Introduction
Lung cancer is the leading cause of cancer deaths in
men and women worldwide (1).
Non-small cell lung cancer (NSCLC) accounts for approximately 85%
of all lung cancers. More than 50% of all patients diagnosed with
lung cancer have advanced disease (2). Despite improvement in cancer
treatment, the prognosis for NSCLC patients is still poor, with
5-year survival rate of approximately 10% (3). Since metastasis remains the cause of
90% of deaths from solid tumors (4), elucidating the mechanisms underlying
lung cancer metastasis appeared to be very important.
Transforming growth factor β (TGF-β) is a ubiquitous
and essential regulator of cellular and physiologic processes, such
as cellular proliferation, migration, invasion and
immunosurveillance (5,6). TGF-β is a double-edged sword in
cancer development and progression. TGF-β signaling pathway plays a
suppressive role in cell growth of early-stage tumors. On the other
hand, it is recognized that metastasis of most tumor cells requires
TGF-β activity, and the active TGF-β signaling became a promoter in
late-stage tumors (7,8). TGF-β-induced phosphorylation of Smad3
is a vital step in the active TGF-β signaling (9) and Smad3 phosphorylation is required
for lung epithelial-to-mesenchymal transition (10), which is a transition process that
provides cancer cells with higher invasive and metastatic abilities
(11).
Decorin (DCN) is a member of the extracellular
matrix small leucine-rich proteoglycan family (12) and DCN regulates multiple processes
in cancer cells (13,14). Accumulative evidence shows that DCN
plays an inhibitory role in tumor growth and metastasis (15–19).
This role can be played through DCN specifically binding to TGF-β
and partially blocking the TGF-β signaling pathway, leading to
growth suppression of distant tumors including lung cancer
(20–22). Although there was evidence
indicating that reduced expression of DCN was associated with
lymphatic metastasis in lung cancer patients (23), the mechanisms by which the reduced
DCN expression causes lung cancer metastasis appear to be
elusive.
Our recent work supported the idea that DNA
methylation could be epigenetically responsible for inactivation of
tumor suppressor gene in NSCLC (24–26).
Moreover, Doi et al and Irizarry et al suggested that
DNA methylation alterations in individual CpG site may have a vital
role in cancer progression (27,28).
Therefore, based on the facts that DCN proximal promoter and
5′-UTR regions harbor several CpG sites and our present study
showing that high-metastatic NSCLC cells presented lower expression
of DCN mRNA compared with low-metastatic NSCLC cells, we
hypothesized that individual CpG methylation epigenetically
functions to affect DCN mRNA expression and TGF-β/Smad
signaling in metastatic NSCLC cells.
Materials and methods
Cell culture and drug treatments
Low- (95C) and high-metastatic (95D) human NSCLC
cell lines were purchased from Shanghai Institute of Biochemistry
and Cell Biology and cultured in RPMI-1640 (Gibco-Invitrogen) with
5% fetal bovine serum (FBS, Gibco-Invitrogen) and incubated at 37°C
in 5% CO2. 95C and 95D cell lines share a similar
genetic background and have different tumor metastatic potential
(29,30). Cell treatment with DNA
demethylating agent, 5-aza-2′-deoxycitidine (5-Aza)
(Sigma-Aldrich), was performed according to the protocol described
previously by us (25) with some
modifications. Culture medium with 5% FBS was always used in the
demethylation process. The concentration of 5-Aza was 10 μM
and the cells were harvested for analysis of DNA methylation on day
5. Drug treatment with 5-Aza is a currently accepted system to
study effects of hypomethylation on gene expression (25).
NSCLC tissue samples
Lung carcinoma tissues were obtained after informed
consent from patients with stage III or IV NSCLC in the First
Affiliated Hospital of Soochow University. Stage III patients (n=5)
had local lymph node metastasis
(T1–4N1–2M0), while stage IV
patients (n=5) had distant organ metastasis
(T1–4N1–2M1). Pathological stages
were determined according to the Revised TNM Staging System for
Lung Cancer. NSCLC patients had received neither chemotherapy nor
radiotherapy before tissue sampling. The tissue samples were
snap-frozen and stored in liquid nitrogen before study. This study
was approved by the Academic Advisory Board of Soochow
University.
Transwell invasion and migration
assays
For invasion assay, BioCoat Matrigel invasion
chambers (BD Biosciences) with 8-μm pore polycarbonate
membranes were pre-treated with serum-free RPMI-1640
(Gibco-Invitrogen) medium at 37°C for 2 h. After removing the
medium, we added 750-μl medium with 10% FBS as
chemoattractant to each lower chamber and then added
5×104 cells in 250-μl serum-free medium to each
upper chamber. After 24-well plates were incubated at 37°C for 24
h, the inserts were removed and non-invasive cells on the upper
surface were removed by a cotton swab. The invasive cells on the
lower surface of the membrane were then fixed in 100% methanol for
15 min, air-dried, and stained with 0.1% crystal violet for 30 min.
The cells were recorded with a digital camera. For migration assay,
a similar procedure to the invasion assay was conducted, but
omitting addition of Matrigel.
Quantitative real-time polymerase chain
reaction (qRT-PCR)
Total RNA was isolated from the cells with or
without treatment of 5-Aza using RNAiso Plus kit (Takara) according
to the manufacturer’s protocol. Synthesis of cDNA with reverse
transcriptase was performed by M-MLV First Strand kit (Invitrogen).
For analysis of mRNA expression of DCN, qRT-PCR analysis was
carried out using Platinum® SYBR® Green qPCR
SuperMix-UDG kits (Invitrogen) according to the kit manual.
Real-time PCR was performed on a Bio-Rad CFX384™
Real-Time PCR system. Primer sequences for DCN mRNA
detection are as follows: 5′-CTGGTGGGCTGG CAGAGCATAA-3′ (forward),
5′-TGTTGTGTCCAGGTGG GCAGAAGT-3′ (reverse). The amplification
conditions are described below: 95°C for 10 min, followed by 40
cycles of 95°C for 30 sec and 66°C for 30 sec and 72°C for 1 min.
As an internal control, β-actin mRNA was measured in the
same reaction conditions. DCN mRNA expression levels were
calculated following normalization to β-actin mRNA
levels.
In silico prediction of functional CpG
sites in DCN proximal promoter and 5′-UTR regions
A 1,230-bp sequence, including DCN proximal
promoter (−200 to +1) and 5′-UTR (+1 to +1,030) regions (RefSeq:
NC_000012.11 and NM_133503.2), was available for CpG sites
selection. Methyl Primer Express® Software v1.0 (Applied
Biosystems) and TFSEARCH (http://mbs.cbrc.jp/research/db/TFSEARCH.html) were
used for the prediction of the putative functional CpG sites.
Clonal bisulfite sequencing
Genomic DNA was extracted from NSCLC cells with or
without treatment of 5-Aza according to standard proteinase K
digestion and phenol-chloroform extraction. The genomic DNA was
modified with bisulfite using EpiTect Bisulfite kit (Qiagen) and
subjected to clonal bisulfite sequencing. Briefly, after the
bisulfite-treated DNA was purified by Qiagen Quickspin columns,
DCN 5′-UTR region containing +58CpG was amplified from
bisulfite-treated DNA by PCR and cloned into pMD19-T vector
(Takara). Twenty clones obtained from each of the PCR products were
randomly selected and sequenced for methylation level of +58CpG
site. The primers designed for the bisulfite-PCR were as follows:
5′-TGTGTGTAAAATATTGTGTAAGGTT-3′ (forward) and
5′-AATACTACTTTCTCCCTCTTCTCTA-3′ (reverse).
Western blot analysis
Cells with or without treatment of 5-Aza were lysed
in RIPA buffer with protease inhibitor (Sigma-Aldrich) and
phosphatase inhibitor cocktail (Sigma-Aldrich) and centrifuged;
cell lysates were resuspended and denatured in SDS buffer, and then
fractionated by SDS-PAGE electrophoresis. Proteins were transferred
to nitrocellulose membranes (Millipore) and blocked with BSA/TBST
buffer. Membranes were incubated with primary antibodies overnight
before incubation with the corresponding HRP-conjugated secondary
antibodies. Detection was performed using ECL kit (Pierce,
Rockford, IL, USA). All experiments were performed in triplicate.
Densitometry values were determined using ImageJ 1.46r software
(NIH). Densitometry values for p-Smad3 level were normalized to
total Smad3, while E-cadherin and AhR was normalized to β-actin.
Antibodies employed in the analysis were: anti-p-Smad3 and
anti-Smad3 (CST), anti-E-cadherin (Abcam), anti-AhR and
anti-β-actin (Santa Cruz), and anti-mouse and anti-rabbit secondary
antibodies (Santa Cruz).
Chromatin immunoprecipitation (ChIP)
assay
ChIP assay was performed using EZ-ChIP™
Chromatin Immunoprecipitation kit (Upstate). Cells grown on 15-cm
dishes were fixed with 1% formaldehyde for 10 min at room
temperature, and the cross-linked cells were washed twice with
phosphate-buffered saline (PBS), and were scrapped in PBS buffer
with protease inhibitors. The cell nuclei were collected by
centrifugation of 700 × g at 4°C and resuspended in SDS lysis
buffer with protease inhibitors, sonicated in ice and the
cross-linked DNA was sheared to 200–1,000 bp, and the insoluble
material was removed by centrifugation at 13,000 x g for 10 min.
Then 100 μl of the supernatant was diluted in 900 μl
buffer with protease inhibitors, and incubated with Protein G
Agarose for 1 h at 4°C. After brief centrifugation, 10 μl
(1%) of the supernatant was saved as the Input at 4°C, and the
remaining was used for the immunoprecipitation and was incubated
with 5 μg anti-AhR antibody (Santa Cruz) overnight at 4°C.
Rabbit IgG antibody was used as a negative control.
Antibody/chromatin complexes were incubated with Protein G Agarose
for 1 h at 4°C, and then Protein G Agarose-antibody/chromatin
complex was collected by brief centrifugation. After washing
followed by centrifugation, we eluted the complex and the Input to
obtain protein-DNA complexes. De-cross-linking was performed to
free DNA by incubating in NaCl, RNase A, EDTA and proteinase K, and
DNA purification was conducted using spin columns. The ChIP DNA
fragments were subjected to PCR, which amplified DCN 5′-UTR
sequence containing +58CpG (positions +2 to +115). Specific ChIP
primers used for PCR amplification were as follows:
5′-GAATAATAAGACACGCCCTGAAG-3′ (forward) and
5′-CACGTTGCTCTTACCTCTTTTAA-3′ (reverse).
Electrophoresis mobility gel shift assay
(EMSA)
Nuclear extracts were prepared from NSCLC cells
using the ProteoJET™ Cytoplasmic and Nuclear Protein
Extraction kit (Fermentas). The cell nuclear extracts and synthetic
double-stranded and 5′ biotin-labeled oligonucleotides
corresponding to the DCN +58 C (unmethylated) and +58 5mC
(methylated) sequences (5′-CAAGCACGCAAAACAAATTG-3′ and
5′-CAAGCA5mCGCAAAACAAATTG-3′,
respectively. The binding sequences of transcriptional factors
AhR/Ar (underlined) were incubated at 25°C for 20 min using the
LightShift Chemiluminescent EMSA kit (Pierce). Each reaction was
performed in a total volume of 20 μl containing 20 fmol
labeled probe, 2 μg nuclear extract and 50 ng Poly (dI·dC).
The reaction mixture was separated on 6% non-denaturing PAGE gel,
and the products were transferred to positive-charged nylon
membrane and then detected by stabilized streptavidin-horseradish
peroxidase conjugate (Pierce). For each competition assay, 200-fold
unlabeled +58 C and +58 5mC probes were added to the reaction
mixture before the addition of biotin-labeled probes.
Construction of luciferase reporter
plasmids containing methylated +58CpG transfection and luciferase
assays
Two fragments of interest containing the
unmethylated and methylated +58CpG (5′-CAAGCACGCAAAACAAATTG-3′ and
5′-CAAGCA5mCGCAAAACAAATTG-3′,
respectively) were directly synthesized and then cloned into the
pGL3-Basic luciferase vector (Promega). Subsequently, the
constructs carrying the unmethylated or methylated +58CpG were
transiently transfected into cells using Lipofectamine 2000
(Invitrogen) and Renilla pRL-TK construct (Promega) was
co-transfected as a normalizing control. Twenty-four hours later,
luciferase activity was measured by the Dual-Luciferase Reporter
Assay kit (Promega) on a TD20/20 Luminometer (Turner Designs). Each
experiment was performed in triplicate. Results are expressed as
relative luciferase activities, which are obtained following
normalization to Renilla luciferase activities.
Enzyme-linked immunosorbent assay
(ELISA)
ELISA was performed to determine expression levels
of DCN protein from cell culture supernatants using Human Decorin
ELISA kit (RayBio). Cell culture supernatants were extracted and
pipetted to each well according to the manufacturer’s instructions.
After adding the ELISA Stop Solution to each well, we read
immediately the absorbance at 450 nm on EL×800™
Microplate Reader (BioTek). Each determination was performed in
triplicate.
Statistical analysis
Results are represented as the means ± standard
deviations (SD) for mRNA or protein expression. Comparisons between
two groups were performed with Student’s t-test for continuous
variables or with χ2 test for categorical variables.
Statistical differences were considered to be significant at
P<0.05. All statistical analyses were performed using GraphPad
Prism 5.02 software.
Results
DNA methylation is involved in DCN mRNA
expression
First, we conducted transwell assay to confirm that
95D cells have higher migration and invasion abilities than 95C
cells (Fig. 1A), the phenotype
differences between the two cell lines are consistent with those
reported previously (29,30). Thus, we examined the expression of
DCN mRNA in low- (95C) and high-metastatic (95D) human NSCLC
cell lines using qRT-PCR analysis, and found that DCN mRNA
expression in 95D cells was lower by far than that in 95C cells
(P<0.01) (Fig. 1B). After
treatment of demethylating agent 5-Aza on 95D cells expressing low
DCN mRNA, DCN mRNA expression was increased by
∼10-fold (P<0.001) (Fig. 1C),
suggesting the involvement of DNA methylation in DCN mRNA
expression.
In silico prediction of functional CpG
sites
Using Methyl Primer Express® software and
TFSEARCH, we identified that one putative functional CpG site at
position +58 in DCN 5′-UTR, close to the transcription start
site, was predicted to locate within a transcription factor
AhR/Ar-binding sequence (5′-AAGCACGCAAAACAAA-3′)
(Fig. 2A). Here we designate this
putative functional CpG site as +58CpG. This prediction provides
support for the notion that methylation of +58CpG may functionally
affect DCN mRNA expression.
Methylation level of +58CpG is higher in
95D than 95C cells, and was associated with DCN mRNA expression in
metastatic NSCLC
To test whether methylation level of +58CpG affects
expression of DCN mRNA, we performed clonal bisulfite
sequencing to compare the differences between 95C and 95D cells. As
a result, we observed that the methylation level of +58CpG was
significantly higher in 95D cells expressing low DCN mRNA
than 95C cells expressing high DCN mRNA (95±5 vs. 65±10%,
P<0.01) (Fig. 2B–D).
Comparably, the results obtained in tumor tissues suggested that
methylation level of +58CpG correlates inversely with DCN
mRNA expression in metastatic NSCLC tissues (Fig. 3).
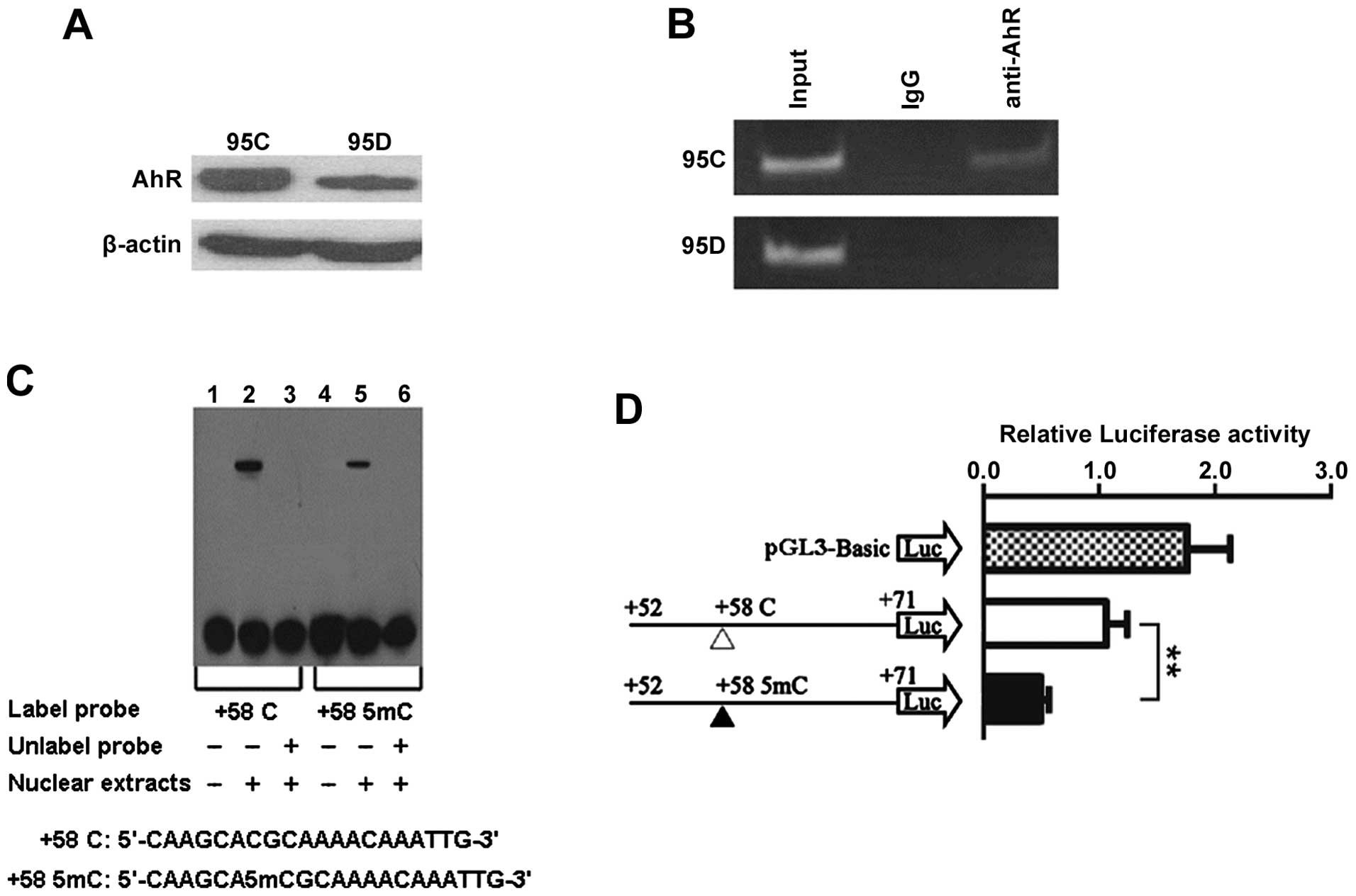
AhR is recruited to DCN 5′-UTR region
containing +58CpG
Aryl hydrocarbon receptor (AhR) was reported to be a
transcriptional activator (31,32)
and +58CpG was in silico predicted to be harbored in
AhR/Ar-binding site at DCN 5′-UTR region (Fig. 2A), leading us to consider the
possibility that AhR can bind to DCN 5′-UTR region
containing +58CpG. First, we performed western blot analysis and
found that AhR was positively expressed in both cell types, and AhR
expression was lower in 95D than 95C cells (Fig. 4A). Subsequently, ChIP analysis
using anti-AhR antibody showed that DCN 5′-UTR sequence
containing +58CpG (positions +2 to +115) was bound by AhR in 95C
cells, but not in 95D cells (Fig.
4B), indicating that there is a correlation between +58CpG and
AhR, and also suggesting that high methylation level of +58CpG
(Fig. 2D) may be one of the
mechanisms accounting for reduced recruitment of AhR to DCN
5′-UTR region in 95D cells.
Methylation of +58CpG decreases binding
ability of transcriptional factors to DCN 5′-UTR region
Next, we performed EMSA to determine whether
methylated +58CpG can affect the ability of binding to
transcriptional factors including AhR/Ar from the nuclear extracts.
As illustrated in Fig. 4C,
although both the probe containing unmethylated +58CpG and the
probe containing methylated +58CpG may combine with specific
DNA-binding proteins from 95D cells, the unmethylated probe has
stronger binding ability than the methylated probe. This effect was
consistently observed in the EMSA experiments, suggesting that the
methylated +58CpG can decrease the ability of binding of
transcriptional factors, including AhR/Ar, to DCN 5′-UTR
region.
Methylation of +58CpG significantly
inhibits luciferase reporter gene transcriptional activity
To further determine whether methylation of +58CpG
alters DCN transcriptional activity by de-recruiting the
transcriptional activator AhR, we therefore constructed luciferase
reporter vectors containing the corresponding unmethylated and
methylated fragments (AhR/Ar-binding sequence) and transiently
transfected them into 95D cells. As illustrated in Fig. 4D, the methylated +58CpG caused a
reduction of ∼50% in transcriptional activity compared with the
unmethylated +58CpG (P<0.01). Taken together with EMSA assay,
the results suggest that methylation of +58CpG may repress
DCN mRNA expression by diminishing the recruitment of AhR to
DCN 5′-UTR region.
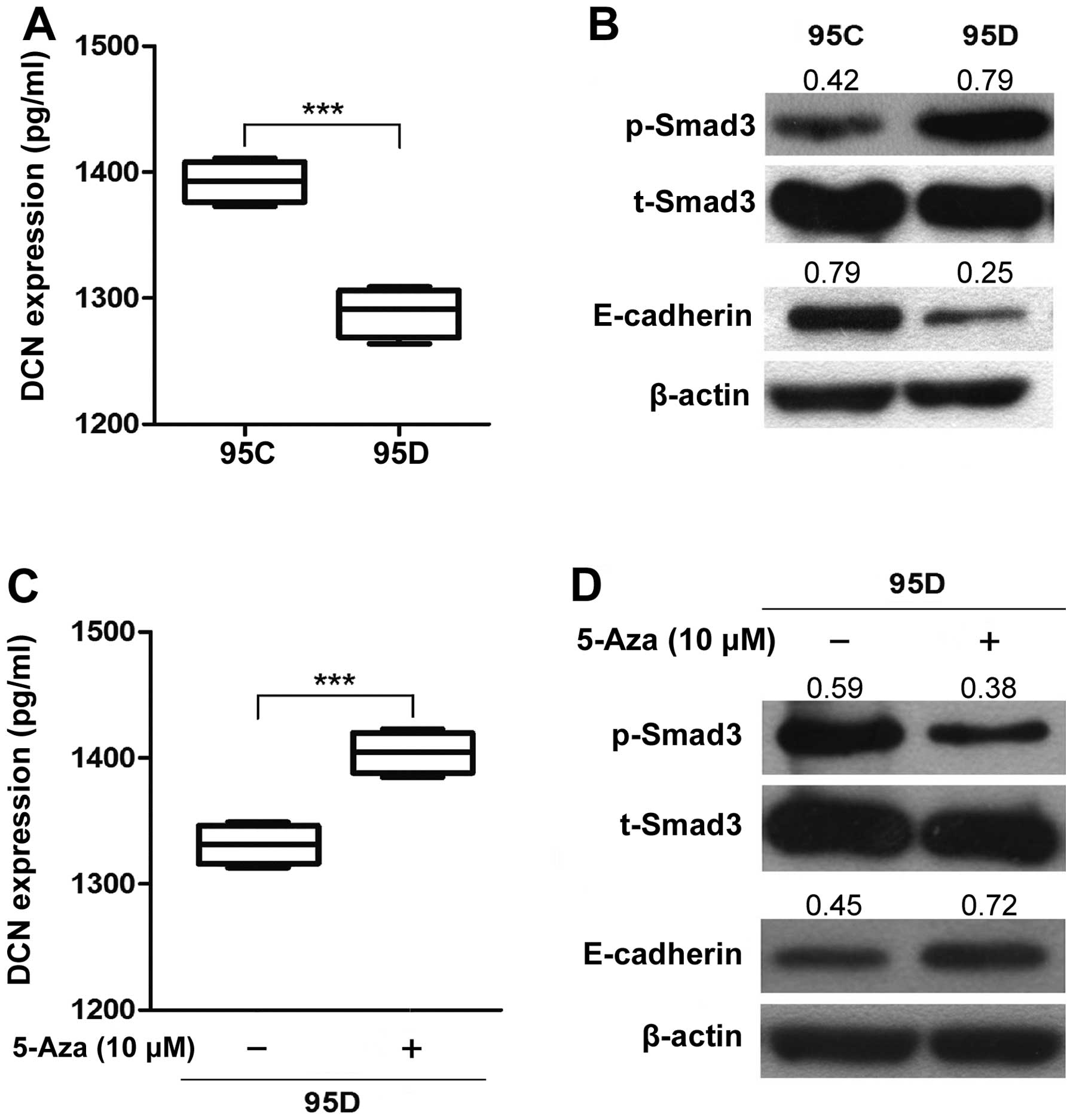
DCN inhibits phosphorylation of Smad3 and
enhances E-cadherin expression in metastatic NSCLC cells
DCN was reported to prevent TGF-β-induced inhibition
of lung morphogenesis (33),
leading us to explore the effect of DCN on TGF-β/Smad signaling.
Thus, we first used ELISA assay to determine the expression of DCN
protein in cell culture supernatants, and found that DCN protein
expression in 95D was significantly lower than that in 95C
(P<0.001) (Fig. 5A). Then we
used 95C and 95D cell lines to detected the level of phosphorylated
Smad3 (p-Smad3), which is an indispensable downstream effector in
canonical TGF-β/Smad signaling (9). At baseline, the level of p-Smad3 was
higher in 95D expressing low DCN (0.79±0.07) than that in 95C
expressing high DCN (0.41±0.05) (P=0.028, Fig. 5B). Correspondingly, 5-Aza treatment
significantly restored expression of DCN protein in 95D cell
culture supernatants (P<0.01) (Fig.
5C) and decreased the level of p-Smad3 in 95D cells (0.37±0.10
vs. 0.55±0.10; P=0.010) (Fig. 5D),
suggesting that demethylation-induced restoration of DCN (Fig. 5C) antagonizes bioactive TGF-β
(33), leading to a reduction in
the p-Smad3 level and an inhibition of TGF-β/Smad signaling.
DCN-mediated inhibition of colorectal cancer
migration is associated with E-cadherin (13,15–19,34).
Here we examined expression of E-cadherin in 95D expressing low DCN
and 95C expressing high DCN, and found that E-cadherin expression
was significantly lower in 95D cells (0.25±0.05) than 95C cells
(0.73±0.07) (P=0.016) (Fig. 5B).
Correspondingly, 5-Aza treatment significantly increased expression
of E-cadherin in 95D cells (0.70±0.07 vs. 0.48±0.10; P=0.013)
(Fig. 5D). The results suggest
that DCN may enhance expression of E-cadherin in metastatic NSCLC
cells.
Discussion
In this study, we have identified the methylated
+58CpG in DCN 5′-UTR associated with reduced expression of
DCN mRNA in high-metastatic NSCLC cells. Our findings reveal
that +58CpG methylation may be one of the mechanisms accounting for
reduced recruitment of transcriptional activator AhR to DCN
5′-UTR, and suggest that this mechanism promotes TGF-β/Smad
signaling by enhancing phosphorylation of Smad3, thereby
downregulates E-cadherin in high-metastatic NSCLC cells.
Compelling evidence has shown that DCN contributes
to inhibition of tumor growth and metastasis (13,15–19,34).
Goldoni et al reported that DCN can display an
anti-metastatic role in breast cancer (16). Bi et al demonstrated that
DCN may act as a tumor suppressor gene (15) and mediate inhibition of colorectal
cancer growth and migration (13).
Among soft tissue tumor patients with recurrent or metastatic
lesions, lower levels of DCN were expressed in secondary lesions
than those in primary lesions (17). Decreased expression of DCN was
found to be significantly associated with lymphatic metastasis of
lung cancer patients (23). In the
present study, we focused on low- (95C) and high-metastatic (95D)
NSCLC cells, which were more genetically similar to each other and
could be an ideal model for cancer metastasis (29,30),
and found that DCN mRNA was significantly lower in 95D cells
compared with that in 95C cells, supporting the idea that
DCN was involved in metastasis of malignant tumors,
including lung cancer. Additionally, demethylating drug 5-Aza
restored DCN mRNA expression in 95D cells, suggesting the
involvement of DNA methylation in DCN expression. Treatment
with 5-Aza is a currently accepted system to study effects of
demethylation on gene expression, albeit this compound is
recognized to be capable of inducing other cellular effects
(25).
To figure out the epigenetic mechanisms underlying
reduced DCN mRNA expression in high-metastatic NSCLC cells,
we examined DNA methylation level of +58CpG that was predicted to
locate within AhR/Ar-binding sequence of 5′-UTR region. Based on
the evidence that AhR/Ar is reported to be a transcriptional
activator (31,32), our findings support the idea that
+58CpG plays a role in reducing DCN mRNA expression by a
methylation mechanism in high-metastatic 95D cells. More recently,
DNA methylation was recognized to suppress epigenetically gene
expression via preventing the binding of TF to methylated CpG
dinucleotides within TF-binding sites (35–37).
This is substantiated by our ChIP, EMSA and luciferase experiments,
which showed that methylated +58CpG can decrease its ability of
binding to AhR and lead to a reduction in transcriptional activity.
The results shed light on the important role of +58CpG methylation
in NSCLC metastasis, and supported the notion that individual CpG
methylation plays a vital role in cancer progression (27,28).
Given that DCN can cause growth inhibition of tumor
cells including NSCLC by blocking TGF-β signaling (20–22),
we are inspired to evaluate the effect of DCN expression on
TGF-β/Smad signaling during NSCLC progression. To reach this goal,
here we examine the level of p-Smad3, which is essential for
activating canonical TGF-β/Smad signaling (9), and the expression of E-cadherin,
which was reported to correlate with DCN-mediated inhibition of
colorectal cancer migration (13),
in 95C expressing high DCN and 95D expressing low DCN. Our results
suggest that DCN inhibited phosphorylation of Smad3 and
enhanced E-cadherin expression in metastatic NSCLC cells.
Interestingly, demethylating drug 5-Aza treatment significantly
decreased the level of p-Smad3 in 95D cells, suggesting that the
blocking of TGF-β/Smad signaling may be caused by
demethylation-induced restoration of DCN. This is consistent with
previous data that DCN can antagonize bioactive TGF-β during lung
growth and differentiation (33).
Moreover, demethylation-induced restoration of DCN via 5-Aza
treatment led to an increased E-cadherin in 95D cells. The results
support the notion that TGF-β/Smad signaling is a promoter in
late-stage tumors, including NSCLC (7,8,11,38),
and DCN-mediated inhibition of colorectal cancer migration is
associated with E-cadherin (12–14).
In conclusion, we identified the methylated +58CpG
in DCN 5′-UTR associated with reduced expression of
DCN mRNA, and revealed that +58CpG methylation may be one of
mechanisms accounting for reduced recruitment of transcriptional
activator AhR to DCN 5′-UTR, and suggest that this mechanism
promotes TGF-β/Smad signaling by enhancing phosphorylation of
Smad3, thereby downregulates E-cadherin in high-metastatic NSCLC
cells.
Acknowledgements
We are grateful for participation and
cooperation from the patients with NSCLC. This study was supported
in part by the grants from National Natural Science Foundation of
China (81171894 and 81372277 to H.-T. Zhang), Program for New
Century Excellent Talents in University (NCET-09-0165 to H.-T.
Zhang), National Natural Science Foundation of China (81201575 to
Z. Liu), Jiangsu Province’s Key Provincial Talents Program
(RC2011106 to J. Zhao), ‘333’ Project of Jiangsu Province
Government (to H.-T. Zhang), Soochow Scholar Project of Soochow
University (to H.-T. Zhang), and Suzhou Key Laboratory for
Molecular Cancer Genetics (SZS201209 to H.-T. Zhang).
References
|
1.
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics, 2008. CA Cancer J Clin. 58:71–96. 2008. View Article : Google Scholar
|
|
2.
|
Ramsey SD, Clarke L, Kamath TV and Lubeck
D: Evaluation of erlotinib in advanced non-small cell lung cancer:
impact on the budget of a U.S. health insurance plan. J Manag Care
Pharm. 12:472–478. 2006.PubMed/NCBI
|
|
3.
|
Feng J, Zhang X, Zhu H, Wang X, Ni S and
Huang J: High expression of FoxP1 is associated with improved
survival in patients with non-small cell lung cancer. Am J Clin
Pathol. 138:230–235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Gupta GP and Massague J: Cancer
metastasis: building a framework. Cell. 127:679–695. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Shi Y and Massague J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Toonkel RL, Borczuk AC and Powell CA:
TGF-beta signaling pathway in lung adenocarcinoma invasion. J
Thorac Oncol. 5:153–157. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Akhurst RJ and Derynck R: TGF-beta
signaling in cancer - a double-edged sword. Trends Cell Biol.
11:S44–S51. 2001.PubMed/NCBI
|
|
8.
|
Xu Y and Pasche B: TGF-beta signaling
alterations and susceptibility to colorectal cancer. Hum Mol Genet.
16(Spec No 1): R14–R20. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Liu X, Sun Y, Constantinescu S, Karam E,
Weinberg R and Lodish H: Transforming growth factor beta-induced
phosphorylation of Smad3 is required for growth inhibition and
transcriptional induction in epithelial cells. Proc Natl Acad Sci
USA. 94:10669–10674. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Velden JL, Alcorn JF, Guala AS, Badura EC
and Janssen-Heininger YM: c-Jun N-terminal kinase 1 promotes
transforming growth factor-beta1-induced epithelial-to-mesenchymal
transition via control of linker phosphorylation and
transcriptional activity of Smad3. Am J Respir Cell Mol Biol.
44:571–581. 2011. View Article : Google Scholar
|
|
11.
|
Yilmaz M, Christofori G and Lehembre F:
Distinct mechanisms of tumor invasion and metastasis. Trends Mol
Med. 13:535–541. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Goldoni S, Owens RT, McQuillan DJ, et al:
Biologically active decorin is a monomer in solution. J Biol Chem.
279:6606–6612. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Bi X, Pohl NM, Qian Z, et al:
Decorin-mediated inhibition of colorectal cancer growth and
migration is associated with E-cadherin in vitro and in mice.
Carcinogenesis. 33:326–330. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Bi XL and Yang W: Biological functions of
decorin in cancer. Chin J Cancer. 32:266–269. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Bi XL, Tong C, Dockendorff A, et al:
Genetic deficiency of decorin causes intestinal tumor formation
through disruption of intestinal cell maturation. Carcinogenesis.
29:1435–1440. 2008. View Article : Google Scholar
|
|
16.
|
Goldoni S, Seidler DG, Heath J, et al: An
antimetastatic role for decorin in breast cancer. Am J Pathol.
173:844–855. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Matsumine A, Shintani K, Kusuzaki K, et
al: Expression of decorin, a small leucine-rich proteoglycan, as a
prognostic factor in soft tissue tumors. J Surg Oncol. 96:411–418.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Nash MA, Deavers MT and Freedman RS: The
expression of decorin in human ovarian tumors. Clin Cancer Res.
8:1754–1760. 2002.PubMed/NCBI
|
|
19.
|
Reed CC, Gauldie J and Iozzo RV:
Suppression of tumorigenicity by adenovirus-mediated gene transfer
of decorin. Oncogene. 21:3688–3695. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Kresse H and Schonherr E: Proteoglycans of
the extracellular matrix and growth control. J Cell Physiol.
189:266–274. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Tralhao JG, Schaefer L, Micegova M, et al:
In vivo selective and distant killing of cancer cells, using
adenovirus-mediated decorin gene transfer. FASEB J. 17:464–466.
2003.PubMed/NCBI
|
|
22.
|
Yamaguchi Y, Mann DM and Ruoslahti E:
Negative regulation of transforming growth factor-beta by the
proteoglycan decorin. Nature. 346:281–284. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Biaoxue R, Xiguang C, Hua L, et al:
Decreased expression of decorin and p57(KIP2) correlates with poor
survival and lymphatic metastasis in lung cancer patients. Int J
Biol Markers. 26:9–21. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Liu Z, Zhao J, Chen XF, et al: CpG island
methylator phenotype involving tumor suppressor genes located on
chromosome 3p in non-small cell lung cancer. Lung Cancer. 62:15–22.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Liu Z, Li W, Lei Z, et al: CpG island
methylator phenotype involving chromosome 3p confers an increased
risk of non-small cell lung cancer. J Thorac Oncol. 5:790–797.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Zhang HT, Chen XF, Wang MH, et al:
Defective expression of transforming growth factor beta receptor
type II is associated with CpG methylated promoter in primary
non-small cell lung cancer. Clin Cancer Res. 10:2359–2367. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Doi A, Park IH, Wen B, et al: Differential
methylation of tissue-and cancer-specific CpG island shores
distinguishes human induced pluripotent stem cells, embryonic stem
cells and fibroblasts. Nat Genet. 41:1350–1353. 2009. View Article : Google Scholar
|
|
28.
|
Irizarry RA, Ladd-Acosta C, Wen B, et al:
The human colon cancer methylome shows similar hypo- and
hypermethylation at conserved tissue-specific CpG island shores.
Nat Genet. 41:178–186. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Lu YL, Huang JX, Li XH, et al: Spontaneous
metastasis of clonal cell subpopulations of human lung giant cell
carcinoma after subcutaneous inoculation in nude mice. Chin J
Oncol. 11:1–7. 1989.PubMed/NCBI
|
|
30.
|
Sun W, Guo C, Meng X, et al: Differential
expression of PAI-RBP1, C1orf142, and COTL1 in non-small cell lung
cancer cell lines with different tumor metastatic potential. J
Investig Med. 60:689–694. 2012.PubMed/NCBI
|
|
31.
|
Shang Y, Myers M and Brown M: Formation of
the androgen receptor transcription complex. Mol Cell. 9:601–610.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Sogawa K and Fujii-Kuriyama Y: Ah
receptor, a novel ligand-activated transcription factor. J Biochem.
122:1075–1079. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Zhao J, Sime PJ, Bringas P Jr, Gauldie J
and Warburton D: Adenovirus-mediated decorin gene transfer prevents
TGF-beta-induced inhibition of lung morphogenesis. Am J Physiol.
277:L412–L422. 1999.PubMed/NCBI
|
|
34.
|
Koninger J, Giese NA, di Mola FF, et al:
Overexpressed decorin in pancreatic cancer: potential tumor growth
inhibition and attenuation of chemotherapeutic action. Clin Cancer
Res. 10:4776–4783. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Cairns P: Gene methylation and early
detection of genitourinary cancer: the road ahead. Nat Rev Cancer.
7:531–543. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Lu F and Zhang HT: DNA methylation and
nonsmall cell lung cancer. Anat Rec. 294:1787–1795. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Luczak MW and Jagodzinski PP: The role of
DNA methylation in cancer development. Folia Histochem Cytobiol.
44:143–154. 2006.PubMed/NCBI
|
|
38.
|
Jeon HS and Jen J: TGF-beta signaling and
the role of inhibitory Smads in non-small cell lung cancer. J
Thorac Oncol. 5:417–419. 2010. View Article : Google Scholar : PubMed/NCBI
|