Introduction
Hepatocellular carcinoma (HCC) is the second leading
cause of cancer-related deaths worldwide, with the average annual
incidence on the rise both in China and elsewhere (1). The increasing knowledge in the
molecular pathogenesis of HCC, as well as the introduction of
molecular targeted therapies in oncology, has created an
encouraging trend in the management of this malignancy. Most HCC
treatments are designed to abrogate signaling pathways related to
cancer cell proliferation, cell survival, angiogenesis, invasion
and metastasis (2,3). Signal transducer and activator of
transcription 3 (STAT3), a major transducer to mediate the signal
from interleukin-6 (IL-6) to the nucleus, may be involved in
oncogenesis, cell proliferation, angiogenesis, immune evasion and
apoptotic resistance (4–8). IL-6 induces STAT3 phosphorylation at
tyrosine residue 705 through Janus-activated kinase (JAK) (9). STAT3 phosphorylation results in
homodimerization or heterodimerization of STAT3, enabling nuclear
localization and DNA binding, and regulating downstream genes
involved in controlling cell cycle progression and programmed cell
death (e.g., Bcl-2, cyclin D1, Mcl-1 and survivin) (10), and in the regulation of
angiogenesis [e.g., vascular endothelial growth factor (VEGF) and
hypoxia-inducible factor-1α (HIF-1α)] (4). The inhibition of aberrant STAT3
activation by genetic or pharmacological approaches has repeatedly
been demonstrated to result in growth inhibition, apoptosis in
vitro (11,12), as well as tumor growth and
metastasis inhibition in vivo (12–14)
in HCC. STAT3 is significantly correlated with the prognosis of HCC
patients (8,15), indicating that IL-6/STAT3 signaling
pathway might be a therapeutic target.
The potential biological significance of hydrogen
sulfide (H2S) has attracted growing interest in recent
years. A number of studies have investigated the role of
H2S in triggering cell death and evidence has been
presented that this gas can affect cancer cell survival using
sulfide salts as donor agents (16,17).
The slow-releasing H2S donor, GYY4137, exhibits
anti-cancer activity by releasing H2S down slowly
(18). It exhibits anticancer
activity by a combination of cell cycle arrest and promoting
apoptosis, inhibits tumor growth (18), however, the precise mechanism(s)
involved remain unclear. Recent study reported that GYY4137
exhibited anti-inflammatory effect in vivo and in
vitro (18,19). We propose that the anticancer
activity of GYY4137 may be relative with its suppression of
IL-6/STAT3 pathway.
We therefore sought to investigate whether GYY4137
is efficacious for treatment of HCC with a particular focus on its
suppression of IL-6/STAT3 pathway. We found that GYY4137 blocked
IL-6-induced STAT3 cascade leading to the suppression of cell
growth, induction of cell apoptosis and cell cycle arrest. GYY4137
also inhibited tumor growth and STAT3 activation in a subcutaneous
xenograft model with HepG2 cells in vivo. These results
suggest that GYY4137 may be a candidate for HCC therapy through
blocking constitutive STAT3 signaling.
Materials and methods
Materials
GYY4137 was obtained from the National Institute for
the Control of Pharmaceutical and Biological Products (Beijing,
China). STAT3, p-STAT3 (Y705), p-STAT3 (S727), JAK, p-JAK2, Bcl-2,
Mcl-1, cyclin D1, survivin, HIF-1α, VEGF, cleaved-poly(ADP-ribose)
polymerase (PARP), cleaved-caspase-9 and cleaved-caspase-3
antibodies were purchased from Cell Signaling Technology (Beverly,
MA, USA). β-actin antibody was purchased from Sigma (St. Louis, MO,
USA). 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium
bromide (MTT), RNase and sodium-orthovanadate and propidium iodide
(PI) was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell lines and cultures
HCC cell lines HepG2, and Bel7402, and
hepatocellular LO2 cells were obtained from the Cell Bank of the
Shanghai Institute of Biochemistry and Cell Biology were maintained
in RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with
10% fetal bovine serum (FBS) (Invitrogen). All cells were cultured
in a humidified atmosphere with a 5% CO2 incubator at
37°C.
Cell viability assay
HepG2, Bel7402 and LO2 cells were incubated in
triplicate in a 96-well plate at a density of 1×104
cells with 100 μl culture medium per well in the presence or
absence of indicated concentrations of GYY4137 for 24, 48 and 72 h.
After which, cell viability was determined by the MTT dye uptake
method as described earlier (20).
Western blot analysis
HepG2 and Bel7402 cells were cultured in 6-well
plates at a density of 5×105 cells/ml in a
CO2 incubator overnight and treated with GYY4137. The
total protein was prepared as previously described (21). The equalized amounts of proteins
from each sample were subjected to sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE) followed by transfer
to polyvinylidene fluoride (PVDF) membranes. The membranes were
blocked in 1% (w/v) bovine serum albumin (BSA) for 2 h, and then
incubated with the primary antibody overnight at 4°C. Primary
antibodies were STAT3, p-STAT3 (Y705), p-STAT3 (S727), JAK2, p-JAK2
(Y1007/1008), Bcl-2, Mcl-1, cyclin D1, survivin, HIF-1α, VEGF,
cleaved-PARP, cleaved-caspase-9, cleaved-caspase-3 (Cell Signaling
Technology) and β-actin (Sigma). Then the membranes were incubated
with secondary antibody conjugated with IgG horseradish peroxidase
(HRP) for 1 h at room temperature and immune complexes were
detected by the enhanced chemiluminescence system. β-actin served
as a loading control.
Cell cycle analysis
HepG2 and Bel7402 cells were incubated in 6-well
plates in the presence of different concentrations of GYY4137 for
24 h. Thereafter, treated cells were fixed and incubated with RNase
and propidium iodide (PI) in PBS. Cell cycle distribution was
analyzed with a FACScan laser flow cytometer (FACSCalibur,
Becton-Dickinson, Franklin Lakes, NJ, USA). The data were analyzed
using the software CELL Quest.
Apoptosis assay
Apoptosis was measured with caspase-3/7 assay
(Promega, Madison, WI, USA) according to the manufacturer’s
protocol. Briefly, cells were seeded into a 96-well plate. After
the treatment, 100 ml of Apo-One Caspase-3/7 reagent was added to
each well and was incubated at 37°C for 30 min. The fluorescence
was measured at an excitation wavelength of 485 nm and an emission
wavelength of 530 nm.
Enzyme-linked immunosorbent assay
(ELISA)
For the measurement of VEGF, 5×105 U266
cells/well were seeded in 12-well plates and grown to 75–80%
confluence, then cells were switched to fresh serum-free mediumin
the presence or absence of rosiglitazone and ATRA and incubated for
another 12 h. Cell-free culture supernatants were harvested and
assayed for secreted VEGF using commercially available ELISA kits
(R&D Systems, Minneapolis, MN, USA).
Animal experiments
Male BALB/c-nu/nu mice (4–6 weeks old, Slac
Laboratory Animal, Shanghai, China) were kept in the animal
facilities at the Nanjing Medical University and maintained under
specific pathogen-free conditions. All animal procedures were
conducted according to the guidelines approved by the China
Association of Laboratory Animal Care. Cultured HepG2 cells
(5×106) suspended in 0.2 ml PBS were injected into the
right flank of mice. The mice were kept in a pathogen-free
environment, 5–7 days later when the tumor volume reached 100
mm3, the mice were divided into four groups (control
group and three-dose GYY4137 groups) in a manner to equalize the
mean tumor among the four groups (n=7 each). GYY4137 at 50, 20 and
10 mg/kg dose suspended in 0.5% carboxymethyl cellulose (CMC) was
given as gavage to mice daily for 4 weeks, and mice of control
group were given 0.1 ml 0.5% CMC solution. The tumor size was
measured in two orthogonal directions using calipers every three
days, and the tumor volume (mm3) was estimated using the
equation length × (width)2 × 0.5. Four weeks later, the
mice were sacrificed and the tumors were resected. Part of tumor
tissues were homogenized and subjected to western blot assay. The
primary antibodies were STAT3, p-STAT3 (Y705), p-STAT3 (S727),
Bcl-2, Mcl-1, cyclin D1, survivin, HIF-1α, VEGF (Cell Signaling
Technology) and β-actin (Sigma).
Immunohistochemical (IHC) analysis
Immunohistology analysis was carried out using
paraffin section. Paraffin section were incubated in a blocking
solution (10% donkey serum + 5% non-fat dry milk + 4% BSA + 0.1%
Triton X-100) for 10 min and then hydrated sections were incubated
at 4°C overnight with anti-p-STAT3 (Y705) and p-STAT3 (S727) or
anti-STAT3 antibody, respectively. After washing with PBS, the
sections were incubated with diluted (1:200) biotinylated secondary
antibody for 30 min. Subsequently, the slides were washed again in
PBS and incubated for 30 min with the preformed avidin-horseradish
peroxidase macromolecular complex. Development of peroxidase
reaction was achieved by incubation in 0.01% 3,3-diaminobenzidine
tetrahydrochloride (DAB) in PBS containing 0.01% hydrogen peroxide
for approximately 5 min at room temperature. Sections were then
washed thoroughly in tap water, counterstained in haematoxylin,
dehydrated in absolute alcohol, cleared in xylene and mounted in
synthetic resin for microscopic examination.
Statistical analysis
Statistical difference was analyzed by two-way
Student’s t-test. P<0.05 was considered to indicate statistical
significance. The values are expressed as the mean ± SD. Three or
more separates experiments were performed.
Results
GYY4137 inhibits IL-6-induced STAT3
phosphorylation in HCC cells
To examine whether the antitumor activity of GYY4137
may be due to the inhibition of IL-6/STAT3 pathway, we evaluated
the effects of GYY4137 on IL-6-induced STAT3 phosphorylation in
human HCC cells. HepG2 and Bel7402 cell lines with higher IL-6 and
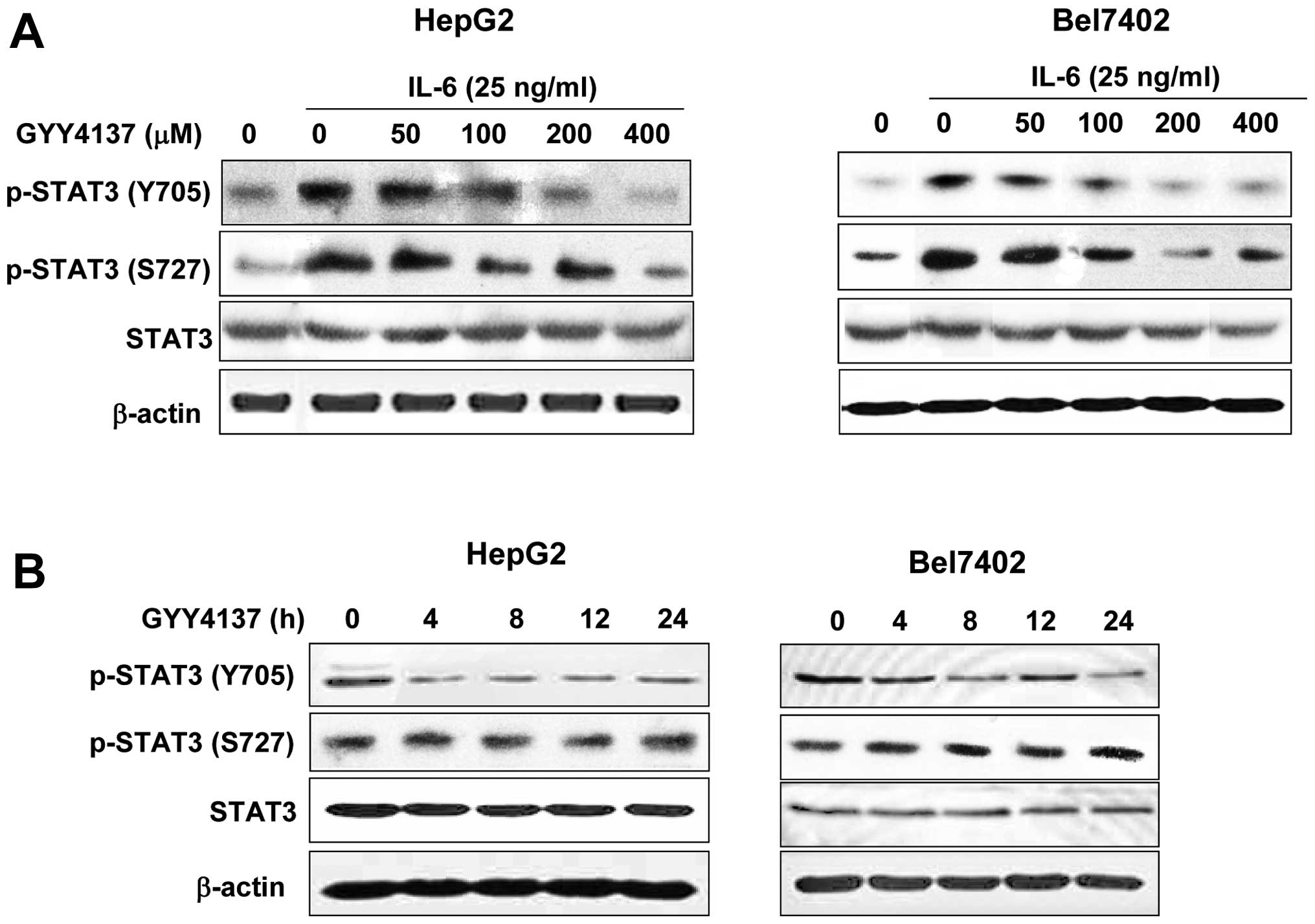
phosphorylated STAT3 were treated with GYY4137 for 24 h. As shown
in Fig. 1A, IL-6 significantly
activated STAT3 phosphorylation at Y705 both in HepG2 and Bel7402
cells. GYY4137 inhibited the IL-6-induced p-STAT3 (Y705) in a
dose-dependent manner, whereas it had no effect on p-STAT3 (S727)
and total STAT3 (Fig. 1A).
Furthermore, inhibition was evident as early as 4 h after treatment
and lasted for 24 h (Fig. 1B) in
HCC cell lines. These findings indicated that GYY4137 inhibited
IL-6-induced STAT3 activation in human HCC cells.
GYY4137 suppresses constitutive JAK2
activation and STAT3 downstream gene expression
JAK2 is considered as one of the most common
activators of STAT3. To explore the mechanism of the inhibitory
effects on p-STAT3 (Y705), the levels of JAK2 phosphorylation were
evaluated. P-JAK2 (Y1007/1008) was suppressed in HepG2 and Bel7402
cell lines with GYY4137 treatment, whereas basic JAK2 was not
detectable (Fig. 2A). To further
investigate whether GYY4137 would affect STAT3 downstream genes, we
used western blot analysis to examine the expression of Bcl-2,
Mcl-1, cyclin D1 and survivin. As shown in Fig. 2B, GYY4137 treatment downregulated
the expression of these proteins both in HepG2 and Bel7402 cell
lines.
GYY4137 causes cell cycle arrest,
suppresses cell viability and induces cell apoptosis
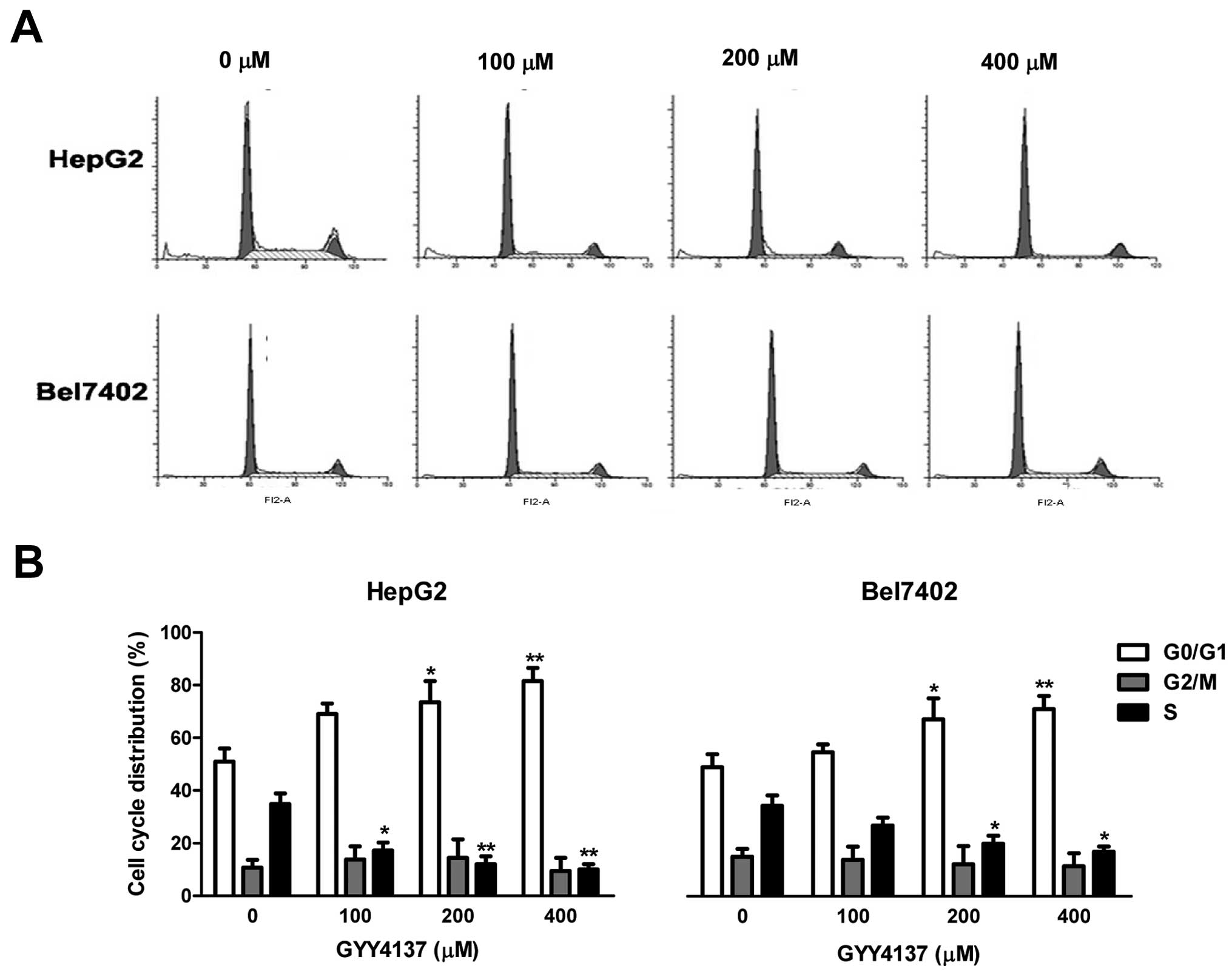
To examine the growth inhibitory effect of GYY4137,
we first assessed cell cycle distribution by flow cytometric
analysis. As shown in Fig. 3,
GYY4137 increased the number of cells in the G0/G1 phase in HepG2
and Bel7402 cells. The results are consistent with the inhibitory
effect of GYY4137 on cyclin D1 in these cells.
Since IL-6/STAT3 targets certain genes such as
Bcl-2, Mcl-1 and survivin involved in anti-apoptosis, we next
examined the effect of GYY4137 on apoptosis induction. As shown in
Fig. 4A, GYY4137 promoted the
cleavage of proapoptotic 116 kDa full-length PARP in an 89 kDa
fragment, a process known to impair genomic integrity before
apoptosis. We also observed that the levels of cleaved caspase-9
and caspase-3 were increased by treatment with GYY4137 in a
dose-dependent manner (Fig. 4A).
Flow cytometric data revealed that treatment with 200 and 400
μM of GYY4137 increased the number of cells in the subG1
phase (13–20%), indicating that the HCC cells were undergoing
apoptosis (Fig. 4B). MTT assay
showed that GYY4137 inhibited tumor cell viability time- and
dose-dependently (Fig. 4C), while
it showed weak inhibition on normal hepatocellular LO2 cells only
after 400 μM GYY4137 treatment for 72 h (data not shown).
These results showed that GYY4137 was able to induce apoptosis in
HCC cells through regulating the caspase pathway.
GYY4137 inhibits expression of VEGF and
HIF-1α in HCC cells
A low oxygen level is a characteristic feature of
solid tumors and a negative prognostic factor for cancer patient
survival. The response of cancer cells to hypoxia not only drives
neo-angiogenesis, but also enhances cancer cell survival and
malignant phenotype development. Hypoxia is known to increase the
expression of HIF-1α, a major regulator of pro-angiogenic
signaling. Since HIF-1α is the major transcriptional modulator of
angiogenic factors such as VEGF, we evaluated the effect of GYY4137
on hypoxia induced expression of HIF-1α and VEGF in HCC cells. The
cells were treated with various concentrations of GYY4137 under
hypoxia mimicking condition induced by 100 μM
CoCl2 for 6 h. As shown in Fig. 5A, GYY4137 suppressed the expression
of HIF-1α, which was increased after exposure to hypoxia. To
further confirm the effect of GYY4137 on hypoxia-induced VEGF, an
immediate downstream target gene of HIF-1α, VEGF protein level
secreted to media was measured by ELISA in HepG2 cells. A notable
increase of VEGF was observed under the hypoxic condition, whereas
the treatment of GYY4137 suppressed VEGF production
dose-dependently (Fig. 5B).
GYY4137 inhibits tumor growth in a
subcutaneous HepG2 xenograft model
To determine the antitumor activity of GYY4137 in
vivo, we evaluated its effect in a nude mouse xenograft model
of HepG2 cells. GYY4137 was administered for 4 weeks. After 4
weeks, the mice were sacrificed and the tumor mass was dissected.
The high dose of GYY4137 (50 mg/kg/d) inhibited tumor growth
significantly (P<0.05) after administration for 13 days, and the
low dose (10 mg/kg) of GYY4137 exhibited no significant inhibitory
effect on tumor growth (Fig. 6A).
Body weight of the mice was observed during administration of
GYY4137. There was no significant weight loss in mice treated with
GYY4137 of middle and high dose groups, while significant weight
loss was detected in mice treated with GYY4137 of low dose or
saline groups (Fig. 6B).
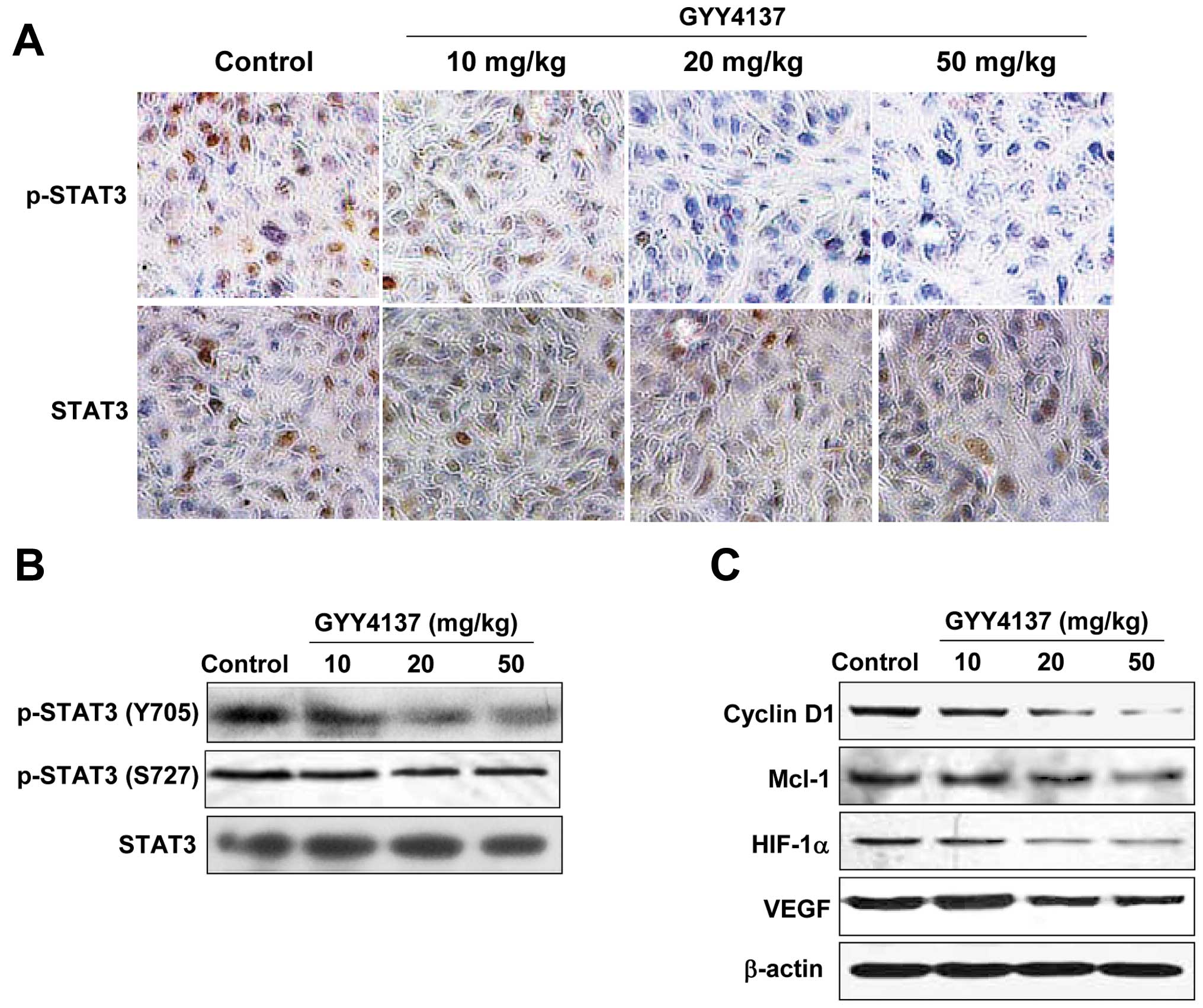
To confirm whether the inhibition of GYY4137 on
xenograft tumor growth was mediated by blocking STAT3 signaling, we
investigated the expression levels of p-STAT3 and STAT3 in
xenograft tumor of mice treated with GYY4137 by IHC analysis. As
shown in Fig. 7A and B, the level
of p-STAT3 (Y705) was decreased in xenograft tumors of mice treated
with GYY4137 compared with the model xenograft tumors, whereas the
level of STAT3 had no significant change in the model group or the
GYY4137 treated group. In concert with the decreased expression of
p-STAT3, a clear downregulation in the target gene expression of
STAT3, such as cyclin D1, Mcl-1, HIF-1α and VEGF, were found in the
xenograft tumor tissue of the GYY4137-treated mice, in a GYY4137
dose-dependent manner (Fig. 7C).
Results in vivo further confirmed that the antitumor effect
of GYY4137 was partly mediated by blocking the STAT3 signaling
pathway.
Discussion
Clinical studies have indicated that constitutive
STAT3 activation plays a critical role in tumor formation and
development in a variety of primary human cancers and cell lines,
including HCC. Therefore, STAT3 signaling has emerged as an
important therapeutic target for anticancer drug development
(12,22). Aberrant activation of STAT3 may
promote tumor cell proliferation through the upregulation of cyclin
D1 and may suppress apoptosis through the upregulation of Bcl-2,
Mcl-1 and survivin (10).
Therefore, suppression of constitutively activated STAT3 not only
induces apoptosis in cancer cells but also overcomes
chemoresistance and radioresistance (23). The aim of this study was to
investigate whether GYY4137 exerts its anticancer effect through
inhibiting STAT3 signaling in HCC. We found that GYY4137 had
anti-HCC effects and the underlying mechanism was associated with
the inhibition of STAT3 signaling that led to the suppression of
cancer cell proliferation by induction of cell cycle arrest,
increased apoptosis and inhibition of angiogenesis in vitro
and in vivo.
Phosphorylation of STAT3 at Y705 enables its
dimerization, nuclear translocation, DNA binding and gene
transcription (23), whereas
phosphorylation of another conserved STAT3 residue, S727, enhances
STAT3 transcriptional activity (24). Cooperation of tyrosine and serine
phosphorylation is necessary for the full activation of STAT3
(25). Here, we showed that
GYY4137 could inhibit IL-6-induced STAT3 activation in HCC cells by
blocking JAK2 phosphorylation. STAT3 was constitutively
phosphorylated at Y705 and S727 in HCC and GYY4137 inhibited
phosphorylation of Y705 among HCC cells and HepG2 xenografts of
nude mice that exhibited different molecular or genetic
characteristics. The inhibition was evident as early as 4 h after
treatment and lasted for 24 h, suggesting that the actions of
GYY4137 are relatively rapid and prolonged. Moreover, we showed
that GYY4137 downregulated STAT3 mediated downstream gene products
involved in controlling cell cycle progression and programmed cell
death (e.g., cyclin D1, Mcl-1, Bcl-2 and survivin) in vitro
and in vivo. Constitutively active STAT3 is associated with
the induction of resistance to apoptosis possibly through the
expression of Bcl-2 and cyclin D1 (26–28).
Overexpression of Mcl-1 is found in a great percentage of HCC.
Targeting Mcl-1 is a promising novel approach in HCC therapy
(29). Therefore, downregulation
of the expression of cyclin D1, Mcl-1, Bcl-2 and survivin was
likely correlated with the ability of GYY4137 to facilitate
apoptosis in HCC cells.
Apoptosis is a process of programmed cell death that
serves as a major mechanism for the precise regulation of cell
numbers and as a defense mechanism to remove unwanted cells
(30). Mitochondria mediate
apoptotic signaling via activation of the cell death initiator
procaspase-9 (30,31). Activated caspase-9 in turn cleaves
executioner caspase-3. The activated caspase-3 then cleaves PARP, a
116 kDa nuclear protein related to the process of programmed cell
death (32). In addition, we
observed that the antitumor activity of GYY4137 was also associated
with the induction of apoptosis in HCC cells as indicated by in
vitro evidence of increased cleaved caspase-9, caspase-3 and
PARP cleavage. These results indicated that GYY4137 inhibited cell
growth by regulating cell cycle progression and induced apoptosis
in HCC cells.
VEGF and HIF-1α are prominent transcriptional
targets for STAT3 among the angiogenesis factors (10,33).
HIF-1α activation appears to be a very early event in
carcinogenesis and this protein is expressed before histological
evidence of angiogenesis or invasion (34,35).
In regards to HCC, overexpression of HIF-1α has been reported,
which has been associated with a poor prognosis (36). Recently, both HIF-1α and VEGF were
identified to be involved in the malignant transformation of
dysphasic liver nodules and additionally a hypoxia-independent
overexpression of HIF-1α has been shown to be involved in a model
of mouse hepatocarcinogenesis (37). Suppression of HIF-1α and STAT3
activity was demonstrated to inhibit the growth in HCC, ultimately
leading to a reduction of tumor growth and vascularization in
vivo (38). In the present
study, we found that GYY4137 effectively inhibited VEGF and HIF-1α
expression in HCC cells and HepG2 xenografts of nude mice, which
may be related with its inhibition of STAT3 activation. Therefore,
downregulation of VEGF and HIF-1α by GYY4137 may be of great
significance in suppressing tumor cell invasion and metastasis.
In conclusion, GYY4137 blocks the STAT3 signaling
pathway. The inhibition of STAT3 (Y705) phosphorylation triggered
downregulation of gene products related to cell survival, cell
cycle progression, thus in turn triggered cell apoptosis and cell
cycle arrest. Importantly, GYY4137 exhibited antitumor effect in
the HepG2 xenograft nude mouse model. These results suggested that
GYY4137 is a potential candidate for treatment of HCC.
Acknowledgements
This study was supported by a grant
from the Priority Academic Program of Jiangsu Higher Education
Institutions (JX10231801).
References
|
1.
|
Bruix J and Sherman M; Practice Guidelines
Committee; American Association for the Study of Liver Diseases:
Management of hepatocellular carcinoma. Hepatology. 42:1208–1236.
2005. View Article : Google Scholar
|
|
2.
|
Villanueva A, Newell P, Chiang DY,
Friedman SL and Llovet JM: Genomics and signaling pathways in
hepatocellular carcinoma. Semin Liver Dis. 27:55–76. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Llovet JM and Bruix J: Novel advancements
in the management of hepatocellular carcinoma in 2008. J Hepatol.
48(Suppl 1): S20–S37. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Niu G, Wright KL, Huang M, et al:
Constitutive Stat3 activity up-regulates VEGF expression and tumor
angiogenesis. Oncogene. 21:2000–2008. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Real PJ, Sierra A, De Juan A, Segovia JC,
Lopez-Vega JM and Fernandez-Luna JL: Resistance to chemotherapy via
Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer
cells. Oncogene. 21:7611–7618. 2002. View Article : Google Scholar
|
|
6.
|
Wang T, Niu G, Kortylewski M, et al:
Regulation of the innate and adaptive immune responses by Stat-3
signaling in tumor cells. Nat Med. 10:48–54. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Schindler C, Levy DE and Decker T:
JAK-STAT signaling: from interferons to cytokines. J Biol Chem.
282:20059–20063. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Alvarez JV, Febbo PG, Ramaswamy S, Loda M,
Richardson A and Frank DA: Identification of a genetic signature of
activated signal transducer and activator of transcription 3 in
human tumors. Cancer Res. 65:5054–5062. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Bollrath J, Phesse TJ, von Burstin VA, et
al: gp130-mediated Stat3 activation in enterocytes regulates cell
survival and cell-cycle progression during colitis-associated
tumorigenesis. Cancer Cell. 15:91–102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Garcia R, Bowman TL, Niu G, et al:
Constitutive activation of Stat3 by the Src and JAK tyrosine
kinases participates in growth regulation of human breast carcinoma
cells. Oncogene. 20:2499–2513. 2001. View Article : Google Scholar
|
|
11.
|
Liu Y, Liu A, Li H, Li C and Lin J:
Celecoxib inhibits interleukin-6/interleukin-6 receptor-induced
JAK2/STAT3 phosphorylation in human hepatocellular carcinoma cells.
Cancer Prev Res (Phila). 4:1296–1305. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Yang J, Cai X, Lu W, et al: Evodiamine
inhibits STAT3 signaling by inducing phosphatase shatterproof 1 in
hepatocellular carcinoma cells. Cancer Lett. 328:243–251. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Gu FM, Li QL, Gao Q, et al: Sorafenib
inhibits growth and metastasis of hepatocellular carcinoma by
blocking STAT3. World J Gastroenterol. 17:3922–3932. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Rajendran P, Ong TH, Chen L, et al:
Suppression of signal transducer and activator of transcription 3
activation by butein inhibits growth of human hepatocellular
carcinoma in vivo. Clin Cancer Res. 17:1425–1439. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Calvisi DF, Ladu S, Gorden A, et al:
Ubiquitous activation of Ras and Jak/Stat pathways in human HCC.
Gastroenterology. 130:1117–1128. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Hu LF, Wong PT, Moore PK and Bian JS:
Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation
by inhibition of p38 mitogen-activated protein kinase in microglia.
J Neurochem. 100:1121–1128. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Rose P, Moore PK, Ming SH, Nam OC,
Armstrong JS and Whiteman M: Hydrogen sulfide protects colon cancer
cells from chemopreventative agent beta-phenylethyl isothiocyanate
induced apoptosis. World J Gastroenterol. 11:3990–3997.
2005.PubMed/NCBI
|
|
18.
|
Lee ZW, Zhou J, Chen CS, et al: The
slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel
anti-cancer effects in vitro and in vivo. PLoS One. 6:e210772011.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Li L, Fox B, Keeble J, et al: The complex
effects of the slow-releasing hydrogen sulfide donor GYY4137 in a
model of acute joint inflammation and in human cartilage cells. J
Cell Mol Med. 17:365–376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Takada Y, Ichikawa H, Badmaev V and
Aggarwal BB: Acetyl-11-keto-beta-boswellic acid potentiates
apoptosis, inhibits invasion, and abolishes osteoclastogenesis by
suppressing NF-kappa B and NF-kappa B-regulated gene expression. J
Immunol. 176:3127–3140. 2006. View Article : Google Scholar
|
|
21.
|
Isomoto H, Mott JL, Kobayashi S, et al:
Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to
SOCS-3 epigenetic silencing. Gastroenterology. 132:384–396. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Won C, Lee CS, Lee JK, et al: CADPE
suppresses cyclin D1 expression in hepatocellular carcinoma by
blocking IL-6-induced STAT3 activation. Anticancer Res. 30:481–488.
2010.PubMed/NCBI
|
|
23.
|
Aggarwal BB, Kunnumakkara AB, Harikumar
KB, et al: Signal transducer and activator of transcription-3,
inflammation, and cancer: how intimate is the relationship? Ann NY
Acad Sci. 1171:59–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Lufei C, Koh TH, Uchida T and Cao X: Pin1
is required for the Ser727 phosphorylation-dependent Stat3
activity. Oncogene. 26:7656–7664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Aziz MH, Manoharan HT, Church DR, et al:
Protein kinase Cepsilon interacts with signal transducers and
activators of transcription 3 (Stat3), phosphorylates Stat3Ser727,
and regulates its constitutive activation in prostate cancer.
Cancer Res. 67:8828–8838. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Selvendiran K, Kuppusamy ML, Ahmed S, et
al: Oxygenation inhibits ovarian tumor growth by downregulating
STAT3 and cyclin-D1 expressions. Cancer Biol Ther. 10:386–390.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Ai T, Wang Z, Zhang M, et al: Expression
and prognostic relevance of STAT3 and cyclin D1 in non-small cell
lung cancer. Int J Biol Markers. 27:e132–e138. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Zhang K, Pang B, Xin T, et al: Increased
signal transducer and activator of transcription 3 (STAT3) and
decreased cyclin D1 in recurrent astrocytic tumours compared with
paired primary astrocytic tumours. J Int Med Res. 39:2103–2109.
2011. View Article : Google Scholar
|
|
29.
|
Sieghart W, Losert D, Strommer S, et al:
Mcl-1 overexpression in hepatocellular carcinoma: a potential
target for antisense therapy. J Hepatol. 44:151–157. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Bossy-Wetzel E and Green DR: Apoptosis:
checkpoint at the mitochondrial frontier. Mutat Res. 434:243–251.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Estaquier J, Vallette F, Vayssiere JL and
Mignotte B: The mitochondrial pathways of apoptosis. Adv Exp Med
Biol. 942:157–183. 2012. View Article : Google Scholar
|
|
32.
|
Wurstle ML, Laussmann MA and Rehm M: The
central role of initiator caspase-9 in apoptosis signal
transduction and the regulation of its activation and activity on
the apoptosome. Exp Cell Res. 318:1213–1220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Wei D, Le X, Zheng L, et al: Stat3
activation regulates the expression of vascular endothelial growth
factor and human pancreatic cancer angiogenesis and metastasis.
Oncogene. 22:319–329. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Zhu H, Feng Y, Zhang J, et al: Inhibition
of hypoxia inducible factor 1alpha expression suppresses the
progression of esophageal squamous cell carcinoma. Cancer Biol
Ther. 11:981–987. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Matsuyama T, Nakanishi K, Hayashi T, et
al: Expression of hypoxia-inducible factor-1alpha in esophageal
squamous cell carcinoma. Cancer Sci. 96:176–182. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Yasuda S, Arii S, Mori A, et al:
Hexokinase II and VEGF expression in liver tumors: correlation with
hypoxia-inducible factor 1 alpha and its significance. J Hepatol.
40:117–123. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Tanaka H, Yamamoto M, Hashimoto N, et al:
Hypoxia-independent overexpression of hypoxia-inducible factor
1alpha as an early change in mouse hepatocarcinogenesis. Cancer
Res. 66:11263–11270. 2006. View Article : Google Scholar
|
|
38.
|
Moser C, Lang SA, Mori A, et al:
ENMD-1198, a novel tubulin-binding agent reduces HIF-1alpha and
STAT3 activity in human hepatocellular carcinoma (HCC) cells, and
inhibits growth and vascularization in vivo. BMC Cancer. 8:2062008.
View Article : Google Scholar : PubMed/NCBI
|