Introduction
Fibroblast growth factor-inducible-14 (Fn14),
originally identified as one of the growth factor-inducible genes
present in murine fibroblasts, is the smallest member of the tumor
necrosis factor (TNF) superfamily of receptors that lacks the
cytoplasmic death domain (1–3). The
Fn14 gene is expressed in many tissues, including the heart,
placenta, kidneys, lungs, and pancreas, and is located on human
chromosome 6 (2). In the GENT
database, Fn14 is upregulated in many tumor tissues compared to
normal tissues (4) and has also
been reported to be elevated in various cancers, including GC
(2,5,6).
Addition of TNF-like weak inducer of apoptosis (TWEAK), a ligand
for Fn14, activates Fn14 and increases cell survival, growth, and
migration in some cancers (5,6).
Inhibition of Fn14 expression decreases migration and invasion,
whereas ectopic expression of Fn14 increases the invasive activity
of prostate cancer cells (7). The
levels of Fn14 expression also affect the growth of GC cells
(6). A recent study reported that
immunotoxins targeting the Fn14 receptor induce melanoma cell
necrosis, which leads to growth inhibition (8), suggesting that Fn14 may be an
appropriate therapeutic target for cancer.
NF-κB is activated in response to various
inflammatory agents, carcinogens, tumor promoters, and growth
factors and regulates the expression of various genes that are
involved in immunity, inflammation, cell survival, or oncogenesis
(9,10). NF-κB is an inhibitor of apoptotic
cell death and activates several target genes that are involved in
preventing the induction of apoptosis (11). NF-κB-mediated anti-apoptotic
factors contain cIAPs, c-FLIP, and members of the Bcl-2 family
(11). NF-κB is constitutively
active in cancer cells and is highly resistant to antitumor drugs
or ionizing radiation, while inhibition of NF-κB activity greatly
promotes sensitivity to such treatments (12). TWEAK/Fn14 signaling is one of the
important upstream stimulators of NF-κB activation resulting in
induction of the expression of NF-κB-regulated anti-apoptotic genes
(13,14). These anti-apoptotic genes are
associated with the inhibition of apoptosis and the resistance to
anticancer drugs, resulting in poor prognosis and shortened
survival (15), suggesting that
the TWEAK/Fn14/NF-κB cascade may be involved in
chemoresistance.
Identification of the major molecules that regulate
cancer resistance and the development of new strategies to treat
the disease are required to find an effective cure for cancer.
Gastric cancer is a common malignancy that is associated with a
high mortality rate. It is the fourth most commonly diagnosed
cancer in the world and the third most common cause of
cancer-related deaths (16).
Although many antitumor drugs have been used to treat patients with
cancer, drug resistance has become a serious problem. Overcoming
the problem of drug resistance could improve the efficacy of
antitumor drugs. 5-FU is a chemotherapeutic agent that is widely
used for the treatment of GC. 5-FU exerts its toxic effects on
cancer cells by inducing apoptosis (17,18).
However, the mechanism by which 5-FU activates the apoptotic
process is not fully understood in GC. Clarification of the
underlying mechanism may help to decrease the frequency of
resistance to 5-FU-induced cell death in cancer. In this study, we
investigated how the Fn14 molecule contributes to 5-FU
chemoresistance in GC cells.
Materials and methods
Cell lines
Gastric cancer cell lines were cultured in complete
RPMI-1640 medium. 293T cells were maintained in complete DMEM
media. All cell lines were obtained from the Korean Cell Line Bank
(http://cellbank.snu.ac.kr/index.htm),
and all complete media contained 10% fetal bovine serum (Hyclone,
Logan, UT, USA), 100 U/ml of penicillin/streptomycin (Invitrogen,
Carlsbad, CA, USA), 2 mM L-glutamine, and 0.5 mM HEPES.
Lentiviral packaging and transduction of
Fn14 small hairpin RNA (shRNA)
A non-targeting shRNA control vector (catalog no.
SHC002) and shRNA lentiviral vectors targeting human Fn14 mRNA
(catalog no. TRCN0000072451) were purchased from Sigma-Aldrich. For
lentivirus production, the shRNA vector was cotransfected with the
lentiviral packaging mix (Sigma-Aldrich, Taufkirchen, Germany) into
293T cells using FuGENE HD (Roche Diagnostics, Indianapolis, IN,
USA) according to the manufacturer’s protocol. SNU-216 and MKN-1
cells were infected and selected using 1 μg/ml of puromycin.
Fn14 protein reduction was assessed using western blot
analysis.
Fn14 stable cell lines
Fn14 expression plasmids (6) were transfected into AGS cells using
Lipofectamine Plus reagent (Invitrogen) according to the
manufacturer’s protocol. Transfected cells were cultured for 2 days
prior to selection in the presence of 1 mg/ml of G418 in complete
medium. Fn14 protein levels were assessed using western blot
analysis.
Transfection and luciferase reporter
assays
SNU-216 cells were seeded into a 24-well plate 1 day
prior to transfection. The cells were transiently transfected with
the NF-κB luciferase reporter gene (Clontech, Palo Alto, CA, USA)
using Lipofectamine Plus reagent (Invitrogen) according to the
manufacturer’s protocol and were treated with either various
amounts of 5-FU, 20 μg of 5-FU alone, or 20 μg of
5-FU in combination with 20 μM PS1145 for 24 h. Forty-eight
hours after transfection, the cells were harvested, and luciferase
activity was determined using the luciferase assay system (Promega,
Madison, WI, USA) with a luminometer. Luciferase activities were
normalized to β-galactosidase activity using the β-galactosidase
enzyme assay system (Promega) according to the manufacturer’s
instructions.
siRNA transfection
A non-targeting siRNA control (catalog no. SN1003)
and siRNAs targeting human Bcl-xL (catalog no. 1011922) or p65 mRNA
(catalog no. 1128166) were purchased from Bioneer (Daejeon, Korea).
For siRNA transfection, SNU-216 and MKN-1 cells were transfected
with siRNAs (100 nM) using Lipofectamine Plus (Invitrogen), and the
knockdown of Bcl-xL and p65 was quantified 48 h post-transfection
using qRT-PCR and western blotting. To assess the effects of Bcl-xL
and p65 siRNA on cell growth, SNU-216 and MKN-1 cells were seeded
in 96-well plates and transfected with siRNAs (100 nM) using
Lipofectamine plus (Invitrogen). Cell growth was measured 24 and 48
h after 5-FU treatment using the CCK-8 reagent (Dojindo, Kumamoto,
Japan).
Real-time RT-PCR
Total RNA was isolated from GC tissues using an
RNeasy kit (Qiagen, Valencia, CA, USA) according to the
manufacturer’s instructions prior to treatment with DNase I
(Promega). DNase-treated RNA was reverse transcribed using
Superscript II reverse transcriptase (Invitrogen). qRT-PCR was
performed using an Exicycler Quantitative Thermal Block (Bioneer).
The resulting cDNA was amplified using 2X SYBR Premix EX Taq
(Takara, Shiga, Japan). β-actin was used as a loading control. The
fold change in the expression of each gene was calculated using the
ΔΔCt method (19). The primer
sequences used were as follows: human p65,
5′-GCGAGAGGAGCACAGATACC-3′ (forward) and 5′-CTGATAGCCTGCTCCAGGTC-3′
(reverse); human Fn14, 5′-TTT CTG GCT TTT TGG TCT GG-3′ (forward)
and 5′-CTT GTG GTT GGA GGA GCT TG-3′ (reverse); Bcl-xL, 5′-CTG AAT
CGG AGA TGG AGA CC-3′ (forward) and 5′-TGG GAT GTC AGG TCA CTG
AA-3′ (reverse); and human β-actin, 5′-CAA GAG ATG GCC ACG GCT
GCT-3′ (forward) and 5′-TCC TTC TGC ATC CTG TCG GCA-3′
(reverse).
Western blot analyses
The cells were pelleted and lysed in NP-40 lysis
buffer (20 mM Tris-HCl, pH 8.0, 140 mM NaCl, 10% glycerol, 1%
NP-40, 2 mM EDTA, and protease inhibitor). Protein levels were
quantified using the Bio-Rad protein assay method. The cell lysates
were separated using SDS-PAGE. After electrophoresis, the proteins
were transferred to nitrocellulose membranes, blotted with the
relevant antibodies, and detected using ECL reagent (Amersham
Bioscience, Buckinghamshire, UK). The primary antibodies used
included Fn14 (Cell Signaling, Beverly, MA, USA), Bcl-xL (Cell
Signaling), p65 (Santa Cruz Biotechnology, Santa Cruz, CA, USA),
phosphor-IκB (Cell Signaling), IκB (Santa Cruz Biotechnology), and
α-tubulin (Sigma).
Caspase-3/7 assay
SNU-216 and MKN-1 cells were seeded in 96-well
plates and treated with 5-FU. After 48 h of treatment, the cells
were washed once with 1X PBS, trypsinized, washed again with 1X
PBS, and then resuspended in PBS. The cells were mixed with
caspase-Glo-3/7 reagent (Promega) and incubated for 1 h at room
temperature with agitation. The luciferase activity was measured
using a luminometer and then normalized according to protein
concentration.
Cell growth assay
Cells from different groups were plated into 96-well
plates at 1×104 cells/well and maintained at 37°C in a
humidified incubator. The cells were treated with various amounts
of 5-FU for 48 h. To assess the effect of TWEAK on cell survival,
the cells were treated with 50 μg (SNU-216) or 20 μg
(MKN-1) of 5-FU alone or in combination with TWEAK (100 ng/ml) for
24 h. Cell growth was measured using the cell counting kit (CCK)-8
(Dojindo).
Statistical analyses
Statistical analyses of differences between groups
were performed using Student’s t-test. A P<0.05 was considered
to be significant.
Results
5-FU treatment upregulates the levels of
Fn14 mRNA and protein in gastric cancer cells
NF-κB is known to be activated by 5-FU, resulting in
increased resistance to this drug (20), and the Fn14 promoter is known to
contain NF-κB binding sites, allowing for positive feedback between
Fn14 and NF-κB to occur (21). The
results of our previous studies also revealed that Fn14
overexpression increased NF-κB transcriptional activity in gastric
cancer cells (6). Therefore, we
investigated whether Fn14 was involved in chemoresistance in
general and in 5-FU resistance in particular. We first examined
Fn14 expression after 5-FU treatment of gastric cancer cells.
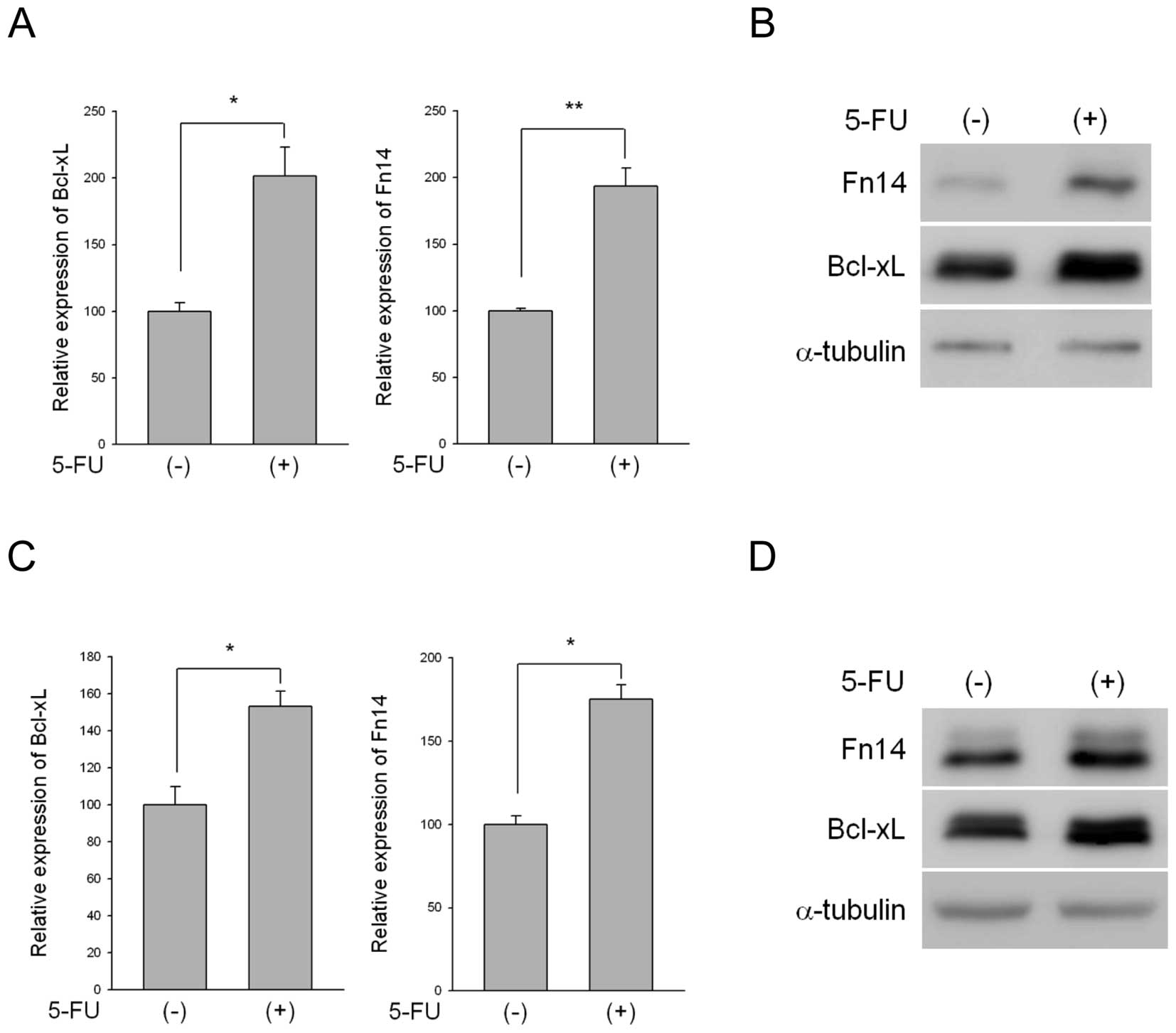
Treatment with 5-FU upregulated Fn14 expression at both the mRNA
and protein levels in SNU-216 and MKN-1 cells (Fig. 1). We also examined Bcl-xL
expression because Bcl-xL is regulated by Fn14 and NF-κB (6) and the expression of Bcl-xL is induced
by 5-FU. Bcl-xL is also known to be upregulated in 5-FU-resistant
colon cancer cells (22). As
predicted, Bcl-xL was upregulated following 5-FU treatment in both
cell lines (Fig. 1).
5-FU upregulates Fn14 gene expression via
NF-κB activation
We investigated the underlying mechanism by which
Fn14 gene expression was upregulated following 5-FU treatment. We
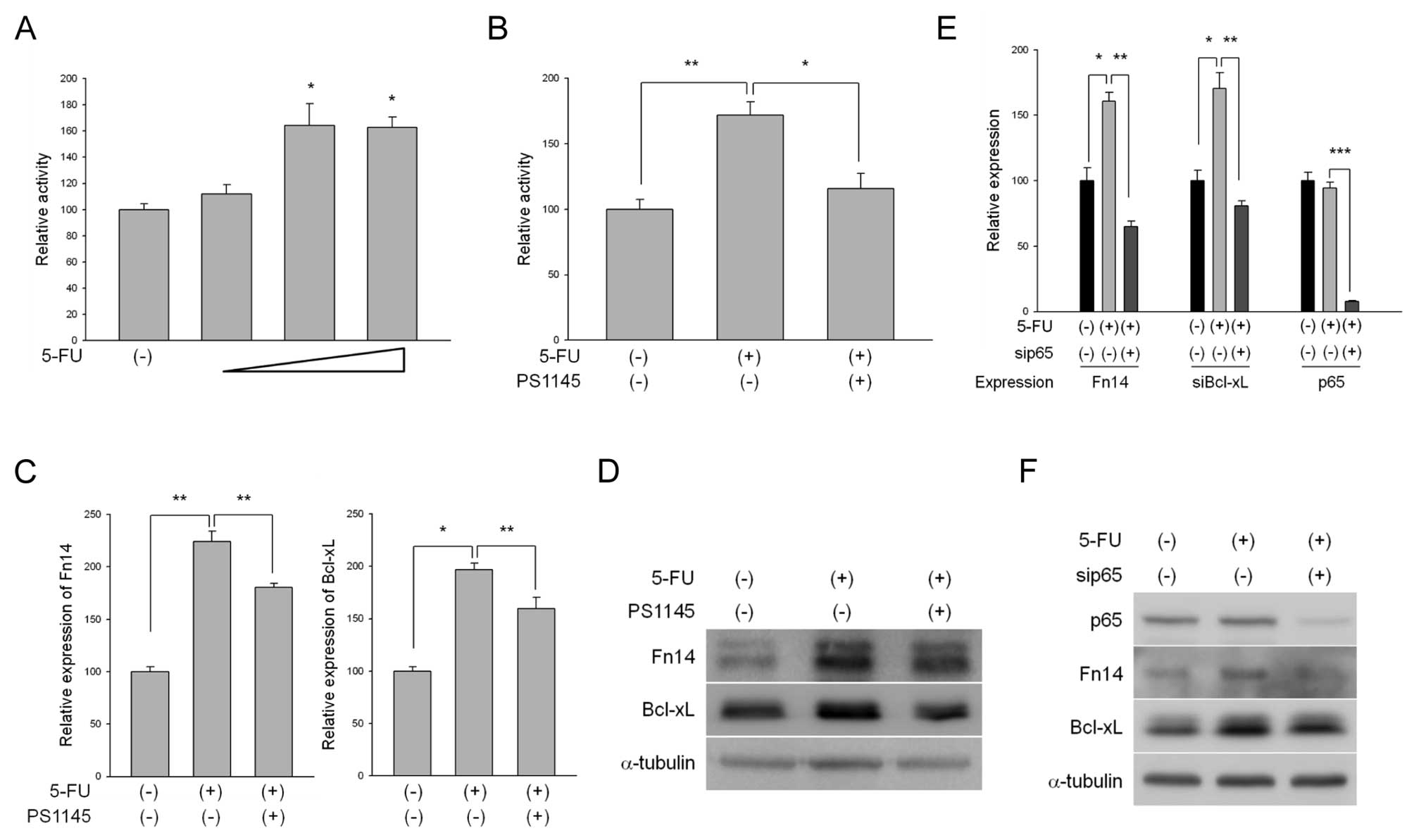
first performed NF-κB luciferase assays to examine 5-FU-mediated
alterations in NF-κB transcriptional activity. 5-FU treatment
increased the activity of the NF-κB reporter gene in a
dose-dependent manner (Fig. 2A).
We further confirmed that inhibition of NF-κB activity was
increased by 5-FU using the IκB kinase (IKK) inhibitor PS1145
(Fig. 2B). We then inspected the
expression levels of Fn14 after treatment of the cells with 5-FU
alone or in combination with PS1145. As a result of 5-FU treatment,
the increased levels of Fn14 expression were significantly reduced
by PS1145 treatment at both the mRNA and protein levels (Fig. 2C and D). Bcl-xl also displayed
similar results to those observed for Fn14 (Fig. 2C and D). In an effort to confirm
the effects of NF-κB on Fn14 expression following 5-FU treatment,
we used p65 siRNA to directly inhibit p65 expression. Similar to
the results obtained following PS1145 treatment, knockdown of p65
reduced the increased expression levels of Fn14 as well as Bcl-xL
following 5-FU treatment (Fig. 2E and
F), indicating that the 5-FU-mediated upregulation of Fn14
expression occurred as a result of NF-κB activation. We further
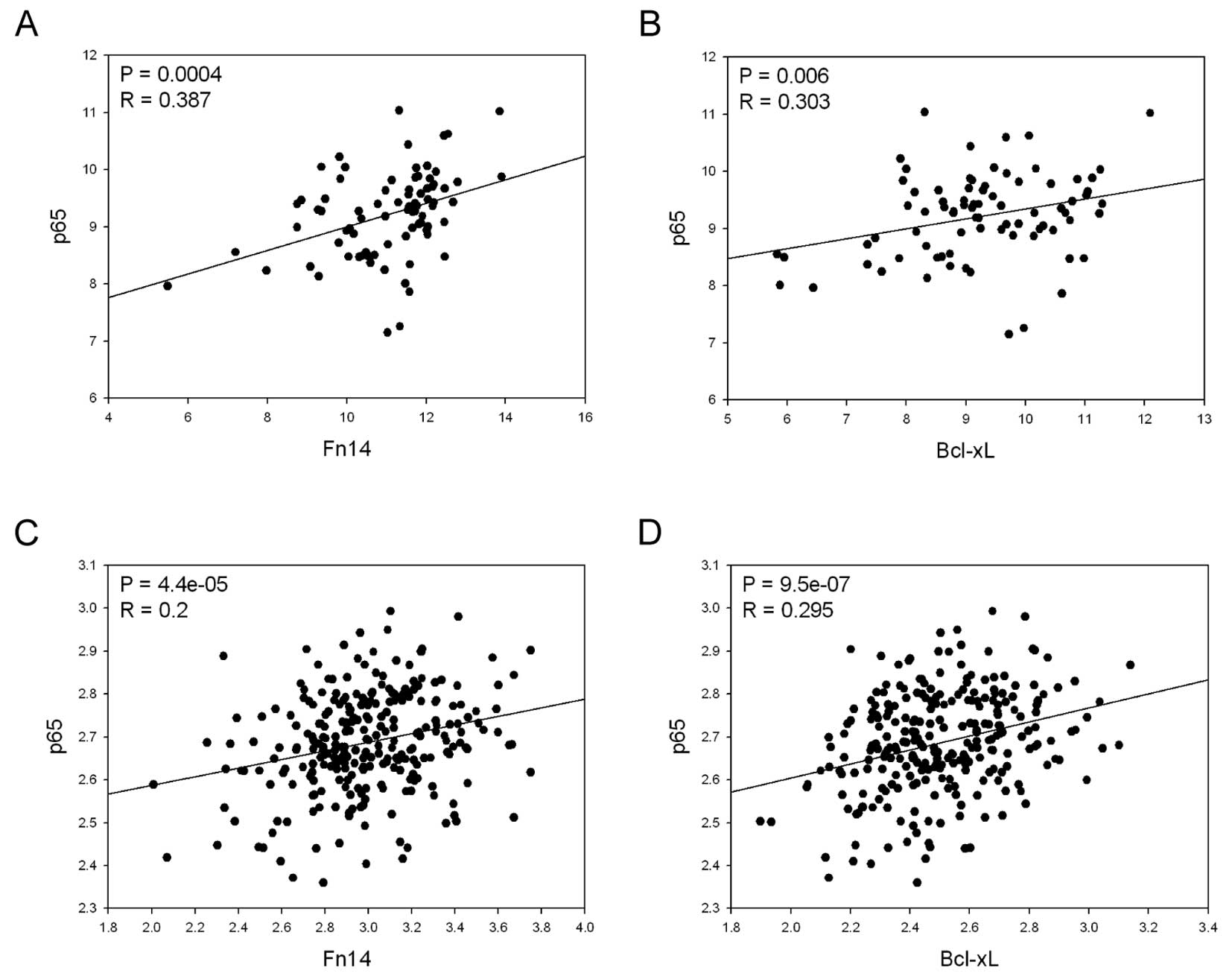
examined the relationship between the expression levels of p65 and
Fn14, as well as those of p65 and Bcl-xL, using the GENT database
in gastric cancer cell lines and tissues (4). We observed significant positive
correlations between the expression levels of those genes in
gastric cancer cell lines and tissues (Fig. 3), further supporting the notion
that strong connections between these genes exist in gastric
cancer.
Fn14 restrains 5-FU-induced
apoptosis
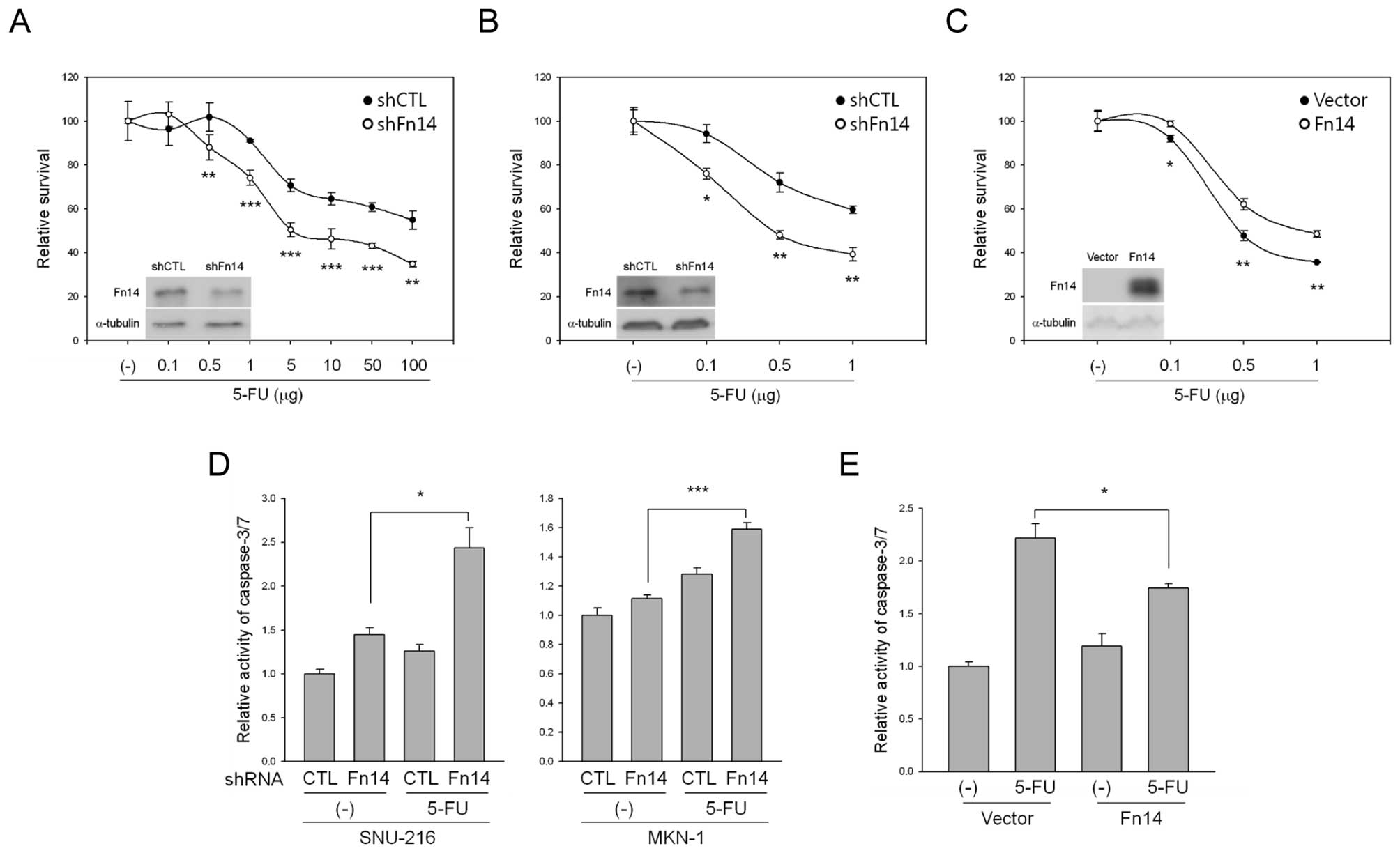
To investigate whether Fn14 expression had an effect
on 5-FU-induced cell death, we transduced Fn14 shRNA into SNU-216
and MKN-1 cells using a lentiviral system. Transduction with Fn14
shRNA resulted in reduced levels of Fn14 protein compared to
transduction with control shRNA (Fig.
4A and B). We then examined whether reduced levels of Fn14
expression promoted sensitivity to 5-FU. As shown in Fig. 3A and B, the reduction of Fn14
expression by shRNA accelerated 5-FU-mediated inhibition of cell
growth in both SNU-216 and MKN-1 cells. In contrast, overexpression
of Fn14 by stable Fn14 DNA transfection in ASG cells weakened
5-FU-mediated inhibition of cell growth (Fig. 4C). We next exposed the cells to
5-FU and analyzed caspase-3/7 activity using caspase-3/7 reagent.
As shown in Fig. 4D,
downregulation of Fn14 significantly increased caspase-3/7 activity
compared to control shRNA. In contrast, overexpression of Fn14
decreased caspase-3/7 activity compared to empty vector-transfected
cells (Fig. 4E), indicating that
Fn14 is an important determinant of apoptotic sensitivity following
5-FU treatment in these cells.
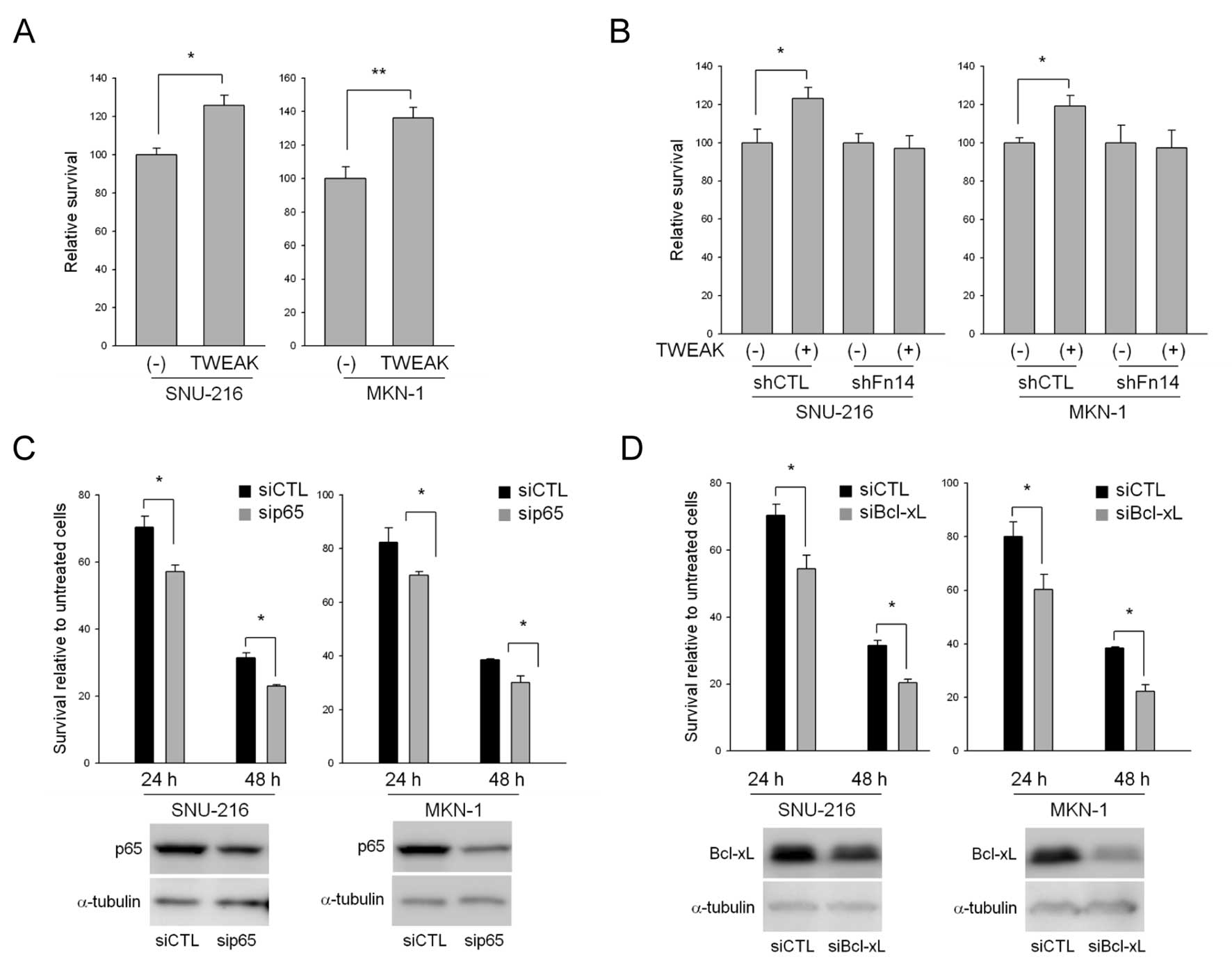
TWEAK stimulation also protects gastric
cancer cells against 5-FU-induced apoptosis
TWEAK transactivates the Bcl-xL promoter via NF-κB
activation and increases resistance to cytotoxic therapy-induced
apoptosis in glioma cells (23).
Therefore, we treated cells with 5-FU alone or in combination with
TWEAK and then examined the effects of TWEAK on 5-FU-induced cell
death. TWEAK treatment increased the rate of cell survival compared
to 5-FU treatment alone in both SNU-216 and MKN-1 cells (Fig. 5A). Although Fn14 is known to be a
TWEAK receptor, TWEAK can mediate the differentiation of RAW264.7
cells, which do not express Fn14, into osteoclasts, indicating that
other TWEAK receptors exist in cells (24). To investigate whether these effects
of TWEAK on 5-FU resistance were mediated through the Fn14
receptor, we treated cells expressing normal (shCTL) or reduced
levels of Fn14 (shFn14) with 5-FU alone or in combination with
TWEAK and then examined the rates of cell survival. TWEAK had no
effect on 5-FU-induced apoptosis in shFn14-transduced cells
compared to control cells (Fig.
5B).
The presumption of NF-κB involvement in
TWEAK/Fn14-mediated resistance to 5-FU was ascertained using p65
siRNA. We first transiently transfected p65 siRNA into SNU-216 and
MKN-1 cells and confirmed the reduced levels of p65 expression at
the protein level via western blotting in both cell lines (Fig. 5C). The survival rate of p65
siRNA-transfected cells was compared with control siRNA-transfected
cells after 5-FU treatment. The results indicated that knock-down
of p65 promoted sensitivity to 5-FU (Fig. 5C). We then examined the involvement
of Bcl-xL in 5-FU resistance because Bcl-xL is a target gene of
NF-κB as well as Fn14, as mentioned previously. Knock-down of
Bcl-xL using siRNA also accelerated 5-FU sensitivity compared to
control siRNA (Fig. 5D).
Bcl-xL and Fn14 are also upregulated by
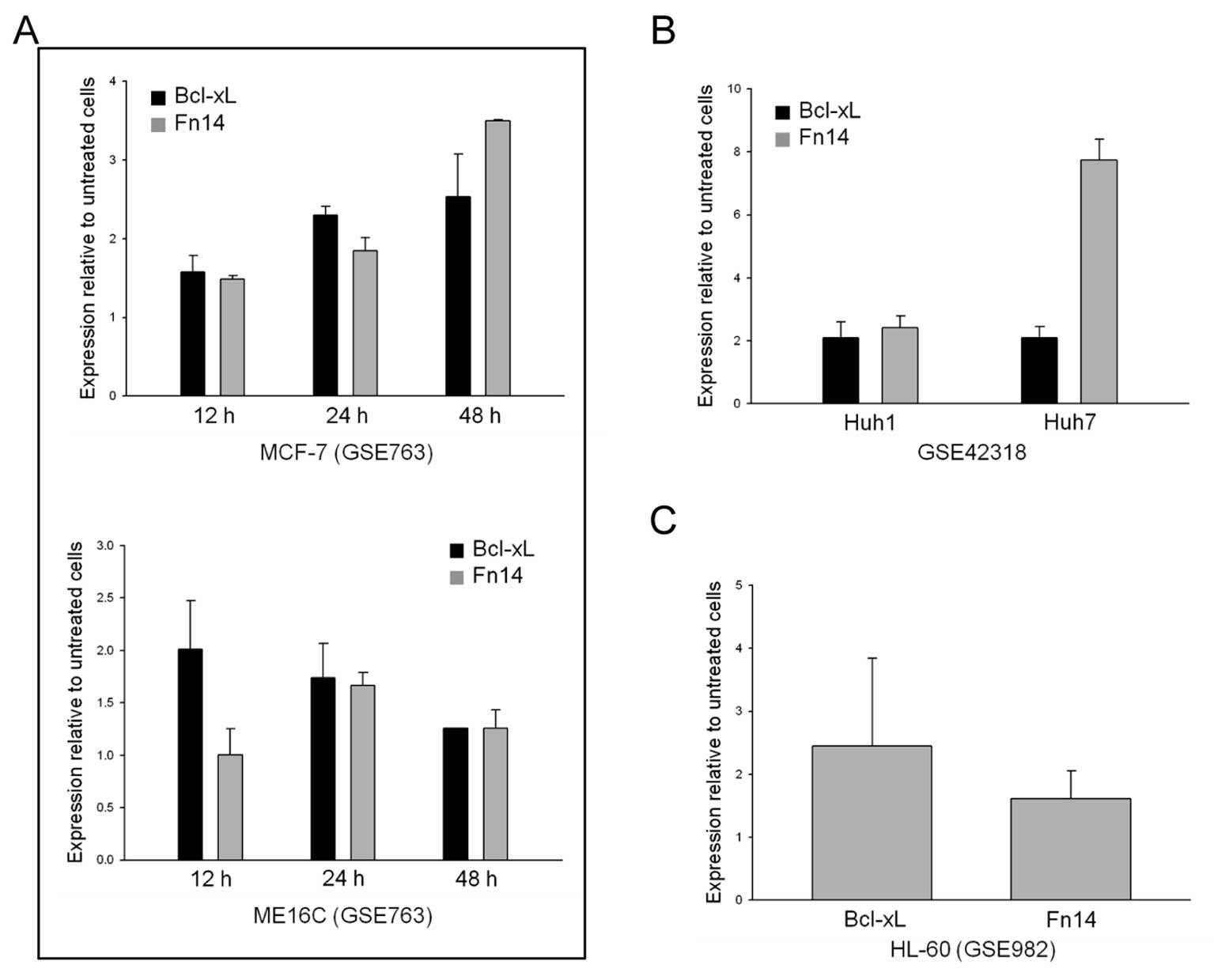
5-FU in various cancer cell lines
We wondered whether the upregulation of Fn14
expression by 5-FU was limited to gastric cancer cells. To
investigate whether the effects of 5-FU on Fn14 expression occurred
in other cancer cell lines, we downloaded gene expression
microarray data from the NCBI Gene Expression Omnibus (GEO) and
examined the expression of Fn14 in various cancer cell lines. We
discovered that various cell lines displayed Bcl-xL and Fn14
upregulation after 5-FU treatment (Fig. 6). Interestingly, Fn14 expression
was upregulated by 5-FU treatment in both solid (Fig. 6A and B) and liquid (Fig. 6C) tumor cells, indicating that the
relationship between Fn14 and Bcl-xL expression and 5-FU is
universal in cancer.
Discussion
NF-κB plays an important role in cell proliferation,
apoptosis, and resistance to antitumor agents (25,26).
In the field of chemotherapy, persistent activation of NF-κB
interrupts the effects of antitumor drugs, resulting in increased
resistance to these agents (27).
Therefore, the regulation of NF-κB activity may be a useful
therapeutic strategy for treating cancer because drug resistance is
the major reason for cancer therapy failure. We previously reported
that Fn14 is involved in the growth of gastric cancer cells through
NF-κB activation, leading to upregulation of the anti-apoptotic
protein Bcl-xL, which is an anti-apoptotic member of the Bcl-2
family that is involved in cancer chemoresistance (28), and that high levels of Fn14
expression are associated with poor clinical outcomes among gastric
cancer patients (6). These results
prompted us to examine the relationship between Fn14 and NF-κB in
chemo-resistance in gastric cancer. Although NF-κB is widely known
to be involved in chemoresistance in cancer, additional studies are
necessary to clarify the underlying mechanism by which NF-κB
contributes to chemoresistance in cancer in general and in gastric
cancer in particular.
5-FU-induced apoptosis is mediated predominantly by
the mitochondrial apoptotic pathway, which occurs as a result of
the activation of cytochrome c-mediated caspase-3 (17,18).
Evidence indicates that management of the mitochondrial apoptotic
pathway may, at least in part, contribute to solving the problem of
drug resistance. Bcl-2 and Bcl-xL block the release of cytochrome
c followed by caspase-3 activation through stabilization of
mitochondrial membrane integrity and subsequent regulation of the
mitochondrial apoptotic pathway (18,22).
Indeed, Bcl-xL is overexpressed in 5-FU-resistant cells and
knock-down of Bcl-xL expression promotes sensitivity to 5-FU in
colon cancer (29). In addition,
Bcl-xL shows anti-apoptotic effects on other antitumor drug
(30). Consistent with this
evidence, our present study showed that Bcl-xL was upregulated by
5-FU and that knock-down of Bcl-xL hastened the 5-FU-induced growth
inhibition of gastric cancer cells, indicating that Bcl-xL is one
of the main determinants in chemoresistance.
A previous study demonstrated that TWEAK prevents
glioma cells from TRAIL- or camptothecin-induced cell death through
activation of the NF-κB pathway and that TWEAK increased the
expression of anti-apoptotic proteins (23). A recent report also showed that
Fn14 expression correlates with p-EGFR levels and is upregulated in
EGFR-mutant cells that contain an EGFR TKI-resistant mutation
(31), indicating that the
TWEAK/Fn14 pathway is involved in resistance to anti-tumor drugs.
In our present study, we showed that knock-down or overexpression
of Fn14 led to the promotion of sensitivity or resistance to 5-FU,
respectively. The effects of Fn14 on antitumor drugs are likely
related to activation of the NF-κB pathway. NF-κB is widely known
to be regulated by TWEAK/ Fn14 signaling, indicating that
TWEAK/Fn14 signaling may contribute to constitutive NF-κB activity
in gastric cancer cells and, hence, may lead to resistance to
multiple antitumor drugs.
The HER2 proto-oncogene, which is a transmembrane
tyrosine kinase receptor and a member of the ErbB protein family,
is considered to be a putative Fn14 inducer. Expression of HER2 is
significantly associated with that of Fn14 in breast cancer
(32). Although it is not clear
whether HER2 directly regulates Fn14 expression, it is of interest
to note that HER2 can activate NF-κB (33) and, hence, may lead to upregulation
of Fn14 gene expression. HER-2-overexpressing breast cancer cells
are less responsive to antitumor drugs than cells that normally
express HER-2, and ectopic expression of HER-2 promotes resistance
to chemotherapies (34,35), suggesting that activation of the
NF-κB/Fn14 cascade by HER-2 may be involved in chemoresistance.
Fn14 activation is also elicited by proinflammatory cytokines,
including IFN-γ and IL-1β, and treatment with the HMG-CoA reductase
inhibitor atorvastatin, which is known to decrease NF-κB
activation, diminishes cytokine-induced Fn14 expression in vascular
smooth muscle cells (36,37). In addition, increased secretion of
IL-1β by nitric oxide (NO) leads to the induction of
chemoresistance in pancreatic carcinoma cells (38). Our study using public data revealed
that 5-FU treatment upregulated Fn14 expression in various cancer
cell lines independent of tumor type (solid or liquid tumor). Taken
together, these results strongly suggest that Fn14 may be involved
in chemoresistance in many cancers as an up- or down-stream
regulator.
In conclusion, we revealed that Fn14 and Bcl-xL were
upregulated by 5-FU-mediated NF-κB activation and promoted
resistance to 5-FU in gastric cancer cells. In addition, we
demonstrated that Fn14 is a new facilitative determinant in
NF-κB-mediated resistance to 5-FU in gastric cancer. Positive
feedback between TWEAK, Fn14, and NF-κB may give rise to more
synergistic restraints on chemotherapy. Although it is necessary
for us to elucidate whether Fn14 only limits the function of 5-FU
in gastric cancer, the results presented here support the notion
that Fn14 targeting may elevate the efficacy of current GC
therapies. These results may also apply to other cancers because we
discovered that Fn14 is upregulated by 5-FU in various cancer cell
lines.
Abbreviations:
|
5-FU
|
5-fluorouracil;
|
|
GC
|
gastric cancer;
|
|
CCK-8
|
cell counting kit-8
|
Acknowledgements
This study was supported by grants
from the Genomics Research Program (2012M3A9D1054670), the
Future-Based Technology Development Program (NRF2011-0015710), and
the KRIBB Research Initiative Program that are funded by the Korean
Ministry of Education, Science and Technology.
References
|
1.
|
Meighan-Mantha RL, Hsu DK, Guo Y, et al:
The mitogeninducible Fn14 gene encodes a type I transmembrane
protein that modulates fibroblast adhesion and migration. J Biol
Chem. 274:33166–33176. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Feng SL, Guo Y, Factor VM, et al: The Fn14
immediate-early response gene is induced during liver regeneration
and highly expressed in both human and murine hepatocellular
carcinomas. Am J Pathol. 156:1253–1261. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Wiley SR, Cassiano L, Lofton T, et al: A
novel TNF receptor family member binds TWEAK and is implicated in
angiogenesis. Immunity. 15:837–846. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Shin G, Kang TW, Yang S, Baek SJ, Jeong YS
and Kim SY: GENT: gene expression database of normal and tumor
tissues. Cancer Inform. 10:149–157. 2011.PubMed/NCBI
|
|
5.
|
Tran NL, McDonough WS, Donohue PJ, et al:
The human Fn14 receptor gene is up-regulated in migrating glioma
cells in vitro and overexpressed in advanced glial tumors. Am J
Pathol. 162:1313–1321. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kwon OH, Park SJ, Kang TW, et al: Elevated
fibroblast growth factor-inducible 14 expression promotes gastric
cancer growth via nuclear factor-kappaB and is associated with poor
patient outcome. Cancer Lett. 314:73–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Huang M, Narita S, Tsuchiya N, et al:
Overexpression of Fn14 promotes androgen-independent prostate
cancer progression through MMP-9 and correlates with poor treatment
outcome. Carcinogenesis. 32:1589–1596. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Zhou H, Ekmekcioglu S, Marks JW, et al:
The TWEAK receptor Fn14 is a therapeutic target in melanoma:
immunotoxins targeting Fn14 receptor for malignant melanoma
treatment. J Invest Dermatol. 133:1052–1062. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Silverman N and Maniatis T: NF-kappaB
signaling pathways in mammalian and insect innate immunity. Genes
Dev. 15:2321–2342. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Karin M and Lin A: NF-kappaB at the
crossroads of life and death. Nat Immunol. 3:221–227. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Wang CY, Cusack JC Jr, Liu R and Baldwin
AS Jr: Control of inducible chemoresistance: enhanced anti-tumor
therapy through increased apoptosis by inhibition of NF-kappaB. Nat
Med. 5:412–417. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Saitoh T, Nakayama M, Nakano H, Yagita H,
Yamamoto N and Yamaoka S: TWEAK induces NF-kappaB2 p100 processing
and long lasting NF-kappaB activation. J Biol Chem.
278:36005–36012. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Sevilla L, Zaldumbide A, Pognonec P and
Boulukos KE: Transcriptional regulation of the bcl-x gene encoding
the anti-apoptotic Bcl-xL protein by Ets, Rel/NFkappaB, STAT and
AP1 transcription factor families. Histol Histopathol. 16:595–601.
2001.PubMed/NCBI
|
|
15.
|
Johnstone RW, Ruefli AA and Lowe SW:
Apoptosis: a link between cancer genetics and chemotherapy. Cell.
108:153–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
17.
|
Sun Y, Tang XM, Half E, Kuo MT and
Sinicrope FA: Cyclooxygenase-2 overexpression reduces apoptotic
susceptibility by inhibiting the cytochrome c-dependent apoptotic
pathway in human colon cancer cells. Cancer Res. 62:6323–6328.
2002.PubMed/NCBI
|
|
18.
|
Martinou JC, Desagher S and Antonsson B:
Cytochrome c release from mitochondria: all or nothing. Nat Cell
Biol. 2:E41–E43. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Fukuyama R, Ng KP, Cicek M, et al: Role of
IKK and oscillatory NFkappaB kinetics in MMP-9 gene expression and
chemoresistance to 5-fluorouracil in RKO colorectal cancer cells.
Mol Carcinog. 46:402–413. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Tran NL, McDonough WS, Savitch BA, et al:
Increased fibroblast growth factor-inducible 14 expression levels
promote glioma cell invasion via Rac1 and nuclear factor-kappaB and
correlate with poor patient outcome. Cancer Res. 66:9535–9542.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Konishi T, Sasaki S, Watanabe T, Kitayama
J and Nagawa H: Overexpression of hRFI inhibits
5-fluorouracil-induced apoptosis in colorectal cancer cells via
activation of NF-kappaB and upregulation of BCL-2 and BCL-XL.
Oncogene. 25:3160–3169. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Tran NL, McDonough WS, Savitch BA, Sawyer
TF, Winkles JA and Berens ME: The tumor necrosis factor-like weak
inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14
(Fn14) signaling system regulates glioma cell survival via NFkappaB
pathway activation and BCL-XL/BCL-W expression. J Biol Chem.
280:3483–3492. 2005. View Article : Google Scholar
|
|
24.
|
Polek TC, Talpaz M, Darnay BG and
Spivak-Kroizman T: TWEAK mediates signal transduction and
differentiation of RAW264.7 cells in the absence of Fn14/TweakR.
Evidence for a second TWEAK receptor J Biol Chem. 278:32317–32323.
2003.PubMed/NCBI
|
|
25.
|
Garg A and Aggarwal BB: Nuclear
transcription factor-kappaB as a target for cancer drug
development. Leukemia. 16:1053–1068. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Escarcega RO, Fuentes-Alexandro S,
Garcia-Carrasco M, Gatica A and Zamora A: The transcription factor
nuclear factor-kappa B and cancer. Clin Oncol (R Coll Radiol).
19:154–161. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Dolcet X, Llobet D, Pallares J and
Matias-Guiu X: NF-κB in development and progression of human
cancer. Virchows Arch. 446:475–482. 2005.
|
|
28.
|
Reed JC: Bcl-2 family proteins: regulators
of apoptosis and chemoresistance in hematologic malignancies. Semin
Hematol. 34:9–19. 1997.PubMed/NCBI
|
|
29.
|
Zhu H, Guo W, Zhang L, et al: Bcl-XL small
interfering RNA suppresses the proliferation of
5-fluorouracil-resistant human colon cancer cells. Mol Cancer Ther.
4:451–456. 2005.PubMed/NCBI
|
|
30.
|
Lei X, Huang Z, Zhong M, Zhu B, Tang S and
Liao D: Bcl-XL small interfering RNA sensitizes cisplatin-resistant
human lung adenocarcinoma cells. Acta Biochim Biophys Sin.
39:344–350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Whitsett TG, Cheng E, Inge L, et al:
Elevated expression of Fn14 in non-small cell lung cancer
correlates with activated EGFR and promotes tumor cell migration
and invasion. Am J Pathol. 181:111–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Willis AL, Tran NL, Chatigny JM, et al:
The fibroblast growth factor-inducible 14 receptor is highly
expressed in HER2-positive breast tumors and regulates breast
cancer cell invasive capacity. Mol Cancer Res. 6:725–734. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Merkhofer EC, Cogswell P and Baldwin AS:
Her2 activates NF-kappaB and induces invasion through the canonical
pathway involving IKKalpha. Oncogene. 29:1238–1248. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Tsai CM, Yu D, Chang KT, et al: Enhanced
chemoresistance by elevation of p185neu levels in
HER-2/neu-transfected human lung cancer cells. J Natl Cancer Inst.
87:682–684. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Gusterson BA, Gelber RD, Goldhirsch A, et
al: Prognostic importance of c-erbB-2 expression in breast cancer.
International (Ludwig) Breast Cancer Study Group J Clin Oncol.
10:1049–1056. 1992.PubMed/NCBI
|
|
36.
|
Munoz-Garcia B, Martin-Ventura JL,
Martinez E, et al: Fn14 is upregulated in cytokine-stimulated
vascular smooth muscle cells and is expressed in human carotid
atherosclerotic plaques: modulation by atorvastatin. Stroke.
37:2044–2053. 2006. View Article : Google Scholar
|
|
37.
|
Ortego M, Bustos C, Hernandez-Presa MA, et
al: Atorvastatin reduces NF-kappaB activation and chemokine
expression in vascular smooth muscle cells and mononuclear cells.
Atherosclerosis. 147:253–261. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Muerkoster S, Wegehenkel K, Arlt A, et al:
Tumor stroma interactions induce chemoresistance in pancreatic
ductal carcinoma cells involving increased secretion and paracrine
effects of nitric oxide and interleukin-1beta. Cancer Res.
64:1331–1337. 2004. View Article : Google Scholar
|