cells
(Fig. 3A). To determine the effect
of PIs (MG132, PI-1 and EPM) on the apoptotic response of the
colorectal cancer cells to APC, untreated and treated cancer cells
were double-stained with Annexin V and propidium iodide to
distinguish between the different types of cell death and analyzed
by flow cytometry. Annexin V binding combined with PI labeling was
performed to distinguish between early apoptotic (Annexin
V+/propidium iodide−) and necrotic (Annexin
V+/propidium iodide+) cells.
Treatment of SW1116 cells with the combination of
APC and EPM markedly induced apoptosis in the cancer cells (0.1%
early apoptosis, 43.2% late apoptosis and 0.7% necrosis) when
compared with untreated SW1116 cells (0.0% early apoptosis, 4.7%
late apoptosis and 0.4% necrosis), SW1116 cells treated with APC
(0.0% early apoptosis, 26.4% late apoptosis and 0.8% necrosis)
(Fig. 3B), or SW1116 cells treated
with EPM (0.1% early apoptosis, 20.3% late apoptosis and 3.1%
necrosis) (22).
In addition, the combination of APC and MG132
greatly induced the apoptosis of SW1116 cells (0.3% early
apoptosis, 67.7% late apoptosis, and 9.9% necrosis) when compared
with untreated SW1116 cells (0.0% early apoptosis, 4.7% late
apoptosis, and 0.4% necrosis), SW1116 cells treated with APC (0.0%
early apoptosis, 26.4% late apoptosis, and 0.8% necrosis) (Fig. 3B), or SW1116 cells treated with
MG132 (0.0% early apoptosis, 26.4% late apoptosis, and 0.8% late
apoptosis) (22).
The combination of APC and PI-1 also induced the
apoptosis of SW1116 cells (0.1% early apoptosis, 43.7% late
apoptosis, and 1.9% necrosis) when compared with untreated SW1116
cells (0.0% early apoptosis, 4.7% late apoptosis, and 0.4%
necrosis), SW1116 cells treated with APC (0.0% early apoptosis,
26.4% late apoptosis, and 0.8% necrosis) (Fig. 3B) or PI-1-treated SW1116 cells
(0.0% early apoptosis, 11.9% late apoptosis, and 0.8% necrosis)
(22). These results suggest that
the tested PIs markedly increased the APC-mediated lethality in
human colorectal cancer cells. The induction of apoptosis in SW837
cells treated with APC, the tested proteasome inhibitors, and
APC/PI combinations provided similar results to those obtained with
SW1116 cells (data not shown).
mRNA expression of cell cycle and
apoptosis-related genes in cancer cells treated with APC,
proteasome inhibitors and APC/PI combinations
The effects of the combination APC/PI treatment on
the expression/activation of various signaling molecules were
examined to understand the molecular mechanisms underlying the
increase in the APC-mediated lethality in human colorectal cancer
cells upon addition of proteasome inhibitors.
The combined treatment of SW1116 cells with APC and
MG132, PI-1 or EPM markedly downregulated the mRNA expression of
genes related to cell cycle control (Cdk1, Cdk2, Cdk4, Cdk6
and Cdc25A) when compared with the single treatment with APC
(Fig. 4) or PIs (MG132, PI-1, EPM)
(22). On the other hand, the same
combined treatments upregulated the mRNA expression of p15, p19,
p21 and p27 when compared with the single treatments
with APC (Fig. 4A) and the tested
PIs (22).
The combined treatment of SW1116 cells with APC and
the tested proteasome inhibitors upregulated the mRNA expression of
the pro-apoptotic genes, Bax, Bim, apaf1 and caspases 2,
3, 6, 7 and 9. The same combined treatment downregulated
the expression of the anti-apoptotic genes, Bcl2, x-IAP,
c-IAP1 and c-IAP2 when compared with the single
treatment with APC (Fig. 4B) or
the tested PIs (MG132, PI-1 and EPM) (22). The cycle threshold (Ct) values of
the target genes under investigation in this study were in the
range of 20.826-30.981.
To further understand the potential mechanisms of
action of APC, proteasome inhibitors and APC/PI combinations, the
expression of pAkt, pERK and pJNK was evaluated. Treatment of
SW1116 cells with APC (P≤0.024) (Fig.
5A), MG132 (P≤0.001) (22), or
APC/MG132 (P≤0.0001) (Fig. 5A)
resulted in a significant decrease in the levels of pAkt when
compared with untreated cancer cells. The combined treatment of APC
and MG132 produced a significant decrease in the levels of pAkt
when compared with the single treatment with APC (P≤0.003)
(Fig. 5A) or MG132 (P≤0.05)
(22). SW1116 cells treated with
APC, PI-1 (P≤0.002) (22), or
APC/PI-1 (P≤0.0001) (Fig. 5A)
exhibited a significant decrease in the levels of pAkt when
compared with untreated cancer cells. The combined treatment with
APC/PI-1 significantly reduced the levels of pAkt when compared
with the single treatment with either APC (P≤0.0001) (Fig. 5A) or PI-1 (P≤0004) (22). Furthermore, SW1116 cells treated
with a combination of APC/EPM showed a significant growth
inhibition when compared with untreated cells (P≤0.0001) or cells
treated with APC alone (P≤0.0001) (Fig. 5A) and EPM alone (P≤0.014) (22).
SW1116 cells treated with MG132, EPM or PI-1
(23) and combinations (APC/MG132,
APC/EPM or APC/PI-1) showed a significant decrease in the levels of
pERK1/2 (P≤0.0001) when compared with untreated cancer cells.
Additionally, SW1116 cells treated with APC showed a significant
decrease in pERK1/2 levels (P≤0.009) when compared with untreated
cells (Fig. 5B).
The combined treatment with APC/MG132 produced a
significant decrease in the levels of pERK1/2 when compared with
the single treatment with either APC (P≤0.0001) or MG132 (P≤0.0001)
(22). The expression of pERK1/2
significantly decreased following the combined treatment with
APC/PI-1 when compared with the single treatment with APC
(P≤0.0001) or PI-1 (P≤0.036) (22). The combined treatment with APC/EPM
also produced a significant decrease in the levels of pERK1/2 when
compared with those produced by single treatment with either APC
(P≤0.0001) (Fig. 5B) or EPM
(P≤0.002) (22).
SW1116 cells treated with MG132, EPM (22), and the APC/MG132, APC/EPM or
APC/PI-1 combinations showed a significant increase in the levels
of pJNK (P≤0.0001) when compared with untreated cancer cells.
Additionally, the treatment of SW1116 cells with APC or PI-1
resulted in an increase in the expression of pJNK. However,
insignificant (P≤0.129) and significant (P≤0.002) increases in the
levels of pJNK were reported following treatment with APC (Fig. 5C) and PI-1 (22), respectively, when compared with
untreated cells. In addition, SW1116 cells treated with a
combination of APC/MG132, APC/PI-1, and APC/EPM showed a
significant increase (P≤0.0001) in the levels of pJNK when compared
with the single treatment with APC (Fig. 5C), MG132, PI-1 or EPM (22).
Chemosensitization of human colorectal
cancer cells by combined treatment with APC and proteasome
inhibitors
The efficacy of the combined treatment with the
HDACI, APC, and the proteasome inhibitors, MG132, PI-1 or EPM, to
sensitize cancer cells to standard chemotherapeutic drugs and the
type of interaction between the combined treatments and standard
chemotherapeutic drugs were investigated. The results are
summarized in Figs. 6–10 and Tables I–V.
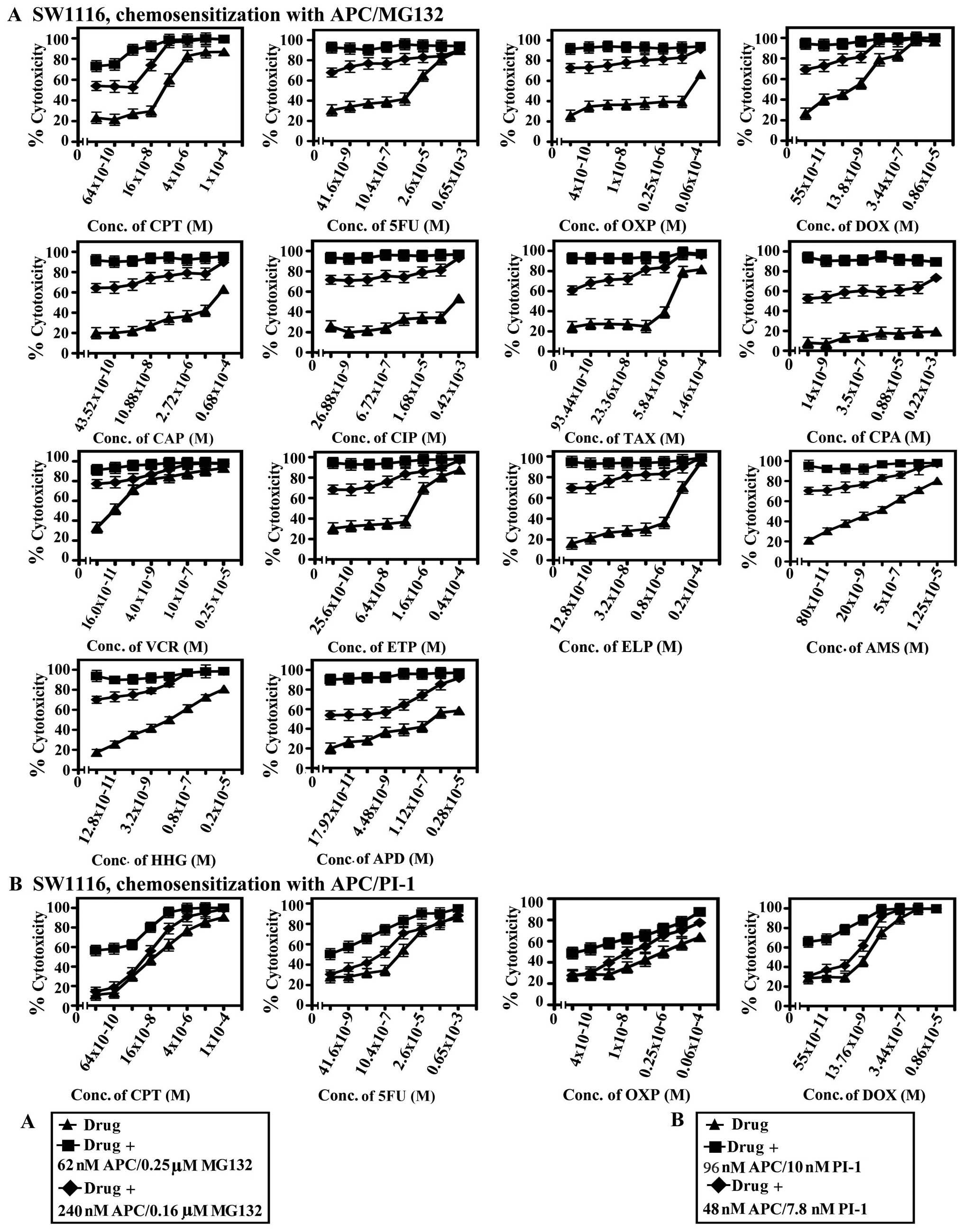 | Figure 6.Chemosensitization of SW1116
colorectal cancer cells by combined treatment with APC and MG132 or
PI-1. SW1116 cells were plated in 96-well plates for 18 h followed
by incubation with various concentrations of CPT, 5FU, OXP, DOX,
CAP, CIP, TAX, CPA, VCR, ETP, ELP, AMS, HHG and APD for 24 h. The
drugs were then removed, and the cells were washed with HBSS and
treated with a combination of APC (240 nM)/MG132 (0.16 nM) and APC
(62 nM)/MG132 (0.25 nM) (A) or APC (96 nM)/PI-1 (10 nM) and APC (48
nM)/PI-1 (7.8 nM) (B). Control cells were treated with vehicle
(DMSO at a final concentration of 0.2%). Cell growth was monitored
by MTT assay. |
 | Figure 10.Chemosensitization of the SW837
colorectal cancer cell line upon combined treatment with APC and
EPM, and the effect of the combined treatment of APC with PIs on
normal human fibroblasts. SW837 cells were plated in 96-well plates
for 18 h. The cells were then treated with various concentrations
of CPT, 5FU, OXP, DOX, CAP, CIP, TAX, CPA, VCR, ETP, ELP, AMS, HHG,
and APD for 24 h. The drugs were removed, and the cells were washed
with HBSS and treated with a combination of APC (125 nM)/EPM (2.4
nM) or APC (250 nM)/EPM (1.4 nM) for 72 h (A). The effect of the
combined treatment with APC (62 nM)/MG132 (0.25 μM), APC
(240 nM)/MG132 (0.16 μM), APC (96 nM)/PI-1 (10 nM), APC (48
nM)/PI-1 (7.8 nM), APC (250 nM)/EPM (1.4 nM) and APC (125 nM)/EPM
(2.8 nM) on the normal human fibroblast cell line, CRL1554 (B).
Control cells were treated with vehicle (DMSO at a final
concentration of 0.2%). Cell growth was monitored by MTT assay. |
 | Table I.IC50-IC80
values, the sensitization ratios and P-values of the combined
treatment with chemotherapeutic drugs and the combination of APC
and MG132 toward human colorectal cancer cell lines. |
Table I.
IC50-IC80
values, the sensitization ratios and P-values of the combined
treatment with chemotherapeutic drugs and the combination of APC
and MG132 toward human colorectal cancer cell lines.
| Combined treatment
with chemotherapeutic drugs and the combination of APC and
proteasome inhibitor MG132 in SW1116 | IC60
(M)a SW1116 | SRb SW1116 | Pc |
|
| APD
(0.28×10−5 – 17.92×10−11 M) + APC/MG132 (62
nM/0.25 μM) |
6.3×10−9 | 47.6 | 0.0001 |
|
| Combined treatment
with chemotherapeutic drugs and the combination of APC and
proteasome inhibitor MG132 in SW1116 | IC70
(M)a SW1116 | SRb SW1116 | Pc |
|
| TAX
(1.46×10−4 – 93.44×10−10 M) + APC/MG132 (62
nM/0.25 μM) |
26.7×10−9 |
7.86×102 | 0.0001 |
|
| Combined treatment
with chemotherapeutic drugs and the combination of APC and
proteasome inhibitor MG132 in SW1116 | IC80
(M)a SW1116 | SRb SW1116 | Pc |
|
| CPT
(1.0×10−4 – 64×10−10 M) + APC/MG132 (62
nM/0.25 μM) |
2×10−7 | 20 | 0.0001 |
| CPT
(1.0×10−4 – 64×10−10 M) + APC/MG132 (240
nM/0.16 μM) |
8.3×10−9 |
4.8×102 | 0.0001 |
| 5 FU
(0.65×10−3 – 41.6×10−9 M) + APC/MG132 (62
nM/0.25 μM) |
5.5×10−6 | 25.4 | 0.0001 |
| DOX
(0.86×10−5 – 55×10−11 M) + APC/MG132 (62
nM/0.25 μM) |
5.16×10−9 | 16.7 | 0.0001 |
| VCR
(0.25×10−5 – 16×10−11 M) + APC/MG132 (62
nM/0.25 μM) |
0.8×10−9 | 5.0 | 0.0001 |
| ETP
(4.0×10−4 – 25.6×10−10 M) + APC/MG132 (62
nM/0.25 μM) |
0.21×10−6 | 42.8 | 0.0001 |
| ELP
(0.2×10−4 – 12.8×10−10 M) + APC/MG132 (62
nM/0.25 μM) |
1.5×10−8 |
4×102 | 0.0001 |
| AMS
(1.25×10−5 – 80×10−11 M) + APC/MG132 (62
nM/0.25 μM) |
0.43×10−7 | 60.5 | 0.0001 |
| HHG
(0.2×10−5 – 12.8×10−11 M) + APC/MG132 (62
nM/0.25 μM) |
3×10−5 | 133 | 0.0001 |
|
| Combined treatment
with chemotherapeutic drugs and the combination of APC and
proteasome inhibitor MG132 in SW837 | IC50
(M)a SW837 | SRb SW837 | Pc |
|
| CPT
(1.0×10−4 – 64×10−10 M) + APC/MG132 (240
nM/0.16 μM) |
5.8×10−7 | 22.4 | 0.027 |
| CPT
(1.0×10−4 – 64×10−10 M) + APC/MG132 (62
nM/0.25 μM) |
58×10−10 |
2.24×103 | 0.0001 |
| VCR
(0.25×10−5 – 16×10−11 M) + APC/MG132 (240
nM/0.16 μM) |
1.60×10−10 | 18.8 | 0.041 |
| VCR
(0.25×10−4 – 16×10−11 M) + APC/MG132 (62
nM/0.25 μM) |
0.67×10−9 | 4.5 | 0.0001 |
| ETP
(0.4×10−4 – 25.6×10−10 M) + APC/MG132 (240
nM/0.16 μM) |
0.28×10−5 | 14.3 | 0.034 |
| ETP
(0.4×10−4 – 25.6×10−10 M) + APC/MG132 (62
nM/0.25 μM) |
0.34×10−6 | 117.6 | 0.002 |
| ELP
(0.2×10−4 – 12.8×10−10 M) + APC/MG132 (240
nM/0.16 μM) |
0.28×10−5 | 3.7 | 0.173 |
| ELP
(0.2×10−4 – 12.8×10−10 M) + APC/MG132 (62
nM/0.25 μM) |
0.57×10−6 | 17.5 | 0.0001 |
| AMS
(1.25×10−5 – 80×10−11 M) + APC/MG132 (240
nM/0.16 μM) |
0.42×10−6 | 31.0 | 0.019 |
| AMS
(1.25×10−5 – 80×10−11 M) + APC/MG132 (62
nM/0.25 μM) |
0.4×10−8 |
3.25×103 | 0.0001 |
|
| Combined treatment
with chemotherapeutic drugs and the combination of APC and
proteasome inhibitor MG132 in SW837 | IC60
(M)a SW837 | SRb SW837 | Pc |
|
| 5FU
(0.65×10−3 – 41.6×10−9 M) + APC/MG132 (240
nM/0.16 μM) |
2.8×10−5 | 20.7 | 0.216 |
| 5FU
(0.65×10−3 – 41.6×10−9 M) + APC/MG132 (62
nM/0.25 μM) |
21×10−8 |
2.76×104 | 0.0001 |
|
| Combined treatment
with chemotherapeutic drugs and the combination of APC and
proteasome inhibitor MG132 in SW837 | IC70
(M)a SW837 | SRb SW837 | Pc |
|
| DOX
(0.86×10−5 – 55×10−11 M) + APC/MG132 (240
nM/0.16 μM) |
6.88×10−8 | 12.5 | 0.171 |
| DOX
(0.86×10−5 – 55×10−11 M) + APC/MG132 (62
nM/0.25 μM) |
14×10−9 | 61.4 | 0.0001 |
| TAX
(1.46×10−4 – 93.44×10−10 M) + APC/MG132 (240
nM/0.16 μM) |
7.33×10−5 | 2.0 | 0.477 |
| TAX
(1.46×10−4 – 93.44×10−10 M) + APC/MG132 (62
nM/0.25 μM) |
2.93×10−6 | 50.2 | 0.0001 |
| Table V.Percentage means cytotoxicity of
standard chemotherapeutic drugs and their combinations with APC/
PI-1 or EPM on human colorectal cancer cells. |
Table V.
Percentage means cytotoxicity of
standard chemotherapeutic drugs and their combinations with APC/
PI-1 or EPM on human colorectal cancer cells.
| Treatment with
chemotherapeutic drugs and the combinations of APC/PI-1 | Percentage means
cytotoxicitya
| P-valuesb
|
| SW1116 | SW837 | SW1116 | SW837 |
|
| CIP
(0.42×10−3 – 26.88×10−9 M) | - | 2±0.9 | | |
| CIP
(0.42×10−3 – 26.88×10−9 M) + APC/PI-1 (48
nM/7.8 nM) | - | 42±1.6 | - | 0.0001 |
| CIP
(0.42×10−3 – 26.88×10−9 M) + APC/PI-1 (96
nM/10 nM) | - | 67±1.2 | - | 0.0001 |
| CPA
(1×10−4 – 64×10−10 M) | 41±1.2 | 7±1.6 | | |
| CPA
(1×10−4 – 64×10−10 M) + APC/PI-1 (48 nM/7.8
nM) | 42±2.1 | 49±1.6 | 0.814 | 0.0001 |
| CPA
(1×10−4 – 64×10−10 M) + APC/PI-1 (96 nM/10
nM) | 71±1.0 | 70±1.5 | 0.0001 | 0.0001 |
| APD
(0.28×10−5 – 17.92×10−11 M) | - | 13±3.2 | | |
| APD
(0.28×10−5 – 17.92×10−11 M) + APC/PI-1 (48
nM/7.8 nM) | - | 19±3.9 | - | 0.208 |
| APD
(0.28×10−5 – 17.92×10−11 M) + APC/PI-1 (96
nM/10 nM) | - | 39±3.3 | | 0.0001 |
|
| Treatment with
chemotherapeutic drugs and the combinations of APC/EPM | Percentage means
cytotoxicity
| P-values
|
| SW1116 | SW837 | SW1116 | SW837 |
|
| OXP
(0.06×10−4 – 4×10−10 M) | 08±2.9 | 36±2 | | |
| OXP
(0.06×10−4 – 4×10−10 M) + APC/EPM (125
nM)/2.8 nM) | 32±2.9 | 47±2.3 | 0.0001 | 0.0001 |
| OXP
(0.06×10−4 – 4×10−10 M) + APC/EPM (250 nM/1.4
nM) | 60±3 | 69±1.7 | 0.0001 | 0.0001 |
| CAP
(0.68×10−4 – 43.52×10−10 M) | 30±2.2 | 28±2.1 | | |
| CAP
(0.68×10−4 – 43.52×10−10 M) + APC/EPM (250
nM/1.4 nM) | 49±2.3 | 67±2.2 | 0.0001 | 0.0001 |
| CIP
(0.42×10−3 – 26.88×10−9 M) | 61±3 | 29±2.4 | | |
| CIP
(0.42×10−3 – 26.88×10−9 M) + APC/EPM (125
nM)/2.8 nM) | 30±2.8 | 50±2.5 | 0.0001 | 0.0001 |
| CIP
(0.42×10−3 – 26.88×10−9 M) + APC/EPM (250
nM/1.4 nM) | 61±3 | 67±1.76 | 0.0001 | 0.0001 |
| CPA
(1×10−4 – 64×10−10 M) | 0.0 | 0.0 | | |
| CPA
(1×10−4 – 64×10−10 M) + APC/EPM (250 nM/1.4
nM) | 57±3 | 35±1.5 | 0.0001 | 0.0001 |
| APD
(0.28×10−5 – 17.92×10−11 M) | 5±2.4 | 7±1.6 | | |
| APD
(0.28×10−5 – 17.92×10−11 M) + APC/EPM (125
nM)/2.8 nM) | 19±3.5 | 22±2.7 | 0.001 | 0.0001 |
| APD
(0.28×10−5 – 17.92×10−11 M) + APC/EPM (250
nM/1.4 nM) | 55±2.9 | 67±3.3 | 0.0001 | 0.0001 |
The combination of APC and the tested PIs
strikingly increased the sensitivity of colorectal cancers to ETP
(117.6-5.5×103-fold), AMS (60-3.25×103-fold),
and 5FU (84-2.76×104-fold). The combination of APC and
MG132 or PI-1 also resulted in marked increases in the sensitivity
of colorectal cancer cells to TAX (50-4.48×103-fold) and
HHG (50-133-fold). Combining APC with MG132 or EPM increased the
sensitivity of colorectal cancer cells to CPT
(137-2.24×103-fold).
The combination of APC and MG132 showed a 133-fold
increase in the sensitivity of the SW1116 colon cancer cells to
HHG, whereas the combination of APC and PI-1 increased the
sensitivity of SW1116 cells to CIP (1.5×103-fold), APD
(3.71×104-fold), and TAX (4.48×103-fold).
Finally, the combination of APC and PI-1 increased the sensitivity
of the SW837 rectal cancer cells to AMS (1.73×103-fold),
whereas the combination of APC and EPM increased the sensitivity of
SW837 cells to ETP (5.7×102-fold) and CPT
(137-fold).
Collectively, these results clearly indicate the
potential of the APC/proteasome inhibitor combination treatments to
markedly increase the sensitivity of colorectal cancer cells to
standard chemotherapeutic drugs. The data also suggest different
mechanisms of action and sensitivity in a combination- and cancer
subtype-dependent manner. The synergistic and/or additive
interaction between the tested chemotherapeutics and the various
combinations of APC and PIs was dependent on the type of drug
tested, the combination of APC and PIs, and the polymorphism of the
genes encoding the drug-metabolizing enzymes, transporters or drug
targets. The effect of the treatment with APC/MG132, APC/PI-1 and
APC/EPM on normal human fibroblast cells (CRL1554) was also
examined microscopically and by MTT assay. The results shown in
Fig. 10B clearly demonstrated
that these combinations had little effect (≤15%) on CRL1554 cells,
indicating their minimal cytotoxicities.
Discussion
HDACs have been demonstrated to be associated with
oncogenic transformation by promoting the function of transcription
factors in certain hematologic malignancies and solid tumors
(23). Therefore, HDACIs have
emerged as a novel class of anticancer agents for the treatment of
solid and hematological malignancies. Despite the promising results
indicating the usefulness of employing HDACIs as an
epigenetic-targeted therapy, its limited success in specific
cancers as a single drug has prompted further investigation of
combining HDACIs with other anticancer agents. These combination
regimens may enhance the clinical efficacy of HDACIs and provide a
therapeutic advantage over HDACIs as a single drug.
Recent evidence has suggested that the
transcriptional activation induced by HDACIs requires a mechanism
other than chromatin remodeling; such a mechanism is through
histone hyperacetylation, a process that is associated with protein
kinase signaling pathways or acetylation of non-histone proteins,
such as p53 or NF-κB (10). The
NF-κB signaling pathway is appreciated as one of the pivotal
modulators of specific gene expression and differential cellular
responses by HDACIs, as the selective inhibition of the NF-κB
pathway leads to an abrogation of NF-κB-dependent gene expression
by HDACIs and the unresponsiveness of cells to HDACI-induced
apoptosis (24). The
unresponsiveness of HeLa cells to apoptosis following the
inhibition of HDAC activity by apicidin is due to NF-κB activation,
which is mediated by IKK and the IκBα signaling pathway, indicating
that NF-κB activation is associated with the resistance of cancer
cells to the apoptotic potential of HDACIs (25). Interference with HDACI-mediated
NF-κB activation would therefore favor the pro-apoptotic actions of
HDACIs and enhance HDACI-mediated cell death. Proteasome
inhibitors, such as PS-341 and bortezomib, suppress NF-κB activity
by inhibiting IκBα degradation; this activity prevents the nuclear
translocation/acetylation of Rel A and interferes with de
novo expression of NF-κB-dependent genes, including IκBα
(26). The aim of the current
study was to determine whether the small-molecule proteasome
inhibitors could abrogate HDACI (APC)-induced NF-κB activation in
colorectal cancer cells and induce their apoptosis. Furthermore,
the study aimed to elucidate the associated mechanisms and assess
the effects of the combined inhibition of HDACs and the proteasome
on the response of human colorectal cancer cells to
chemotherapy.
New combination therapies for cancer are expected
to produce enhanced efficacy, improved selectivity, and reduced
toxicity. An ideal combination regimen would consist of agents with
different mechanisms of action, leading to complementary antitumor
activities and little cross resistance (27). In the present study, exposure to
submicromolar concentrations of APC in combination with
submicromolar concentrations of MG132 and nanomolar concentrations
of EPM or PI-1 synergistically decreased the survival of cancer
cells when compared with the single treatments (Fig. 1A). MG132 (0.3 μM) increased
the sensitivity of both SW1116 (SR=2.64) and SW837 (SR=2.5) cells
to APC. PI-1 (7.8 nM) increased the sensitivity of SW1116 (SR=2.8)
and SW837 (SR=2.15) to APC, and at 15.6 nM, PI-1 further increased
the sensitivity of SW1116 (SR=4.63) and SW 837 (SR=3.3) cells to
APC. Finally, EPM (2.8 nM) increased the sensitivity of SW1116
(SR=1.91) and SW837 (SR=3.29) cells to APC. Colony formation assays
(Fig. 1B) and morphological
changes (Fig. 1C) confirmed the
results of the inhibition studies. Both APC and the tested
proteasome inhibitors had little toxic effects on normal human
fibroblasts. Cancer cells rely heavily on the proteasome to
eliminate unwanted proteins, most likely due to their rapid protein
turnover rate; therefore, cancer cells are more susceptible to
proteasome inhibition when compared with non-transformed cells
(28). Our results are consistent
with previous studies reported by several groups using diverse
malignant cell types and employing various combinations of HDAC and
proteasome inhibitors (22,25,29).
The rationale to target the proteasome in cancer
cells stems from data indicating that malignant cells accumulate
more misfolded, mutated, or damaged proteins, which are disposed of
by the proteasome; therefore, cancer cells are more dependent on
proteasome activity (30).
Furthermore, the inhibition of the proteasome induces apoptosis and
has been shown to have antitumor effects in several xenograft
models and cancer cell lines representing prostate, pancreas
(31), lymphoma, head and neck,
melanoma, lung, and breast cancers as well as leukemias (32).
The acetylation and deacetylation of histone
proteins control gene transcription, and the processes are
regulated by two families of enzymes, acetyltransferases and HDACs
(33). Increased histone
acetylation is typically associated with transcriptionally active
genes, whereas low levels of acetylation correlate with
transcriptional repression (33).
In the present study, APC treatment resulted in a significant
inhibition (P≤0.0001) of intranuclear deacetylase activity, and the
data were in agreement with the antimitogenic effect of APC on
human colorectal cancer cells (Fig.
2A). These findings are consistent with those reported for
other HDACIs, such as TSA and sodium butyrate, on head and neck
squamous cell cancer cell lines (34). However, data reported by Denlinger
et al indicated that although all intracellular deacetylase
activity was inhibited in three different non-small cell lung
cancer (NSCLC) cell lines, they were resistant to HDAC
inhibitor-mediated cell death (35).
NF-κB, which is thought to be targeted by
proteasome inhibitors, is a transcription factor regulating gene
involved in proliferation, apoptosis, and angiogenesis, and it is
particularly well studied as the main effector of apoptosis. NF-κB
is known to be overexpressed or constitutively active in a number
of human tumor types, possibly conferring resistance to cancer cell
death (36). HDACIs have been
shown to exert pleiotropic effects on NF-κB activity in various
cell types, including producing an increase in activity in some
cells (37) and a decrease in
activity in others (38). Whether
this discrepancy reflects concentration-, drug-, or cell
type-specific differences remains to be determined. In any case, in
the present study, APC significantly increased (P≤0.022) the DNA
binding activity of NF-κB. On the other hand, the MG132 (P≤0.87),
PI-1 (P≤0.419) and EPM (P≤0.352) proteasome inhibitors decreased
the DNA binding activity of NF-κB. This activity was decreased even
more significantly after the combined treatment of APC with MG132
(P≤0.008), PI-1 (P≤0.01) or EPM (P≤0.001) (Fig. 2B), suggesting their synergic
mechanism as negative regulators of the NF-κB pathway. These
findings are consistent with data obtained using HDACIs and PIs in
other malignancies (25,39).
Proteasome inhibitors act by targeting the
catalytic core of the proteasome and inducing the apoptosis of
tumor cells. Among other mechanisms, proteasome inhibitors lead to
the cytoplasmic accumulation of the I-κB protein and reduced NF-κB
activity. Other downstream targets involved in the action of
proteasome inhibitors include the generation of ROS, caspase
activation and accumulation of cellular proteins linked to cell
proliferation and apoptosis (40).
In the present study, the tested proteasome
inhibitors, MG132 (P≤0.001), EPM (P≤0.001), and PI-1 (P≤0.0001), as
well as their combination with APC significantly suppressed
(P≤0.0001) proteasome activity in colorectal cancer cells when
compared with untreated cells (Fig.
2C). Such findings are in agreement with findings in other
malignancies (41). Previous
studies have suggested that the lethal effects of proteasome
inhibitors either administered alone in lung cancer cells (42) or in combination with HDACIs in
Bcr/Abl+ myeloid leukemia cells (43) stem from the generation of ROS.
Studies were therefore performed to determine
whether a similar mechanism might underlie the lethality of APC/PI
combination treatment in colorectal cancer cells. The combined
treatment with APC/MG132, APC/EPM and APC/PI-1 showed a pronounced
generation of ROS (P≤0.0001) when compared with the untreated
control and the single treatments with APC and the tested
proteasome inhibitors (Fig. 2D).
These observations are consistent with previous findings in other
malignancies that have attributed HDAC and proteasome
inhibitor-mediated cytotoxicities to increased ROS generation
(25,44). Co-administration of the
antioxidant, L-NAC (15 mM), substantially blocked the
APC/PI-mediated increase in ROS levels (data not shown). Increased
ROS production has been reported to result in protein, lipid and
DNA-based oxidation with the latter forming apurinic/apyrimidic
sites (among other types of DNA damage). These findings indicate
that the generation of ROS plays a significant role in the
APC/PI-mediated lethality in colorectal cancer cells (45).
The combined treatment of APC with MG132, PI-1, or
EPM resulted in a prominent arrest of the human colorectal cancer
cells in G1-G0/S-, G2-M/S-, or
S-phases, respectively (Fig. 3A).
Exposure to other combinations of HDAC and proteasome inhibitors
was also reported to induce a G2/M cell cycle arrest in
different types of malignancies (25). Furthermore, APC interacted
synergistically with MG132, PI-1 or EPM to induce apoptosis when
compared with single treatments with APC, MG132, PI-1 and EPM or
with untreated colorectal cancer cells (Fig. 3B). Such findings are in accord with
those reported for other combinations of HDAC and proteasome
inhibitors and in other types of malignancies (22,46).
We also evaluated the molecular events that
potentially led to the effectiveness of the combination of APC with
the proteasome inhibitors by analyzing several signaling pathways
linked to cell proliferation and apoptosis. Human colorectal cancer
cells exposed to APC and MG132, PI-1, or EPM displayed a marked
downregulation in the mRNA expression of genes related to cell
cycle control, including Cdk1, Cdk2, Cdk4, Cdk6 and
Cdc25A, when compared with the single treatments with APC,
MG132, PI-1 and EPM. However, the same combined treatments resulted
in the upregulation of the cell cycle-dependent kinase inhibitor
genes, p15, p19, p21 and p27, when compared with the
single treatments (Fig. 4). These
results are consistent with findings in other malignancies using
various HDAC and proteasome inhibitors (25,44,47,48).
Increased levels of p15, p19, p21 and p27 and
maintenance of the key cell cycle regulatory proteins that counter
proliferative signals, eventually leading to apoptosis (49), are possible mechanisms of
colorectal cancer cell death induced by the combined treatment of
APC with proteasome inhibitors.
Co-exposure of human colorectal cancer cells to APC
and MG132, PI-1, or EPM resulted in the overexpression of a number
of pro-apoptotic genes, including Bim, Bax, Apaf1 and
caspases 2, 3, 4, 7 and 9. The same combined
treatments exhibited reduced mRNA expression of anti-apoptotic
genes, including Bcl2, x-IAP and c-IAP-1 (Fig. 4). These results are in agreement
with those reported in other malignancies using various
combinations of HDACIs and proteasome inhibitors (25,39,44,50).
Bax promotes apoptosis through its interaction with
the anti-apoptotic members in the mitochondria, such as Bcl-2. Such
interactions have been shown to activate caspases, which cleave
many vital cellular substrates, thereby contributing to cell death
and the apoptotic phenotype. Caspase 9 is an initiator caspase in
the apoptotic process, and its function is to activate the effector
caspases 6, 7, and 3 (51).
The upregulation of pro-apoptotic genes in
conjunction with the downregulation of anti-apoptotic genes as well
as the increased production of ROS in colorectal cancer cells
co-exposed to APC and proteasome inhibitors may serve to shift the
balance from pro-survival to pro-apoptotic. The APC/PI combination
regimens thus lower the threshold of colorectal cancer cells to
pro-death signals, thereby accounting for the lethality of the
combination regimens.
The effects of single and combined treatments of
APC with proteasome inhibitors were examined in relation to
perturbations of cytoprotective and stress-related signaling
modules in the colorectal cancer cells. The combined APC/proteasome
inhibitor treatments downregulated the levels of the phosphorylated
forms of AKT and ERK (P≤0.0001) in the cancer cells. On the other
hand, these combined treatments markedly augmented JNK
phosphorylation (P≤0.0001) (Fig.
5). Similar results have been reported for other malignancies
using various combinations of HDAC and proteasome inhibitors
(25,39,44).
Several lines of evidence have suggested that the
enhanced lethality of APC in conjunction with the tested proteasome
inhibitor combinations stem, at least in part, from a redirection
of signals away from cytoprotection and toward the stress-related
cascade. For example, in PC12 cells, the net output of the JNK and
ERK pathways determines whether cells live or die in response to
growth factor deprivation. Furthermore, JNK activation has been
implicated in events associated with mitochondrial damage and
cytochrome c release (52). For
these reasons, the activation of stress-related cascades is one of
the factors thought to be involved in proteasome inhibitor-mediated
lethality (53). Thus, in
colorectal cancer cells, the combined downregulation of ERK and AKT
may be considerably more lethal than the disruption of either
pathway alone.
Several of the signaling pathways affected by HDAC
and proteasome inhibitors, such as the disruption of Akt signaling
and the hyperactivation of JNK signaling, have been implicated in
Bax regulation. Direct phosphorylation of Bax by Akt on
ser184 inhibits Bax translocation to mitochondria in
neutrophils (54). Alternatively,
phosphorylation of Bax by JNK stimulates mitochondrial
translocation (55). The effects
on Bax-interacting proteins may also contribute to the effects
induced by HDAC and proteasome inhibitors. Ku70 was initially
identified as a component of the machinery that repairs DNA strand
breaks, but it has also been shown to have an important role in
sequestering Bax in the cytoplasm. Sequestration of Bax by Ku70
depends on the acetylation of several key residues within the
c-terminal linker domain that is adjacent to the Bax interaction
domain (54). Treating cells with
an HDAC inhibitor is therefore expected to result in an
accumulation of acetylated Ku70, which may be unable to sequester
Bax in the cytoplasm to suppress apoptosis. Thus, it is likely that
the multiple cellular changes induced by the HDACI, APC and PIs
(MG132, PI-1 and EPM) in human colorectal cancer cells contribute
to the activation of Bax and the subsequent induction of
apoptosis.
The mechanisms by which HDAC and proteasome
inhibitors exert their cytotoxic effects have not been fully
delineated and are likely to be multifactorial and cell
line-specific. Numerous genes are affected by proteasome inhibitors
or APC, and it is therefore likely that a combination of these
effects may lead to the synergy observed between the two classes of
agents. Both proteasome inhibitors and APC generate ROS, either via
mitochondrial injury or by disrupting antioxidant systems. When
paired, these two compounds increase oxidative stress, leading to
apoptosis. High levels of ROS can cause damage to proteins, a
process that contributes to ER stress. Inhibiting the proteasome
also results in aggregates of ubiquitin-conjugated proteins that
were originally targeted for degradation by the proteolytic
complex. HDACIs disrupt aggresomes, leading to ER stress (8). Thus, the combination of proteasome
inhibitors (MG132, PI-1 and EPM) and the HDACI, APC, leads to
increased cellular stress and apoptosis.
Both oxidative stress and disruption of aggresome
formation, leading to ER stress, are important pathways that
contribute to the synergy observed between proteasome inhibitors
and APC. However, because both of these types of drugs have
pleiotropic effects, other mechanisms may also be involved. Both
HDAC and proteasome inhibitors influence many cellular processes;
thus, further studies examining their interactions may reveal
additional overlapping mechanisms that have not yet been identified
as contributing to their synergism.
Cancer patients often develop chemoresistance, and
increasing the concentrations of cytotoxic drugs fails to
significantly improve the therapeutic response. One hypothesis for
the development of chemoresistance is an acquired resistance of the
tumor cells to apoptosis (56),
allowing tumors to withstand high levels of chemotherapy. Tumor
cells that are resistant to apoptosis also exhibit increased
proliferative capacity.
In this study, we showed that the combined
treatment of APC with the proteasome inhibitors, MG132, PI-1 or
EPM, markedly reduced the apoptotic threshold of the colorectal
cancer cells. These results prompted us to examine the potential of
the combined treatments to augment the sensitivity of human
colorectal cancer cells to standard chemotherapeutic drugs. Our
data clearly indicated that the APC/proteasome inhibitor (MG132,
PI-1 or EPM) combinations led to a marked increase in the
sensitivity of colorectal cancer cells to standard chemotherapies
in an APC/proteasome inhibitor- and colorectal cancer
subtype-dependent manner (Figs.
6–10, Tables I–V). These results are in line with
findings in other malignancies (22,56–58).
Given the pleiotropic actions of both HDAC and
proteasome inhibitors, it is unlikely that an interruption of NF-κB
signaling is the sole basis for the synergistic interactions
between such agents and the enhanced chemosensitization potential
of the combination treatment. In all probability, additional
mechanisms are involved. Cytotoxicity and chemosensitization
potentials of the APC/proteasome inhibitor combination treatment
are most likely associated with multiple interacting mechanisms,
including proteasome and NF-κB inhibition, ROS generation, cell
cycle perturbation, downregulation of anti-apoptotic and
cytoprotective molecules, and upregulation of pro-apoptotic and
stress-related molecules. Although clinical studies of HDAC and
proteasome inhibitors as single agents have yielded only modest
results (59), the finding that
very low concentrations of APC and a proteasome inhibitor (MG132,
PI-1 or EPM) interact synergistically to kill colorectal cancer
cells and potentiate their sensitivity to standard chemotherapeutic
drugs suggests the need to develop novel combination therapies for
treating malignant diseases, including colorectal cancer. These
combinations of drugs should function through complementary
mechanisms of action with a manageable toxicity and low cross
resistance. In vivo studies using animal models are
necessary to confirm the validity of the combinational strategy for
the treatment of colorectal cancer. Furthermore, testing this
strategy with a larger number of cancer cell lines would increase
the value of this study.
Combination treatments may lead to better efficacy
and utility of HDACIs in the clinic. However, the effects of these
combinations on dose-limiting toxicities have not been thoroughly
evaluated. Combination studies may therefore provide an opportunity
to use lower doses and reduce dose-limiting toxicities, including
the fatigue, vomiting, nausea and diarrhea (among others) that have
been observed with the use of HDACIs as single agents (60). Although there are concerns that
using combinations of agents may result in increased toxicities,
preliminary data from current clinical trials indicate that these
combinations are safe and well tolerated.
Many questions remain unanswered with respect to
HDACI specificities for particular tumor subtypes and the molecular
mechanisms underlying HDACI-induced cell differentiation, cell
cycle arrest and apoptosis. The mechanisms that regulate specific
gene expression and recruitment of HDAC complexes to specific
promoter sites are also unknown. It is important to distinguish the
HDAC specificity of HDACIs for the development of selective
therapies at the molecular level.
Acknowledgements
This study was supported by Kuwait
University, Research Grant No. [SL 05/05].
References
|
1.
|
Heider U, Rademacher J, Lamottke B, et al:
Synergistic interaction of the histone deacetylase inhibitor SAHA
with the proteasome inhibitor bortezomib in cutaneous T cell
lymphoma. Eur J Haematol. 82:440–449. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Marks PA and Breslow R: Dimethyl sulfoxide
to vorinostat: development of this histone deacetylase inhibitor as
an anticancer drug. Nat Biotechnol. 25:84–90. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Frew AJ, Johnstone RW and Bolden JE:
Enhancing the apoptotic and therapeutic effects of HDAC inhibitors.
Cancer Lett. 280:125–133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Andrews KT, Walduck A, Kelso MJ, Fairlie
DP, Saul A and Parsons PG: Anti-malarial effect of histone
deacetylation inhibitors and mammalian tumor cytodifferentiating
agents. Int J Parasitol. 30:761–768. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Im JY, Park H, Kang KW, Choi WS and Kim
HS: Modulation of cell cycles and apoptosis by apicidin in estrogen
receptor (ER)-positive and -negative human breast cancer cells.
Chem Biol Interact. 172:235–244. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kim MS, Son MW, Kim WB, Park YI and Moon
A: Apicidin, an inhibitor of histone deacetylase, prevents
H-ras-induced invasive phenotype. Cancer Lett. 157:23–30. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Kwon SH, Ahn SH, Kim YK, et al: Apicidin,
a histone deacetylase inhibitor, induces apoptosis and Fas/Fas
ligand expression in human acute promyelocytic leukemia cells. J
Biol Chem. 277:2073–2080. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Choi JH, Lee JY, Choi AY, et al: Apicidin
induces endoplasmic reticulum stress- and mitochondrial
dysfunction-associated apoptosis via phospholipase Cγ1- and
Ca2+-dependent pathway in mouse Neuro-2a. Apoptosis.
17:1340–58. 2012.PubMed/NCBI
|
|
9.
|
Kobayashi H, Tan EM and Fleming SE:
Acetylation of histones associated with the p21WAF1/Cip1 gene
transcription in human colorectal adenocarcinoma cells. Int J
Cancer. 109:207–213. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Xu Y: Regulation of p53 responses by
post-translational modifications. Cell Death Differ. 10:400–403.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Tang G, Minemoto Y, Dibling B, Purcell NH,
Li Z, Karin M and Lin A: Inhibition of JNK activation through NF-κB
target gene. Nature. 414:313–317. 2001.
|
|
12.
|
Verma IM, Stevenson JK, Schwartz EM, Van
Antwerp D and Miyamoto S: Rel/NF-κB family: intimate tales of
association and dissociation. Genes Dev. 9:2723–2735. 1995.
|
|
13.
|
Huang TT, Kudo N, Yoshida M and Miyanoto
S: A nuclear export signal in the N-terminal regulatory domain of
IκBα controls cytoplasmic localization of inactive NF-κB/IκBα
complexes. Proc Natl Acad Sci USA. 97:1014–1019. 2000.
|
|
14.
|
Zandi E, Rothwarf DM, Delhase M, Hayakawa
M and Karin M: The IκB kinase complex (IKK) contains two kinase
subunits, IKKα and IKKβ, necessary for IκB phosphorylation and
NF-κB activation. Cell. 91:243–252. 1997.
|
|
15.
|
Ghosh S, May MJ and Kopp EB: NF-κB and Rel
proteins: evolutionarily conserved mediators of immune responses.
Ann Rev Immunol. 16:225–260. 1998.
|
|
16.
|
Chen L, Fischle W, Verdin E and Greene WC:
Duration of nuclear NF-κB action regulated by reversible
acetylation. Science. 293:1653–2657. 2001.
|
|
17.
|
Zhong H, May MJ, Jimi E and Ghosh S: The
phosphorylation status of nuclear NF-κB determines its association
with CBP/ p300 or HDAC-1. Mol Cell. 9:625–636. 2002.
|
|
18.
|
Sterz J, von Metzler I, Hahne JC, et al:
The potential of proteasome inhibitors in cancer therapy. Expert
Opin Investig Drugs. 17:879–895. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Abaza MS, Al-Safar A, Al-Sawan S and
Al-Attiyah R: c-myc anti-sense oligonucleotides sensitize human
colorectal cancer cells to chemotherapeutic drugs. Tumor Biol.
29:287–303. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Abaza MSI: Augmentation of the anticancer
effects of proteasome inhibitors by combination with sodium
butyrate in human colorectal cancer cells. Exp Ther Med. 1:675–693.
2010.
|
|
21.
|
Lam PK, To EW, Chan ES, Liew CT, Lung IW
and King WK: In vitro modulation of head and neck cell growth by
human recombinant interferon α and 13-cis-retinoic acid. Br J
Biomed Sci. 58:226–229. 2001.
|
|
22.
|
Abaza MSI, Bahman AM, Al-Attiyah RJ and
Kollamparambil AM: Synergistic induction of apoptosis and
chemosensitization of human colorectal cancer cells by histone
deacetylase inhibitor, scriptaid and proteasome inhibitors:
potential mechanisms of action. Tumor Biol. 33:1951–1972. 2012.
View Article : Google Scholar
|
|
23.
|
Rosato RR and Grant S: Histone deacetylase
inhibitors in clinical development. Expert Opin Investig Drugs.
13:21–38. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Mayo MW, Denlinger CE, Broad RM, et al:
Ineffectiveness of histone deacetylase inhibitors to induce
apoptosis involves the transcriptional activation of NF-κB through
the Akt pathway. J Biol Chem. 278:18980–18989. 2003.PubMed/NCBI
|
|
25.
|
Dasmahapatra G, Lembersky D, Son MP, et
al: Carfilzomib interacts synergistically with histone deacetylase
inhibitors in mantle cell lymphoma cells in vitro and in vivo. Mol
Cancer Ther. 10:1686–1697. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Savita B, Sriram B, Kevin D, et al: The
histone deacetylase inhibitor PCI-24781 induces caspase and
reactive oxygen species-dependent apoptosis through NFκB mechanisms
and is synergistic with bortezomib in lymphoma cells. Clin Cancer
Res. 15:3354–3365. 2009.PubMed/NCBI
|
|
27.
|
Luu T, Chung C and Somlo G: Combining
emerging agents in advanced breast cancer. Oncologist. 16:760–771.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Masdehors P, Omura S, Merle-Béral H, et
al: Increased sensitivity of CCL-derived lymphocytes to apoptotic
death activation by the proteasome-specific inhibitor lactacystin.
Br J Haematol. 105:752–757. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Frankland-Searby S and Bhaumik SR: The 26S
proteasome complex: An attractive target for cancer therapy.
Biochim Biophys Acta. 1825:64–76. 2012.PubMed/NCBI
|
|
30.
|
Adams J: The proteasome: a suitable
anti-neoplastic target. Nat Rev Cancer. 4:349–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Nawrocki ST, Bruns CJ, Harbison MT, et al:
Effects of the proteasome inhibitor PS-341 on apoptosis and
angiogenesis in orthotopic human pancreatic tumor xenografts. Mol
Cancer Ther. 1:1243–1253. 2002.PubMed/NCBI
|
|
32.
|
Yu C, Rahmani M, Conrad D, Subler M, Dent
P and Grant S: The proteasome inhibitor bortezomib interacts
synergistically with histone deacetylase inhibitors to induce
apoptosis in Bcr/Abl+ cells sensitive and resistant to
ST1571. Blood. 102:3765–3774. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Vigushin DM and Coomes RC: Histone
deacetylase inhibitors in cancer treatment. Anticancer Drugs.
13:1–13. 2002. View Article : Google Scholar
|
|
34.
|
Duan J, Friedman J, Nottingham L, Chen Z,
Ara G and Van Waes C: Nuclear factor-κ p65 small interfering RNA or
proteasome inhibitor bortezomib sensitizes head and neck squamous
cell carcinomas to classic histone deacetylae inhibitors and novel
histone deacetylase inhibitor PZD101. Mol Cancer Ther. 6:37–50.
2007.
|
|
35.
|
Denlinger C, Keller M, Mayo M, Broad RM
and Jones DR: Combined proteasome and histone deacetylase
inhibition in non-small cell lung cancer. J Thorac Cardiovasc Surg.
127:1078–1086. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Tai DI, Tsai SL, Chang YH, et al:
Constitutive activation of nuclear factor kappaB in hepatocellular
carcinoma. Cancer. 89:2274–2228. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Johnstone RW: Histone-deacetylase
inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug
Discov. 1:287–299. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Yin L, Laevsky G and Giardina C: Butyrate
suppression of colonocyte NF-kappa B activation and cellular
proteasome activity. J Biol Chem. 276:44641–44646. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Jiang Y, Wang Y, Su Z, et al: Synergistic
induction of apoptosis in HeLa cells by the proteasome inhibitor
bortezomib and histone deacetylase inhibitor SAHA. Mol Med Rep.
3:613–619. 2010.PubMed/NCBI
|
|
40.
|
Perez-Galan P, Roue G, Villamor N,
Montserrat E, Campo E and Colomer D: The proteasome inhibitor
bortezomib induces apoptosis in mantle-cell lymphoma through
generation of ROS and Noxa activation independent of p53 status.
Blood. 107:257–264. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Heider U, von Metzler I, Kaiser M, et al:
Synergistic interaction of the histone deacetylase inhibitor SAHA
with the proteasome bortezomib in mantle cell lymphoma. Eur J
Haematol. 80:133–142. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Ling YH, Liebes L, Zou Y and Perez-Soler
R: Reactive oxygen species generation and mitochondrial dysfunction
in the apoptotic response to bortezomib, a novel proteasome
inhibitor, in human H460 non-small cell lung cancer cells. J Biol
Chem. 278:33714–33723. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Pei XY, Dai Y and Grant S: Synergistic
induction of oxidative injury and apoptosis in human multiple
myeloma cells by the proteasome inhibitors bortezomib and histone
deacetylase inhibitors. Clin Cancer Res. 10:3839–3852. 2004.
View Article : Google Scholar
|
|
44.
|
Pitts TM, Morrow M, Kaufman SA, Tentler JJ
and Eckhardt SG: Vorinostat and bortezomib exert synergistic
antiproliferative and proapoptotic effects in colon cancer cells
models. Mol Cancer Ther. 8:342–349. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Perez-Galan P, Roue G, Villamor N,
Montserrat E, Campo E and Colomer D: The BH3-mimetic GX15-070
synergizes with bortezomib in mantle cell lymphoma by enhancing
Noxa-mediated activation of Bak. Blood. 109:4441–4449. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Miller C, Ban K, Dujka ME, et al:
NPI-0052, a novel proteasome inhibitor, induces caspase-8 and
ROS-dependent apoptosis alone and in combination with HDAC
inhibitors in leukemia cells. Blood. 110:267–277. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Yu C, Friday BB, Yang L, et al:
Mitochondrial Bax translocation partially mediates synergistic
cytotoxicity between histone deacetylase inhibitors and proteasome
inhibitors in glioma cells. Neuro Oncol. 10:309–319. 2007.
View Article : Google Scholar
|
|
48.
|
Orlowski RZ: The role of
ubiquitin-proteasome pathway in apoptosis. Cell Death Diff.
6:303–331. 1999. View Article : Google Scholar
|
|
49.
|
Buoncervello M, Borghi P, Romagnoli G, et
al: Apicidin and docetaxel combination treatment drives CTCFL
expression and HMGB1 release acting as potential antitumor immune
response inducers in metastatic breast cancer cells. Neoplasia.
14:855–867. 2012.
|
|
50.
|
Lawen A: Apoptosis - an introduction.
Bioessays. 25:888–896. 2003. View Article : Google Scholar
|
|
51.
|
Lei K, Nimmual A, Zong WX, et al: The Bax
subfamily of Bcl2 related proteins is essential for apoptotic
signal transduction by c-Jun NH2-terminal kinase. Mol Cell Biol.
22:4929–4942. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Tacchini L, Dansi P, Matteucci E,
Bemelli-Zazzera A and Desiderio MA: Influence of proteasome and
redox state on heat shock-induced activation of stress kinases,
AP-1 and HSF. Biochim Biophy Acta. 1538:76–89. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Gardai SJ, Hildeman DH, Frankel SK, et al:
phosphorylation of Bax Ser 184by Akt regulates its activity and
apoptosis in neutrophils. J Biol Chem. 279:21085–21095. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Catley L, Weisberg E, Kiziltepe T, et al:
Aggresome induction by proteasome inhibitor bortezomib and
α-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor
LBH589 are synergistic in myeloma cells. Blood. 108:3441–3449.
2006.
|
|
55.
|
Baldini E, Gardin G, Giannessi P, et al: A
randomized trial of chemotherapy with or without estrogenic
recruitment in locally advanced breast cancer. North-West Oncology
Group (GONO) study, Italy. Tumori. 83:829–833. 1997.PubMed/NCBI
|
|
56.
|
Hwang JJ, Kim YS, Kim T, et al: A novel
histone deacetylase inhibitor, CG2007745, potentiates anticancer
effect of docetaxel in prostate cancer via decreasing Mcl-1 and
Bcl-(XL). Invest New Drugs. 30:1434–1442. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Linares A, Dalenc F, Balaguer P, Boulle N
and Gavailles V: Manipulating protein acetylation in breast cancer:
a promising approaching in combination with hormonal therapies? J
Biomed Biotechnol. 2011:1–15. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Munster PN, Thurn KT, Thomas S, et al: A
phase II study of the histone deacetylase inhibitor vorinostat
combined with tamoxifen for the treatment of patients with hormone
therapy-resistant breast cancer. Br J Cancer. 104:1828–1835. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
O’Connor OA, Heaney ML, Schwartz L, et al:
Clinical experience with intravenous and oral formulations of the
novel histone deacetylase inhibitor suberoylanilide hydroxamic acid
in patients with advanced hematologic malignancies. J Clin Oncol.
24:166–173. 2005.
|
|
60.
|
Vansteenkiste J, Van Cutsem E, Dumez H, et
al: Early phase II trial of oral vorinostat in telapsed or
refractory breast, colorectal, or non-small cell lung cancer.
Invest New Drugs. 26:483–488. 2008. View Article : Google Scholar : PubMed/NCBI
|



















