Introduction
Neoplastic transformation is associated with
alterations in DNA methylation, including both global
hypomethylation and gene-specific hypermethylation (1–3).
Hypomethylation may result in aberrant or inappropriate expression
of genes that contribute to neoplastic transformation,
tumorigenesis or cancer progression (oncogenes) (4). In addition, genome-wide loss of
methylation contributes to chromosomal instability by destabilizing
pericentromeric regions of certain chromosomes (5–7).
Gene-specific hypermethylation typically reflects hypermethylation
of CpG-rich regions within gene promoter sequences that lead to
gene silencing events (1).
Methylation-dependent gene silencing is a mutation-independent
mechanism for inactivation of tumor suppressor genes (and other
negative mediators of neoplastic development) in cancer (8). Methylation-dependent gene silencing
is a common epigenetic modification present in breast cancer cells
contributing to the initiation, development and progression of
breast cancer (9–11). Recently, we identified a subset of
breast cancer cell lines and primary breast cancers that exhibit
aberrant DNA hypermethylation that results in concurrent epigenetic
silencing of multiple methylation-sensitive genes secondary to DNA
methyltransferase enzyme hyperactivity associated with
overexpression of DNMT3b (12).
Breast cancers that exhibit this aberrant DNA hypermethylation are
substantially enriched for the basal-like molecular subtype
(12). The close association of
aberrant DNA hypermethylation with the basal-like molecular subtype
of breast cancer strongly suggests that dysfunction of the
epigenome represents a fundamental biological property that
contributes to the clinical behavior of this form of breast
cancer.
While the expression of the DNMT3b-related aberrant
DNA hypermethylation among basal-like breast cancers is now well
established, the molecular mechanism governing the hyper
methylation defect has not been examined in primary breast cancer.
Several studies in the literature have shown that DNMT3b is often
overexpressed in different types of cancers including breast cancer
(12–16). However, unlike other genes that are
overexpressed in cancer as a result of genetic mutations and/or
gene amplifications, the mechanisms accounting for overexpression
of DNMT3b does not involve these changes (17). Likewise, inappropriate or increased
trans-activation does not account for the overexpression of DNMT3b
in cancer (17). Numerous studies
have now demonstrated that DNMT3b is negatively
post-transcriptionally regulated by microRNAs (miRs), which are
small endogenous non-coding RNAs (19–25 nucleotide long) that have
emerged as key players in regulation of gene expression (18). Post-transcriptional regulation of
gene expression by miRs occurs through sequence-specific targeting
of mRNAs as a result of recognition of complementary sites, most
often in the 3’-untranslated region (UTR) of the target mRNA,
producing either translational repression or degradation of the
target mRNA (19–23). miRs are expressed in a
tissue-specific manner and have been implicated in the regulation
of myriad of biological processes, including cellular
proliferation, differentiation, apoptosis and development (24–27).
Altered miR expression is associated with several types of human
cancer, including breast cancer (28–31).
The dysregulated pattern of expression of miRs between normal and
cancerous tissues in breast cancer has been extensively studied.
The expression patterns of different miRs have been correlated with
tumor stage, estrogen and progesterone receptor expression,
proliferation index, vascular invasion, epithelial to mesenchymal
transition, metastasis and neovascularization (29,32–34).
Studies have shown that DNMT3a and DNMT3b are
directly targeted by members of the miR-29 family (miR-29a, miR-29b
and miR-29c) in lung cancer (35)
and acute myeloid leukemia (36).
Similarly, DNMT3b is regulated by the miR-148 family
(miR-148a and miR-148b) in cell lines of multiple origin, including
the MCF-7 breast cancer cell line (37). In recent investigations, we found
that the mechanism accounting for overexpression of DNMT3b among
breast cancer cell lines that express aberrant DNA hypermethylation
is related to concurrent loss of microRNAs (miRs) that
post-transcriptionally regulate DNMT3b mRNA, including
miR-29c, miR-148a, miR-148b, miR-26a, miR-26b and miR-203 (38).
In the current study, we investigated loss of
miR-mediated post-transcriptional regulation of DNMT3b in
primary invasive breast cancers as a molecular mechanism governing
the overexpression of DNMT3b that drives aberrant DNA
hypermethylation in basal-like breast cancer. We analyzed 70
paraffin-embedded human primary invasive breast cancers (36 luminal
A-like, 13 luminal B-like, 5 HER2-enriched and 16 basal-like) and
18 normal mammoplasty tissues for differential expression of
regulatory miRs. The results show that i) significantly reduced
expression of miR-29c distinguishes basal-like breast cancers from
the other breast cancer clinical subtypes; ii) miR expression
patterns revealed two groups among the basal-like breast cancers
corresponding to those with diminished expression and those
expressing normal levels of regulatory miRs; iii) loss of
combinations of miR-29a, miR-29b, miR-148a, miR-148b, miR-26a and
miR-26b is associated with expression of aberrant DNA
hypermethylation among primary invasive breast cancers; and iv)
basal-like breast cancers that exhibit aberrant DNA
hypermethylation express diminished levels of miRs that
post-transcriptionally regulate DNMT3b.
Materials and methods
Primary breast cancers and normal
mammoplasty tissues
A total of 70 paraffin-embedded human primary breast
tumors and 18 normal mammoplasty tissues were obtained from the
paraffin archives of the UNC Lineberger Comprehensive Cancer Center
at the University of North Carolina, School of Medicine (Chapel
Hill, NC, USA). Clinical classification of primary breast cancers
was accomplished by immunohistochemistry for ER, PR, HER2, CK5/6
and EGFR (39–41). The cohort of primary breast cancers
evaluated included examples from each of the intrinsic molecular
subtypes based upon their immunohistochemical surrogate: 36 luminal
A-like (ER+/PR+/HER2−), 13 luminal
B-like (ER+/PR+/HER2+), 5
HER2-enriched (ER−/PR−/HER2+), and
16 basal-like (ER−/PR−/HER2− plus
CK5/6+ or EGFR+) (42–44).
Protection of patient privacy and handling of specimens followed
strict policies of the Institutional Review Board of the University
of North Carolina School of Medicine. The current study was
reviewed by the Institutional Review Board of the University of
North Carolina School of Medicine and was formally declared exempt
based upon the use of existing data and existing tissue specimens
that were stripped of all identifying information. Hence, patient
consent was not required and was not sought.
RNA isolation from paraffin-embedded
tissues
The paraffin blocks were blinded (in terms of
clinical subtypes) before selection for analysis and up to 35 mg of
tissue core samples were obtained from the Translational Pathology
Laboratory core facility (Department of Pathology and Laboratory
Medicine, University of North Carolina School of Medicine). H&E
stained sections from each paraffin block were evaluated to select
areas of the blocks to be cored. This selection ensured that the
cores consisted of cancer tissue or normal breast epithelium (and
not stroma/fat) from primary breast cancer and reduction
mammoplasty tissues, respectively. Total RNA was isolated from
breast cancers and normal breast epithelium using Recover All™
Total Nucleic Acid Isolation Kit for FFPE according to the
manufacturer’s instructions (cat no. AM1975, Ambion/Life
Technologies, Carlsbad, CA, USA). Tissue cores were crushed and
ground in liquid nitrogen, and then deparaffinized using a series
of washes with Slide Brite (part no. SBT G1, Biocare Medical,
Concord, CA, USA) and ethanol. Nucleic acid samples were purified
using the Qiagen RNeasy mini kit (cat no. 74104, Qiagen, Valencia,
CA, USA). Isolated RNA was quantified after extraction using a
Nanodrop 2000 Spectrophotometer (NanoDrop Technologies, Wilmington,
DE, USA).
MicroRNA expression analysis
Members of miR-29 (miR-29a, miR-29b and miR-29c) and
miR-148 (miR-148a and miR-148b) family were selected for
examination based upon available literature linking them to direct
post-transcriptional regulation of DNMT3b in lung cancer
(35), acute myeloid leukemia
(36) and cell lines of multiple
origin (37). In addition,
candidate miR regulators of DNMT3b were identified (38) using the computational tools of
target prediction programs and resources from publicly available
databases, including Miranda (http://www.microRNA.org/), TargetScan (http://www.targetscan.org/vert_42/), miRGen
(http://www.diana.pcbi.upenn.edu/miRGen/v3/miRGen.html),
PicTar (http://pictar.mdc-berlin.de/), and
miRBase (http://microrna.sanger.ac.uk/sequences/). Target
predictions were made using Gene symbol DNMT3b (Entrez Gene ID 1789
and Ensembl Gene ID ENSG00000088305). Based on high stringency
in silico selection criteria that included PicTar score
(indicative of HMM maximum likelihood fit), highly conserved miRs,
and good mirSVR scores (indicative of seed-site pairing, site
context, free-energy and conservation), we identified miRs that
potentially target DNMT3b (38). The candidate miRs were prioritized
based on the available literature and/or their recognition as
potential candidates by multiple target prediction programs. miRs
that were differentially expressed among breast cancer cells in
primary cancers (29) and cell
lines (45) were considered for
further analysis. Based upon this computational analysis, nine miRs
were selected for examination: miR-29a, miR-29b, miR-29c, miR-148a,
miR-148b, miR-26a, miR-26b, miR-203 and miR-222.
miR expression analysis was accomplished by
real-time PCR utilizing an ABI 7500 Real Time PCR System (Applied
Biosystems/Life Technologies, Carlsbad, CA, USA) according to
TaqMan miRNA assay protocol (Applied Biosystems). TaqMan MiRNA
Reverse Transcription Kit (part no. 4366596, Applied
Biosystems/Life Technologies) was employed to reverse transcribe
the total RNA samples (10 ng) using the TaqMan miRNA specific
primers (Applied Biosystems/Life Technologies) according to the
manufacturer’s protocol. Real-time primers and probes for miR-29a
(assay ID 000412), miR-29b (assay ID 000413), miR-29c (assay ID
000415), miR-148a (assay ID 000470), miR-148b (assay ID 000471),
miR-26a (assay ID 000405), miR-26b (assay ID 000407), miR-203
(assay ID 000507), miR-222 (assay ID 002276) and RNU66 (assay ID
001002) were purchased from Applied Biosystems/Life Technologies.
These assays specifically detect mature miRNAs (not pre-miRNAs).
All real-time PCR reactions were performed in triplicate using
TaqMan Universal PCR Master mix (cat no. 4324018, Applied
Biosystems/Life Technologies) in 20 μl volume containing 10
μl TaqMan Universal PCR Master mix, 1 μl of primers
and probe mix of the miR-specific TaqMan MicroRNA Assay (Applied
Biosystems/Life Technologies), 1.33 μl of RT product, and
7.67 μl of nuclease free water and the following
amplification conditions: 95°C for 10 min, 40 cycles of 95°C for 15
sec and 60°C for 1 min. Relative expression levels for each miR
were calculated based upon the expression of RNU66 and differences
in gene expression were determined relative to normal breast
tissues from reduction mammoplasties using the comparative Ct
method described in the ABI Prism 7700 User Bulletin #2 (Applied
Biosystems/Life Technologies).
Gene expression analysis
Gene expression analysis was accomplished by
real-time PCR utilizing an ABI 7500 Real Time PCR System (Applied
Biosystems/Life Technologies). Total RNA samples (2 μg) were
reverse transcribed using the High Capacity cDNA Reverse
Transcription Kit (part no. 4368814, Applied Biosystems/Life
Technologies) according to the manufacturer’s protocol. Real-time
primers and probes for CEACAM6 (Hs00366002_m1), CDH1
(Hs00170423_m1), CST6 (Hs00154599_m1), ESR1
(Hs00174860_m1), GNA11 (Hs01588833_m1), MUC1
(Hs00159357_m1), MYB (Hs00920554_m1), SCNN1A
(Hs00168906_m1), TFF3 (Hs00173625_m1) and β-actin
(Hs99999903_m1) were purchased from Applied Biosystems/Life
Technologies. All real-time PCR reactions were performed in
triplicate using TaqMan Universal PCR Master mix (cat no. 4324018,
Applied Biosystems/Life Technologies) in 20 μl volume (10
μl TaqMan Universal PCR Master mix, 1.0 μl TaqMan
real-time primers and probes, and 9 μl cDNA and
nuclease-free water) and the following amplification conditions:
95°C for 10 min, 40 cycles of 95°C for 15 sec and 60°C for 1 min.
Relative expression levels for each gene were calculated based upon
the expression of β-actin for each sample and differences in
gene expression were determined relative to normal breast tissue
from reduction mammoplasties for primary tumors using the
comparative Ct method described in the ABI Prism 7700 User Bulletin
#2 (Applied Biosystems/Life Technologies).
Statistical analysis
The values for the mean and standard error of the
mean (SEM) were calculated using the statistical function of
Microsoft Excel 2007. Statistical significance was determined using
an unpaired t-test (two-tailed). Error bars depicted in bar graphs
represent SEM of 3–6 independent experiments.
Results
Breast cancers with aberrant DNA
hypermethylation express diminished levels of regulatory miRs
Our previous investigations identified aberrant DNA
hypermethylation (characterized by concurrent methylation-dependent
gene silencing events) that is significantly associated with the
basal-like subtype of breast cancer (12). The aberrant DNA hypermethylation
occurs secondary to DNMT hyperactivity and overexpression of DNMT3b
(12). In this study, we
investigated possible molecular mechanisms governing DNMT3b
overexpression driving aberrant DNA hypermethylation, with a focus
on miR-mediated regulation of DNMT3b in basal-like breast
cancers. Hence, we examined the levels of expression of select miRs
that are known or predicted to regulate DNMT3b (miR-26a,
miR-26b, miR-29a, miR-29b, miR-29c, miR-148a, miR-148b, miR-203 and
miR-222) among primary breast cancers. We utilized a cohort of 70
primary human breast cancers of known clinical classification
representing each of the intrinsic molecular subtypes (36 luminal
A-like, 13 luminal B-like, 5 Her2-enriched and 16 basal-like) and
18 normal mammoplasty tissues to analyze expression of microRNAs
that contribute to regulation of DNMT3b. Average miR
expression for breast cancers reflecting each of the clinical
classifications is shown in Table
I. Among the miRs evaluated, breast cancers associated with
specific clinical classifications displayed distinguishing levels
of expression. Significantly reduced average expression of miR-29c
distinguished basal-like breast cancers from other clinical
subtypes (Table I). Likewise,
HER2-enriched breast cancers expressed miR-29b at lower levels and
the luminal A-like breast cancers expressed miR-203 at lower levels
compared to the other breast cancer subtypes (Table I).
 | Table I.Average miR expression levels among
breast cancers representing clinical subtypes. |
Table I.
Average miR expression levels among
breast cancers representing clinical subtypes.
| miR species | Luminal A-like
(n=36) | Luminal B-like
(n=13) | HER2-enriched
(n=5) | Basal-like
(n=16) |
|---|
| miR-29a | 2.4±0.3 | 2.4±0.6 | 2.0±0.5 | 2.2±0.6 |
| miR-29b | 7.4±1.2 | 8.0±1.6 | 4.9±1.5 | 8.9±2.7 |
| miR-29c | 5.7±1.1 | 8.5±3.4 | 3.2±0.7 | 1.9±0.4 |
| miR-148a | 3.6±0.5 | 3.4±0.9 | 2.8±0.6 | 3.7±0.8 |
| miR-148b | 4.1±0.6 | 4.3±0.6 | 3.0±0.7 | 3.7±1.1 |
| miR-26a | 1.8±0.2 | 1.7±0.3 | 1.4±0.5 | 1.1±0.4 |
| miR-26b | 2.6±0.3 | 3.0±0.5 | 1.6±0.5 | 2.1±0.7 |
| miR-203 | 3.7±0.8 | 11.8±3.3 | 18.7±9.6 | 17.7±7.3 |
| miR-222 | 1.4±0.4 | 1.3±0.3 | 2.4±1.0 | 1.7±0.5 |
The methylation status of a subset of 33 breast
cancers (6 luminal A-like, 6 luminal B-like, 5 HER2-enriched, and
16 basal-like) was established through examination of
methylation-sensitive biomarker gene expression. Individual cancers
were classified as having aberrant DNA hypermethylation when their
expression signature reflected diminished levels of ≥7 epigenetic
biomarker genes. Among this cohort of 33 cancers, 11 (33%) were
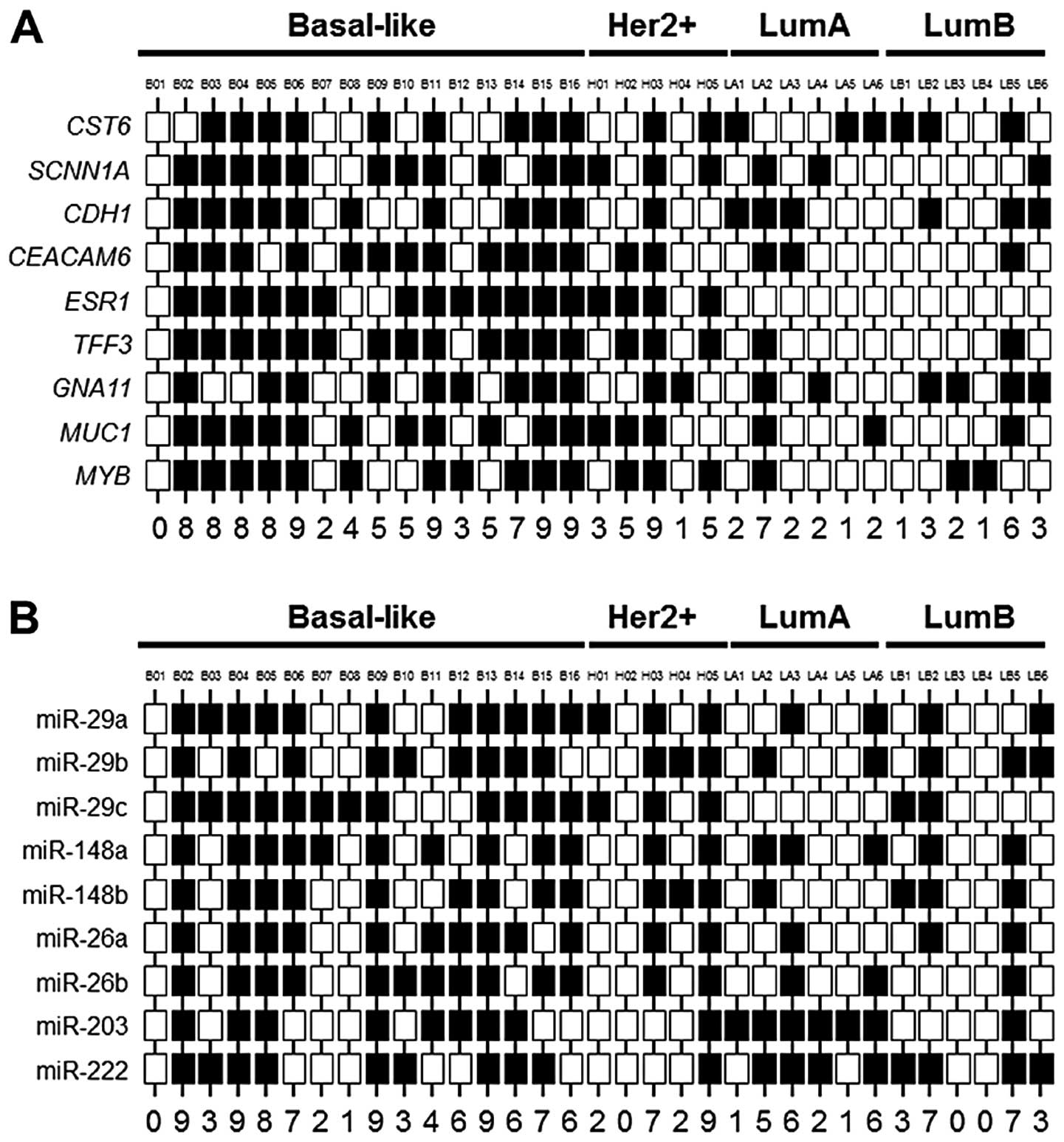
classified as having aberrant DNA hypermethylation (Fig. 1A). A total of 9/11 (82%) breast
cancers exhibiting aberrant DNA hypermethylation corresponded to
the basal-like subtype, and this group contains 56% (9/16) of all
basal-like cancers examined (Fig.
1A). The remaining breast cancers exhibiting aberrant DNA
hypermethylation correspond to the luminal A-like (n=1) and
HER2-enriched (n=1) subtypes. This finding is consistent with the
observation of a large degree of correspondence and overlap between
basal-like breast cancers and breast cancers exhibiting aberrant
DNA hypermethylation. The miR expression status within this group
of 33 breast cancers is shown in Fig.
1B.
Gene expression analysis identified two subsets of
basal-like breast cancers - those exhibiting aberrant DNA
hypermethylation (n=9, 56%) and those lacking aberrant DNA
hypermethylation (n=7, 44%) (Fig.
1A). The basal-like breast cancers exhibiting aberrant DNA
hypermethylation include B02–B06, B11 and B14–B16 (Fig. 1A). While there was variability in
miR expression among the basal-like breast cancers examined, in
general the cancers exhibiting aberrant DNA hypermethylation also
expressed diminished levels of regulatory miRs compared to the
cancers lacking aberrant DNA hypermethylation (Fig. 1B). However, miR-29c did not display
the pattern of expression observed with the majority of regulatory
miRs evaluated. Since loss of miR-29c differentiated the basal-like
cancers from other subtypes of breast cancers (Table I), the absence of differential
expression among the basal-like cancers suggests that loss of
miR-29c to be a feature of this clinical breast cancer subtype,
irrespective of aberrant DNA hypermethylation status.
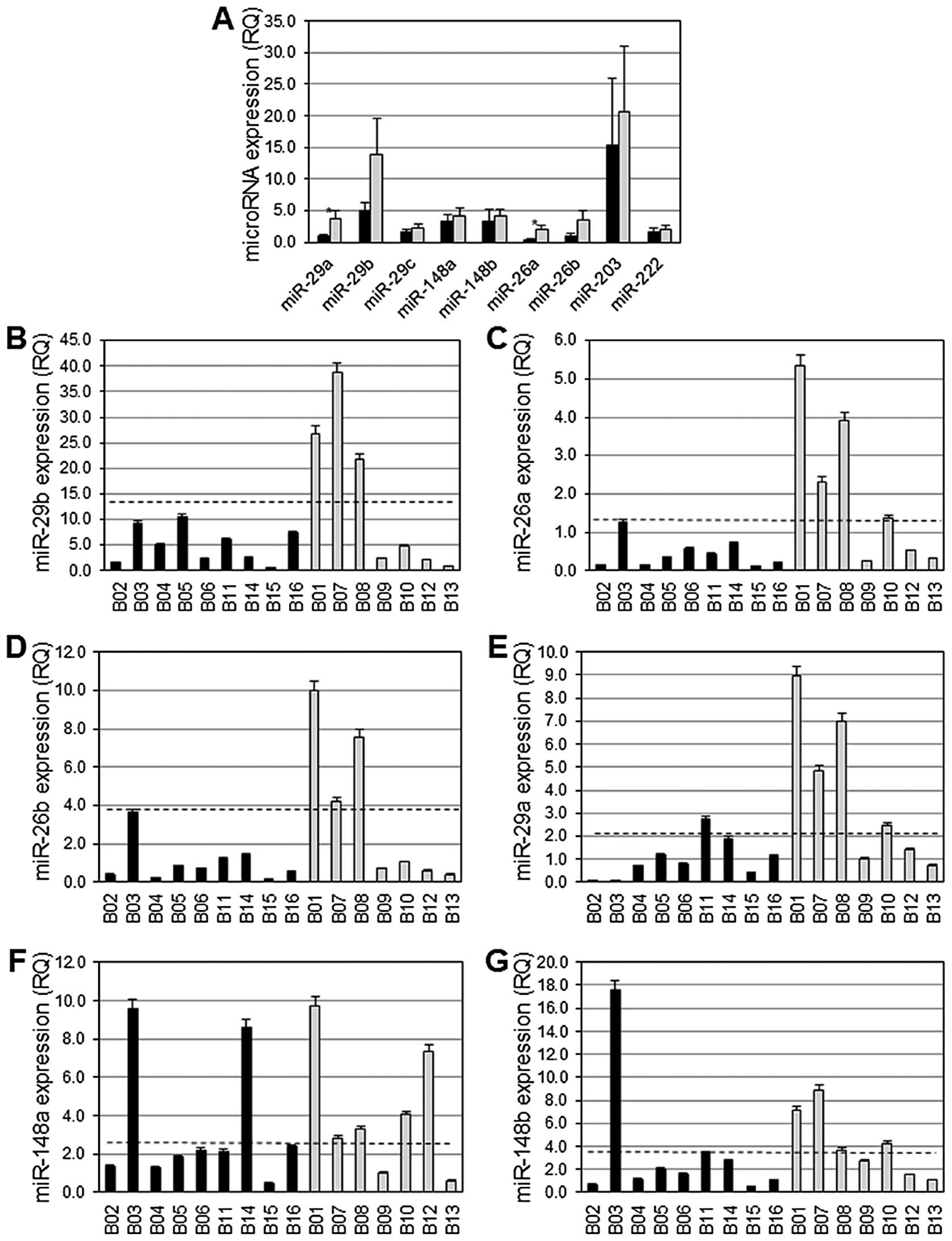
The average expression of miR-29a and miR-26a among
basal-like breast cancers exhibiting aberrant DNA hyper-methylation
was significantly diminished compared to the average expression of
these miRs among basal-like breast cancers lacking aberrant DNA
hypermethylation (p= 0.03) (Fig.
2A). Differences in the average expression level of miR-29b and
miR-26b among basal-like breast cancers exhibiting aberrant DNA
hypermethylation versus those lacking aberrant DNA hypermethylation
did not reach statistical significance (Fig. 2A), although there was a distinct
trend towards lower expression in the basal-like breast with
aberrant DNA hypermethylation (p=0.11 and p=0.08, respectively). A
total of 9/9 (100%) breast cancers exhibiting aberrant DNA
hypermethylation expressed low levels of miR-29b, miR-26a and
miR-26b, and 8/9 (89%) of these expressed low levels of miR-29a
(Fig. 2B–E). However, among breast
cancers lacking aberrant DNA hypermethylation, miR-29a and miR-26a
were normally expressed in 4/7 (57%) cancers, and miR-29b and
miR-26b were expressed at normal levels in 3/7 (43%)
non-hypermethylator cancers (Fig.
2B–E). Interestingly, the three breast cancers lacking aberrant
DNA hypermethylation with low levels of expression of miR-29a
exhibited low levels of expression of miR-26a, miR-29b and miR-26b.
In addition, these three cancers express low levels of miR-148b and
miR-203. A total of 7/9 (78%) and 8/9 (89%) basal-like breast
cancers exhibiting aberrant DNA hypermethylation had diminished
levels of expression of miR-148a and miR-148b, respectively
(Fig. 2F–G). These miRs were
expressed at normal levels in 5/7 (71%, miR-148a) and 4/7 (57%,
miR-148b) basal-like breast cancers that lack aberrant DNA
hypermethylation (Fig. 2F–G). A
total of 7/9 (78%) breast cancers exhibiting aberrant DNA
hypermethylation expressed miR-203 at low levels, while 3/7 (43%)
breast cancers lacking aberrant DNA hypermethylation expressed
miR-203 at easily detectable levels (data not shown). miR-222 was
expressed at low levels in 5/9 (56%) basal-like breast cancers
exhibiting aberrant DNA hyper-methylation, while 6/7 (87%)
basal-like breast cancers lacking aberrant DNA hypermethylation
expressed miR-222 at normal levels (data not shown).
Diminished expression of miR-29a,
miR-29b, miR-26a, miR-26b, miR-148a and miR-148b predict aberrant
DNA hyper-methylation among breast cancers
We observed differential expression of miR-29a,
miR-29b, miR-26a, miR-26b, miR-148a and miR-148b among basal-like
breast cancers with strong trends towards diminished expression in
those that exhibiting aberrant DNA hypermethylation compared to
those that do not. To assess the value of individual miR expression
levels in the prediction of aberrant DNA hypermethylation status of
a certain tumor, a Bayesian analysis was performed. Correct
assignments (CA) were used as a guiding principle to determine the
threshold values for each of the differentially expressed miRs
indicated in Fig. 2. The
expression level of miR-26a (CA, 81%) emerged as the best
individual predictor of aberrant DNA hypermethylation status among
basal-like breast cancers, followed by miR-29a (CA, 75%), miR-29b
(CA, 75%), miR-26b (CA, 75%), miR-148a (CA, 75%) and miR-148b (CA,
75%) (Table II). These miRs
individually displayed excellent sensitivity (range, 78–100%) and
negative predictive value (NPV range, 71–100%), as well as good
specificity (range, 43–71%) and positive predictive value (PPV
range, 69–78%). The remaining miRs displayed poor predictive value
for determination of aberrant DNA hypermethylation status among
breast cancers (CA, 63–69%) (Table
II).
| Table II.Bayesian analyses show that loss of
regulatory miR expression is associated with expression of aberrant
DNA hyper-methylation among primary basal-like breast cancers. |
Table II.
Bayesian analyses show that loss of
regulatory miR expression is associated with expression of aberrant
DNA hyper-methylation among primary basal-like breast cancers.
| miR species | Sensitivity
(%) | Specificity
(%) | Positive predictive
value (%) | Negative predictive
value (%) | Correct assignments
(%) |
|---|
| miR-29a | 89 | 58 | 73 | 80 | 75 |
| miR-29b | 100 | 43 | 69 | 100 | 75 |
| miR-29c | 67 | 43 | 60 | 50 | 63 |
| miR-148a | 78 | 71 | 78 | 71 | 75 |
| miR-148b | 89 | 57 | 73 | 80 | 75 |
| miR-26a | 100 | 57 | 75 | 100 | 81 |
| miR-26b | 100 | 43 | 69 | 100 | 75 |
| miR-203 | 78 | 43 | 64 | 60 | 63 |
| miR-222 | 56 | 86 | 83 | 60 | 69 |
miR scores correlate with
methylation-sensitive gene expression scores among primary breast
cancers
miR scores were generated for each basal-like breast
cancer, reflecting the number of miRs with diminished expression.
miR expression patterns revealed two groups among basal-like breast
cancers corresponding to those with low expression and those with
high expression (Fig. 1B). Low
expression is defined as diminished expression of ≥6 regulatory
miRs (n=11 basal-like breast cancers) and high expression is
defined as normal expression of ≥3 regulatory miRs (n=5 basal-like
breast cancers) (Fig. 1B).
Basal-like breast cancers exhibiting aberrant DNA hypermethylation
frequently express diminished levels of this panel of miRs. A total
of 7/9 (78%) basal-like cancers with aberrant DNA hypermethylation
express ≥6 regulatory miRs at diminished levels (Fig. 1B), resulting in higher miR scores.
Two basal-like breast cancers exhibiting aberrant DNA
hypermethylation expressed diminished levels of all nine miRs
examined (Fig. 1B). In contrast to
these these basal-like breast cancers, basal-like breast cancers
lacking aberrant DNA hypermethylation typically express the
majority of these regulatory miRs at higher levels. A total of 4/7
(57%) basal-like breast cancers lacking aberrant DNA
hypermethylation cancers express ≥7 miRs at higher levels (Fig. 1B), resulting in lower miR scores.
The relationship between aberrant DNA hypermethylation status and
miR score is complicated by the observation of two basal-like
breast cancers exhibiting aberrant DNA hypermethylation that
express most regulator miRs (n=6–7) at normal levels. Likewise, two
basal-like breast cancers lacking aberrant DNA hypermethylation
express all 9 regulatory miRs at reduced levels. Basal-like breast
cancers with aberrant DNA hypermethylation exhibit an average miR
score of 6.6±0.7, whereas, basal-like breast cancers lacking
aberrant DNA hypermethylation exhibit an average miR score of
4.2±1.4 (NS).
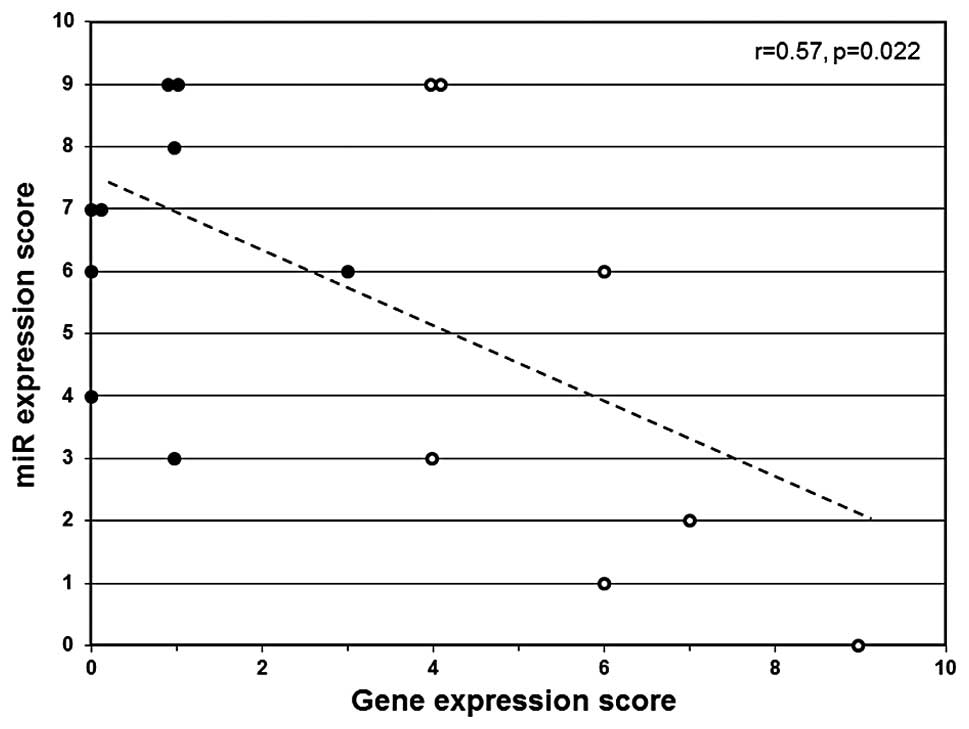
A linear correlation analysis was performed to
determine if miR scores significantly correlate with expression
score among basal-like breast cancers. The expression score
reflects the combined relative gene expression status for
methylation-sensitive biomarker genes associated with aberrant DNA
hypermethylation (CEACAM6, CDH1, CST6,
ESR1, GNA11, MUC1, MYB, TFF3 and
SCNN1A) (12). A
significant correlation (r= 0.57, p= 0.022) was observed between
miR score and gene expression score (Fig. 3). The cancers that exhibit
diminished expression of multiple regulatory miRs (high miR score)
tend to express low levels of methylation-sensitive genes (gene
expression score) and cancers that express higher levels of
regulatory miRs (low miR score) tend to express
methylation-sensitive genes at higher levels (Fig. 3).
Co-regulation of miR expression among
primary breast cancers
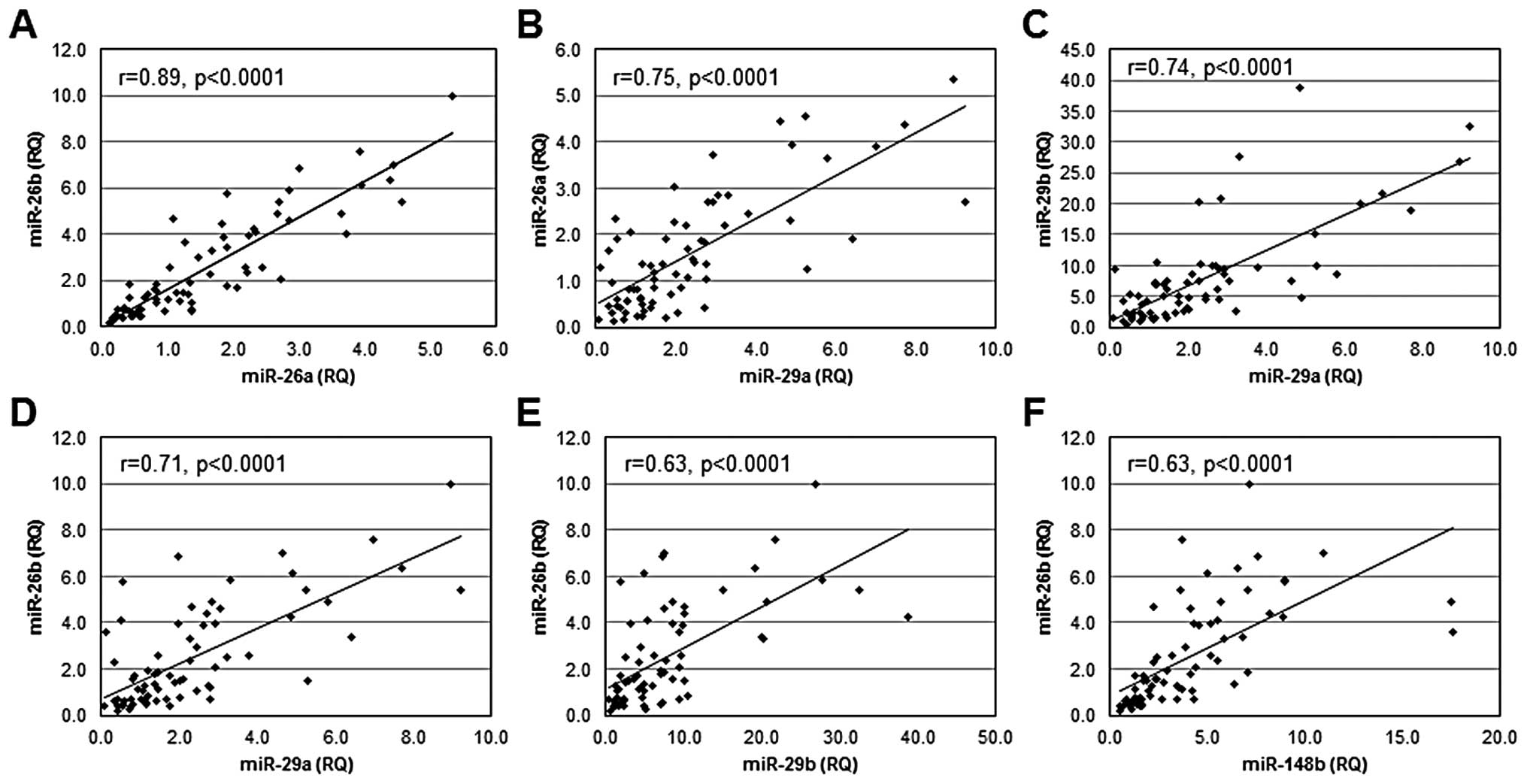
To determine if miRs that regulate DNMT3b are
independently regulated or co-regulated at the level of expression,
a linear correlation analysis was performed to examine patterns of
miR expression among primary breast cancers. Statistically
significant linear relationships were observed between the levels
of expression of several miRs (Fig.
4): miR-26a and miR-26b (r=0.89, p<0.0001); miR-29a and
miR-26a (r= 0.75, p<0.0001); miR-29a and miR-29b (r=0.74,
p<0.0001); miR-29a and miR-26b (r=0.71, p<0.0001); miR-29b
and miR-26b (r=0.63, p<0.0001); and miR-148b and miR-26b (r=
0.63, p<0.0001). In addition, significant linear relationships
were observed for expression of miR-26a and miR-203 (r=0.71,
p=0.0019); miR-26b and miR-203 (r=0.68, p=0.038); miR-26a and
miR-29c (r=0.60, p=0.014), miR-148a and miR-203 (r=0.60, p=0.014),
and miR-26b and miR-148b (r= 0.50, p= 0.04). No significant linear
relationships were observed for expression of miR-26b and miR-29c;
miR-148b and miR-203; or miR-29c and miR-203. Combined, these
observations suggest that the miRs that function in the regulation
of DNMT3b are co-regulated.
Discussion
Epigenetic changes play an important role in normal
regulation of gene expression and loss of regulation can
significantly contribute to cancer initiation, development and
progression (46,47). Epigenetic aberrations such as the
silencing of tumor suppressor genes and other negative mediators of
neoplastic development have been documented in breast
carcinogenesis (11,48,49).
The CpG island methylator phenotype (or CIMP) represents a major
epigenetic mechanism that has been recognized to contribute
significantly to colorectal carcinogenesis as well as to cancers
affecting other tissues (50–52).
In previous studies, we identified a subset of breast cancer cell
lines and primary breast cancers that exhibit aberrant DNA
hypermethylation that results in concurrent epigenetic silencing of
multiple methylation-sensitive genes (including CEACAM6,
CDH1, CST6, ESR1, GNA11, MUC1,
MYB, TFF3 and SCNN1A) secondary to DNA
methyltransferase enzyme hyper activity associated with
overexpression of DNMT3b (12).
Mining of microarray-based expression data identified the gene
expression signature associated with aberrant DNA hyper-methylation
in primary sporadic invasive breast cancers (12). A significant correspondence was
observed between aberrant DNA hypermethylation and the basal-like
molecular subtype of breast cancers (12). Many basal-like breast cancers
exhibit the silencing of genes associated with DNMT3b protein
over-expression. This observation strongly suggests that the unique
features of basal-like breast cancers (poor clinical outcomes,
variable response to chemotherapy and recurrence following
chemotherapy) may be a direct consequence of methylation-dependent
gene silencing events associated with DNMT3b overexpression. This
fundamental observation related to the basal-like breast cancers
underscores the significance of understanding the mechanism
contributing to the overexpression of DNMT3b in these deadly breast
cancers.
Several studies have established that miRs exhibit
altered expression in cancer tissues compared to normal tissues
suggesting that miRs have a role in defining the molecular and
pathological profiles of cancers including breast cancer (29,53,54).
In addition, miRs have been established as key players in
carcinogenesis, with oncogenic or tumor suppressor-like functions
(17). Our results suggest loss of
combinations of miR-29a, miR-29b, miR-148a, miR-148b, miR-26a and
miR-26b is associated with aberrant DNA hypermethylation in primary
invasive breast cancers, consistent with the idea that these miRs
function as negative mediators of DNMT3b-mediated aberrant DNA
hypermethylation. We have also observed a significant concordance
between basal-like breast cancers that exhibit aberrant DNA
hypermethylation and the group of primary cancers with diminished
expression of regulatory miRs. Loss/ reduced levels of these miRs
has been documented in various forms of cancer, supporting the
suggestion that these miRs possess tumor suppressor-like function.
miR-29a and miR-29b are downregulated in chronic lymphocytic
leukemia, acute myeloid leukemia, lung cancers, cholangiocarcinoma
and prostate cancer (35,55–59).
Diminished expression of miR-26a occurs in hepatocellular
carcinoma, oral squamous cell carcinoma, bladder cancer, thyroid
anaplastic carcinoma, Burkitt’s lymphoma, acute myeloid leukemia,
papillary carcinoma, prostate cancer and breast cancer (18,60,61).
miR-26b expression is decreased in Hodgkin’s lymphoma, oral
squamous cell carcinoma and prostate cancers (61). miR-29c expression is depressed in
nasopharyngeal carcinomas, bladder cancers, chronic lymphocytic
leukemia, acute myeloid leukemia, lung cancers, cholangiocarcinoma,
esophageal squamous cell carcinoma and pancreatic ductal
adenocarcinoma (18,55,60–62).
miR-148a is downregulated in breast cancers, papillary thyroid
carcinoma, pancreatic ductal adenocarcinoma, prostate cancer and
colorectal adenocarcinoma (60,61).
miR-148b is expressed at diminished levels in oral squamous cell
carcinoma, papillary thyroid carcinoma, prostate cancer, colorectal
adenocarcinoma and pancreatic ductal adenocarcinoma (61). These studies from the literature
provide evidence of loss or diminished expression of these
regulatory miRs in various forms of cancer, including breast in
some cases.
Multiple mechanisms contribute to miR dysregulation
in cancer, including genetic abnormalities (such as chromosomal
rearrangement, deletion, amplification or sequence mutations) and
epigenetic changes (methylation-dependent silencing of miR
expression or alterations in the miRNA biogenesis machinery)
(18). More than half of the miR
genes (>50%) are located within or in close proximity to
chromosomal fragile sites and other genomic regions associated with
cancer (18). These sites are
prone to genetic alterations and changes in these chromosomal
regions result in dramatic alteration of miR expression levels
(18). Similarly, numerous studies
report promoter hypermethylation as an important mechanism leading
to loss of miR expression in cancer (17). miR-148a and miR148b are susceptible
to methylation-dependent silencing in cancer (17). These observations suggest that loss
of regulatory miR expression leading to DNMT3b dysregulation
could be the result of genetic or epigenetic mechanisms. Further
investigation will be required to establish which of these
potential mechanisms contribute to miR dysregulation in basal-like
breast cancer.
Acknowledgements
This study was supported by grants to
W.B.C. from the Susan G. Komen Breast Cancer Foundation
(BCTR0100-575), the National Cancer Institute (NIH grant CA78343),
Friends for an Earlier Breast Cancer Test (Earlier.org), the
UNC Lineberger Comprehensive Cancer Center, the University Research
Council, and the Medical Alumni Endowment Fund of the University of
North Carolina at Chapel Hill.
References
|
1.
|
Herman JG and Baylin SB: Gene silencing in
cancer in association with promoter hypermethylation. N Engl J Med.
349:2042–2054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Baylin S: DNA methylation and epigenetic
mechanisms of carcinogenesis. Dev Biol (Basel). 106:85–87.
2001.PubMed/NCBI
|
|
3.
|
Baylin SB, Herman JG, Graff JR, Vertino PM
and Issa JP: Alterations in DNA methylation: a fundamental aspect
of neoplasia. Adv Cancer Res. 72:141–196. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Feinberg AP and Vogelstein B:
Hypomethylation of ras oncogenes in primary human cancers. Biochem
Biophys Res Commun. 111:47–54. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Narayan A, Ji W, Zhang XY, Marrogi A,
Graff JR, Baylin SB and Ehrlich M: Hypomethylation of
pericentromeric DNA in breast adenocarcinomas. Int J Cancer.
77:833–838. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Eden A, Gaudet F, Waghmare A and Jaenisch
R: Chromosomal instability and tumors promoted by DNA
hypomethylation. Science. 300:4552003. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Gaudet F, Hodgson JG, Eden A,
Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H and Jaenisch R:
Induction of tumors in mice by genomic hypomethylation. Science.
300:489–492. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Jones PA and Laird PW: Cancer epigenetics
comes of age. Nat Genet. 21:163–167. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Kanai Y and Hirohashi S: Alterations of
DNA methylation associated with abnormalities of DNA
methyltransferases in human cancers during transition from a
precancerous to a malignant state. Carcinogenesis. 28:2434–2442.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Lewis CM, Cler LR, Bu DW, Zochbauer-Muller
S, Milchgrub S, Naftalis EZ, Leitch AM, Minna JD and Euhus DM:
Promoter hypermethylation in benign breast epithelium in relation
to predicted breast cancer risk. Clin Cancer Res. 11:166–172.
2005.PubMed/NCBI
|
|
11.
|
Ai L, Kim WJ, Kim TY, Fields CR, Massoll
NA, Robertson KD and Brown KD: Epigenetic silencing of the tumor
suppressor cystatin M occurs during breast cancer progression.
Cancer Res. 66:7899–7909. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Roll JD, Rivenbark AG, Jones WD and
Coleman WB: DNMT3b overexpression contributes to a hypermethylator
phenotype in human breast cancer cell lines. Mol Cancer. 7:152008.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Girault I, Tozlu S, Lidereau R and Bieche
I: Expression analysis of DNA methyltransferases 1, 3A, and 3B in
sporadic breast carcinomas. Clin Cancer Res. 9:4415–4422.
2003.PubMed/NCBI
|
|
14.
|
Robertson KD, Uzvolgyi E, Liang G,
Talmadge C, Sumegi J, Gonzales FA and Jones PA: The human DNA
methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression
in normal tissues and overexpression in tumors. Nucleic Acids Res.
27:2291–2298. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Mizuno S, Chijiwa T, Okamura T, Akashi K,
Fukumaki Y, Niho Y and Sasaki H: Expression of DNA
methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in
acute and chronic myelogenous leukemia. Blood. 97:1172–1179. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Jin F, Dowdy SC, Xiong Y, Eberhardt NL,
Podratz KC and Jiang SW: Up-regulation of DNA methyltransferase 3B
expression in endometrial cancers. Gynecol Oncol. 96:531–538. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Veeck J and Esteller M: Breast cancer
epigenetics: from DNA methylation to microRNAs. J Mammary Gland
Biol Neoplasia. 15:5–17. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Melo SA and Esteller M: Dysregulation of
microRNAs in cancer: Playing with fire. FEBS Lett. 585:2087–2099.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Giraldez AJ, Mishima Y, Rihel J, Grocock
RJ, Van Dongen S, Inoue K, Enright AJ and Schier AF: Zebrafish
MiR-430 promotes deadenylation and clearance of maternal mRNAs.
Science. 312:75–79. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Bartel B: MicroRNAs directing siRNA
biogenesis. Nat Struct Mol Biol. 12:569–571. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Bagga S, Bracht J, Hunter S, Massirer K,
Holtz J, Eachus R and Pasquinelli AE: Regulation by let-7 and lin-4
miRNAs results in target mRNA degradation. Cell. 122:553–563. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Pillai RS, Bhattacharyya SN, Artus CG,
Zoller T, Cougot N, Basyuk E, Bertrand E and Filipowicz W:
Inhibition of translational initiation by Let-7 MicroRNA in human
cells. Science. 309:1573–1576. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Sevignani C, Calin GA, Siracusa LD and
Croce CM: Mammalian microRNAs: a small world for fine-tuning gene
expression. Mamm Genome. 17:189–202. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Miska EA: How microRNAs control cell
division, differentiation and death. Curr Opin Genet Dev.
15:563–568. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Carleton M, Cleary MA and Linsley PS:
MicroRNAs and cell cycle regulation. Cell Cycle. 6:2127–2132. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Calin GA, Dumitru CD, Shimizu M, Bichi R,
Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L,
Kipps T, Negrini M, Bullrich F and Croce CM: Frequent deletions and
down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in
chronic lymphocytic leukemia. Proc Natl Acad Sci USA.
99:15524–15529. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Iorio MV, Ferracin M, Liu CG, Veronese A,
Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M,
Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I,
Calin GA, Querzoli P, Negrini M and Croce CM: MicroRNA gene
expression deregulation in human breast cancer. Cancer Res.
65:7065–7070. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Lujambio A, Calin GA, Villanueva A, Ropero
S, Sanchez-Cespedes M, Blanco D, Montuenga LM, Rossi S, Nicoloso
MS, Faller WJ, Gallagher WM, Eccles SA, Croce CM and Esteller M: A
microRNA DNA methylation signature for human cancer metastasis.
Proc Natl Acad Sci USA. 105:13556–13561. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Hayashita Y, Osada H, Tatematsu Y, Yamada
H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y and
Takahashi T: A polycistronic microRNA cluster, miR-17-92, is
overexpressed in human lung cancers and enhances cell
proliferation. Cancer Res. 65:9628–9632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Korpal M, Lee ES, Hu G and Kang Y: The
miR-200 family inhibits epithelial-mesenchymal transition and
cancer cell migration by direct targeting of E-cadherin
transcriptional repressors ZEB1 and ZEB2. J Biol Chem.
283:14910–14914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Anand S, Majeti BK, Acevedo LM, Murphy EA,
Mukthavaram R, Scheppke L, Huang M, Shields DJ, Lindquist JN,
Lapinski PE, King PD, Weis SM and Cheresh DA: MicroRNA-132-mediated
loss of p120RasGAP activates the endothelium to facilitate
pathological angiogenesis. Nat Med. 16:909–914. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Ma L, Teruya-Feldstein J and Weinberg RA:
Tumour invasion and metastasis initiated by microRNA-10b in breast
cancer. Nature. 449:682–688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Fabbri M, Garzon R, Cimmino A, Liu Z,
Zanesi N, Callegari E, Liu S, Alder H, Costinean S,
Fernandez-Cymering C, Volinia S, Guler G, Morrison CD, Chan KK,
Marcucci G, Calin GA, Huebner K and Croce CM: MicroRNA-29 family
reverts aberrant methylation in lung cancer by targeting DNA
methyltransferases 3A and 3B. Proc Natl Acad Sci USA.
104:15805–15810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Garzon R, Liu S, Fabbri M, Liu Z, Heaphy
CE, Callegari E, Schwind S, Pang J, Yu J, Muthusamy N, Havelange V,
Volinia S, Blum W, Rush LJ, Perrotti D, Andreeff M, Bloomfield CD,
Byrd JC, Chan K, Wu LC, Croce CM and Marcucci G: MicroRNA-29b
induces global DNA hypomethylation and tumor suppressor gene
reexpression in acute myeloid leukemia by targeting directly DNMT3A
and 3B and indirectly DNMT1. Blood. 113:6411–6418. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Duursma AM, Kedde M, Schrier M, le Sage C
and Agami R: miR-148 targets human DNMT3b protein coding region.
RNA. 14:872–877. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Sandhu R, Rivenbark AG and Coleman WB:
Loss of post-transcriptional regulation of DNMT3b by microRNAs: a
possible molecular mechanism for the hypermethylation defect
observed in a subset of breast cancer cell lines. Int J Oncol.
41:721–732. 2012.PubMed/NCBI
|
|
39.
|
Livasy CA, Karaca G, Nanda R, Tretiakova
MS, Olopade OI, Moore DT and Perou CM: Phenotypic evaluation of the
basal-like subtype of invasive breast carcinoma. Mod Pathol.
19:264–271. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Nielsen TO, Hsu FD, Jensen K, Cheang M,
Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler
L, Akslen LA, Ragaz J, Gown AM, Gilks CB, van de Rijn M and Perou
CM: Immunohistochemical and clinical characterization of the
basal-like subtype of invasive breast carcinoma. Clin Cancer Res.
10:5367–5374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Harvey JM, Clark GM, Osborne CK and Allred
DC: Estrogen receptor status by immunohistochemistry is superior to
the ligand-binding assay for predicting response to adjuvant
endocrine therapy in breast cancer. J Clin Oncol. 17:1474–1481.
1999.PubMed/NCBI
|
|
42.
|
Perou CM, Sorlie T, Eisen MB, van de Rijn
M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA,
Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE,
Borresen-Dale AL, Brown PO and Botstein D: Molecular portraits of
human breast tumours. Nature. 406:747–752. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Sorlie T, Perou CM, Tibshirani R, Aas T,
Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey
SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein
Lonning P and Borresen-Dale AL: Gene expression patterns of breast
carcinomas distinguish tumor subclasses with clinical implications.
Proc Natl Acad Sci USA. 98:10869–10874. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Sorlie T, Tibshirani R, Parker J, Hastie
T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S,
Demeter J, Perou CM, Lonning PE, Brown PO, Borresen-Dale AL and
Botstein D: Repeated observation of breast tumor subtypes in
independent gene expression data sets. Proc Natl Acad Sci USA.
100:8418–8423. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Gaur A, Jewell DA, Liang Y, Ridzon D,
Moore JH, Chen C, Ambros VR and Israel MA: Characterization of
microRNA expression levels and their biological correlates in human
cancer cell lines. Cancer Res. 67:2456–2468. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Feinberg AP and Tycko B: The history of
cancer epigenetics. Nat Rev Cancer. 4:143–153. 2004. View Article : Google Scholar
|
|
47.
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar
|
|
48.
|
Yang X, Yan L and Davidson NE: DNA
methylation in breast cancer. Endocr Relat Cancer. 8:115–127. 2001.
View Article : Google Scholar
|
|
49.
|
Esteller M: CpG island hypermethylation
and tumor suppressor genes: a booming present, a brighter future.
Oncogene. 21:5427–5440. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Toyota M, Ahuja N, Ohe-Toyota M, Herman
JG, Baylin SB and Issa JP: CpG island methylator phenotype in
colorectal cancer. Proc Natl Acad Sci USA. 96:8681–8686. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Shen L, Ahuja N, Shen Y, Habib NA, Toyota
M, Rashid A and Issa JP: DNA methylation and environmental
exposures in human hepatocellular carcinoma. J Natl Cancer Inst.
94:755–761. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Toyota M, Ahuja N, Suzuki H, Itoh F,
Ohe-Toyota M, Imai K, Baylin SB and Issa JP: Aberrant methylation
in gastric cancer associated with the CpG island methylator
phenotype. Cancer Res. 59:5438–5442. 1999.PubMed/NCBI
|
|
53.
|
Nikiforova MN, Tseng GC, Steward D, Diorio
D and Nikiforov YE: MicroRNA expression profiling of thyroid
tumors: biological significance and diagnostic utility. J Clin
Endocrinol Metab. 93:1600–1608. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Corcoran C, Friel AM, Duffy MJ, Crown J
and O’Driscoll L: Intracellular and extracellular microRNAs in
breast cancer. Clin Chem. 57:18–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Garzon R, Calin GA and Croce CM: MicroRNAs
in cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar
|
|
56.
|
Calin GA, Ferracin M, Cimmino A, Di Leva
G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M,
Iuliano R, Palumbo T, Pichiorri F, Roldo C, Garzon R, Sevignani C,
Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M and
Croce CM: A MicroRNA signature associated with prognosis and
progression in chronic lymphocytic leukemia. N Engl J Med.
353:1793–1801. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Yanaihara N, Caplen N, Bowman E, Seike M,
Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, Calin
GA, Liu CG, Croce CM and Harris CC: Unique microRNA molecular
profiles in lung cancer diagnosis and prognosis. Cancer Cell.
9:189–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Garzon R, Volinia S, Liu CG,
Fernandez-Cymering C, Palumbo T, Pichiorri F, Fabbri M, Coombes K,
Alder H, Nakamura T, Flomenberg N, Marcucci G, Calin GA, Kornblau
SM, Kantarjian H, Bloomfield CD, Andreeff M and Croce CM: MicroRNA
signatures associated with cytogenetics and prognosis in acute
myeloid leukemia. Blood. 111:3183–3189. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Mott JL, Kobayashi S, Bronk SF and Gores
GJ: mir-29 regulates Mcl-1 protein expression and apoptosis.
Oncogene. 26:6133–6140. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Visone R and Croce CM: MiRNAs and cancer.
Am J Pathol. 174:1131–1138. 2009. View Article : Google Scholar
|
|
61.
|
Wang Y and Lee CG: MicroRNA and cancer -
focus on apoptosis. J Cell Mol Med. 13:12–23. 2009. View Article : Google Scholar
|
|
62.
|
Sengupta S, den Boon JA, Chen IH, Newton
MA, Stanhope SA, Cheng YJ, Chen CJ, Hildesheim A, Sugden B and
Ahlquist P: MicroRNA 29c is down-regulated in nasopharyngeal
carcinomas, up-regulating mRNAs encoding extracellular matrix
proteins. Proc Natl Acad Sci USA. 105:5874–5878. 2008. View Article : Google Scholar : PubMed/NCBI
|