Introduction
Prostate cancer is the second leading cause of
cancer death among men in the United States, with an estimated
30,000 deaths among American men in 2014 (American Cancer Society,
www.cancer.org). For effective treatment of prostate
cancer it is necessary to identify effective and specific molecular
targets. As cancer cells exhibit various degrees of mitochondrial
dysfunction (1–4), it is of interest to determine whether
therapeutic strategies focused on mitochondria can be developed to
preferentially kill cancer cells.
Mitochondria are dynamic organelles that play
essential roles in cellular metabolism, homeostasis and regulation
of cell death. Increasing evidence has demonstrated that changes in
mitochondrial morphology are important determinants of
mitochondrial function (1,5). Mitochondria range from long
filamentous structures to small punctate spheres in different cell
types and under different conditions in the same cell type
(6,7). Mitochondrial structure is affected by
the interplay between the opposing fission and fusion processes
that occur normally in a cell (8).
Formation of extensive mitochondrial networks (generated by fusion)
is considered important for efficient intracellular energy transfer
into different cell compartments (9), whereas the punctate mitochondria
(arising from fission) seem to assist, in some cases, the induction
of apoptosis (10–14).
Mitochondrial fission involves the large GTPase,
dynamin-related protein 1 (Drp1) and fission 1 (Fis1) (1). Recent discoveries suggest that
mitochondrial fission factor (Mff), MIEF1/MiD51 and Mid49 act as
novel mitochondrial receptors for Drp1 to regulate mitochondrial
dynamics in mammalian cells (15–19).
Mitochondrial fusion of the outer membranes is controlled by
mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) whereas optic atrophy 1
(OPA1) is implicated in fusion of inner mitochondrial membranes
(20,21). Mitofusins are large transmembrane
GTPases of the mitochondrial outer membrane, which facilitate the
fusion of these membranes in a GTPase-dependent manner (1,22–25).
Overexpression of Mfn1 results in the formation of characteristic
networks of interconnected mitochondria (24), whereas loss of mitofusin function
causes mitochondrial fragmentation and dysfunction (1,24,26–28).
In the nervous system, overexpression of Mfn1 can protect against
NO-induced neuronal cell death (29). In yeast, it has been shown that
Mdm30p ligase promotes ubiquitination of Fzo1p, a yeast homologue
of mammalian Mfn1, and its subsequent degradation by the 26S
proteasome (30,31). Recently, March5, an E3 ubiquitin
ligase, has been shown to be involved in mammalian Mfn1 degradation
in HeLa and Chang cells (32).
In recent years, agents that impact mitochondria and
exhibit anticancer activity are gaining attention as possible
cancer therapies. CGP37157 (CGP) inhibits the efflux of
mitochondrial calcium through the sodium/calcium exchanger, thereby
increasing calcium levels in mitochondria and altering their
function. Earlier, we demonstrated that CGP treatment increases the
interaction between Drp1 and Fis1 to induce mitochondrial fission
(14), while CGP when combined
with TNF-α related apoptosis-inducing ligand (TRAIL), sensitizes
TRAIL-resistant prostate cancer cells to undergo apoptosis
(33). We have reported that
androgen upregulates the expression and levels of Drp1, a protein
involved in mitochondrial fission, but does not by itself induce
mitochondrial fission or apoptosis (11). However, treatment of LNCaP cells
with CGP in the presence of androgen increases mitochondrial
fission and apoptosis (11),
suggesting that the androgen-induced increase in Drp1 is not
sufficient to trigger mitochondrial fission and apoptosis but
requires a second signal to affect mitochondrial function and
enhance these processes. Although we demonstrated a concomitant
decrease in the fusion protein Mfn1, the mechanism by which CGP
affected Mfn1 protein expression was not investigated. Therefore,
our objective was to investigate the regulation of the
mitochondrial fusion protein, Mfn1, in response to CGP treatment as
well as its potential role in prostate cancer cell apoptosis.
Materials and methods
Reagents
The mammalian Myc-tagged Mfn1 overexpression plasmid
(Myc-Mfn1; a kind gift from Alexander van der Bliek, University of
California, Los Angeles, CA) was used for ubiquitination studies.
Antibodies used were: anti-Mfn1 (Novus Biologicals, Littleton, CO);
anti-Mfn2, anti-β-actin and anti-chicken IgG (Sigma-Aldrich
Corporation, St. Louis, MO); anti-GAPDH (Chemicon International,
Temecula, CA); anti-COX IV and anti-mouse and anti-rabbit IgG (Cell
Signaling, Danvers, MA); anti-Drp1 (BD Biosciences, San Jose, CA);
anti-ubiquitin and anti-Fis1 (Santa Cruz Biotechnology, Santa Cruz,
CA); and anti-March5 and anti-OPA1 (Abcam Inc., Cambridge, MA). The
chemicals used were: cycloheximide (Sigma- Aldrich);
7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one
or CGP37157 (CGP) (Calbiochem, San Diego, CA); and lactacystin
(Cayman Chemical Company, Ann Arbor, MI). Scrambled siRNA and siRNA
for Mfn1 and March5 were from Qiagen (Valencia, CA). The c-Myc
immunoprecipitation kit was from Thermo Scientific (Rockford,
IL).
Cell cultures and treatment
Prostate cancer cell lines LNCaP, DU145, and PC3
were purchased from ATCC (Manassas, VA). Cells were maintained in
RPMI-1640 (HyClone, Logan, UT) containing 9% FBS (v/v), 0.5%
penicillin-streptomycin (v/v), and 0.1% fungizone (v/v). The CWR-R1
cells (provided by Dr Elizabeth Wilson, University of North
Carolina, Chapel Hill, NC) were grown in Richter’s MEM.
Non-tumorigenic prostate epithelial cells (P69, a gift from Dr
Leland Chung) were maintained in T-medium (Gibco-Invitrogen,
Carlsbad, CA). Cells were treated with cycloheximide (50
μg/ml) or CGP (10–80 μM) for the indicated period of
time. Transfection with siRNA was performed using HiPer-Fect
reagent (Qiagen) for 48 h followed by treatment with various drugs.
Cells were transiently transfected with the myc-tagged Mfn1
overexpression plasmid (Myc-Mfn1) by electroporation using the
Electro Square Porator™ ECM-830 (BTX Inc., San Diego, CA) as
described previously (11). After
electroporation, cells were seeded in normal growth medium for 24 h
and then used in experiments.
Protein extraction and western blot
analysis
Appropriately treated cells were washed once with 1X
PBS followed by addition of lysis buffer [150 mM NaCl, 50 mM
Tris-HCl, pH 8.0, 1 mM EDTA, 0.7% (v/v) Triton X-100, 0.25% (w/v)
sodium deoxycholate, 1 mM NaF, 1 mM sodium pyrophosphate, 100
μM Na3VO4, 1 mM phenylmethylsulfonyl
fluoride, 10 μg/ml leupeptin, 0.7 μg/ml pepstatin,
and 10 μg/ml aprotinin] at 4°C. Cells were incubated on ice
for 30 min and lysates centrifuged at 10,000 × g at 4°C for 10 min.
The supernatants were collected, and the protein concentration was
estimated using a Bio-Rad DC protein reagent (Bio-Rad Laboratories,
Hercules, CA). Proteins (25 μg) were separated on 10 or 12%
(w/v) SDS-polyacrylamide gels and transferred to (PVDF) membranes
(Bio-Rad Laboratories) using a BioRad semi-dry transfer apparatus.
The blots were blocked in 5% (w/v) non-fat dry milk in TBS
containing 0.1% (v/v) Tween-20 and incubated overnight at 4°C with
primary antibody. Immunoreactive bands were visualized using an ECL
detection system (Amersham, Pharmacia Biotech, Arlington Heights,
IL) on an 8900 Alpha Innotech Image Analyzer (San Leandro, CA)
and/or exposed to Hyperfilm and developed. The membranes were
re-probed with antibody against β-actin and/or GAPDH, which were
used as loading controls. Antibodies were diluted in 3% (w/v) BSA
in TBST.
Measurement of apoptosis
Measurement of apoptosis was performed using an M30
Apoptosense kit (Peviva, DiaPharma Group, West Chester, OH) as
described previously (11).
Briefly, total cell lysates (5 μg) were added to 96-well
plates pre-coated with mouse monoclonal M30 antibody; horseradish
peroxidase tracer solution was then added to the wells and
incubated for 4 h. Color was developed by adding tetramethyl
benzidine solution and the optical density was measured at 450 nm
on a Spectra MAX 340 microplate reader (Molecular Devices
Corporation, Sunnyvale, CA). Standard curves were generated as
instructed by the supplier.
Statistical analyses
Data are presented as means ± SEM. We compared group
mean values, as appropriate, using one-way analysis of variance
with Newman-Keuls multiple comparison test (GraphPad Prism).
Significant differences were defined at p<0.01 and
p<0.05.
Results
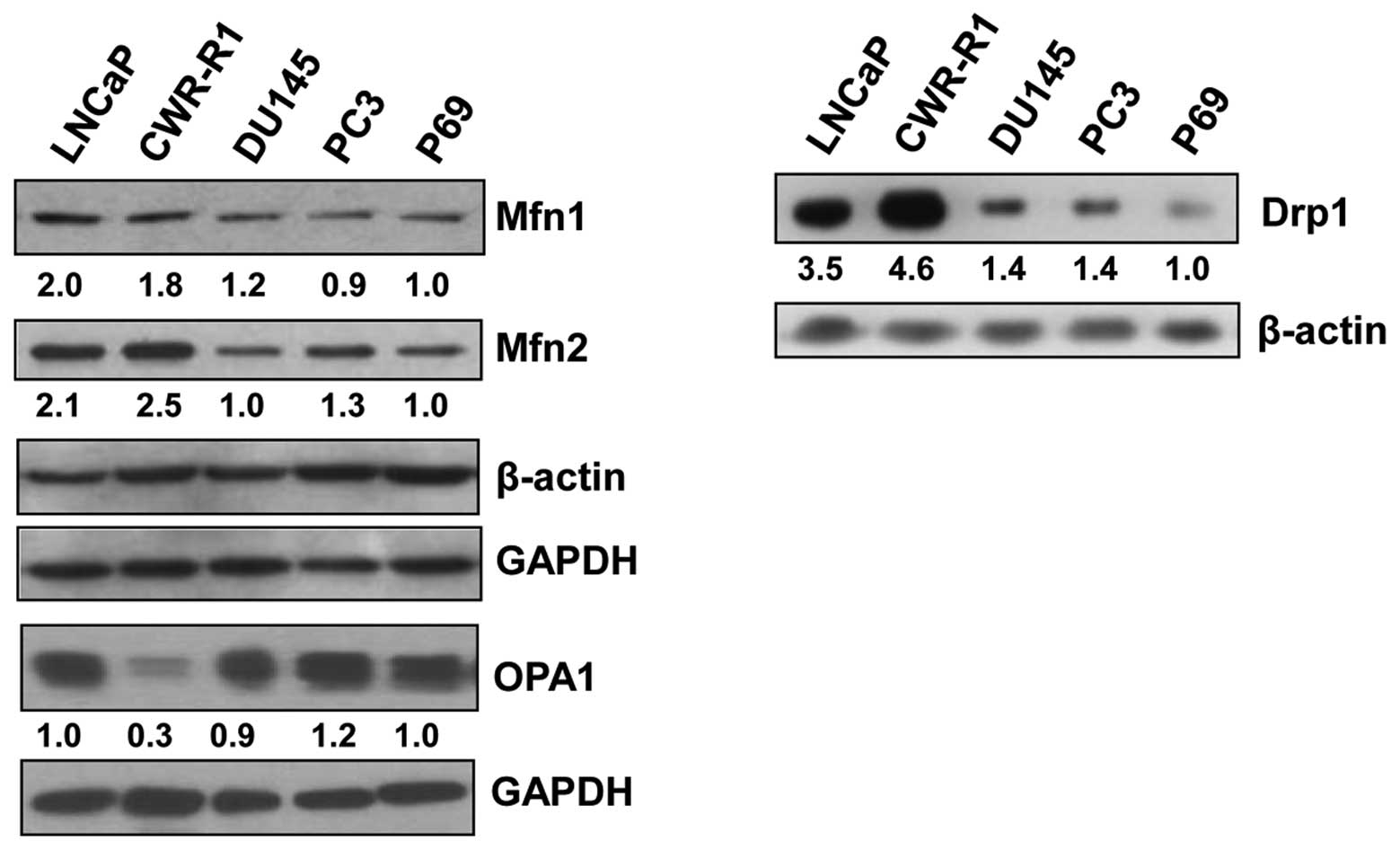
Basal levels of mitofusin 1 in prostate
cancer cells
Western blot analysis to examine the basal levels of
the proteins that regulate mitochondrial fusion in prostate cancer
cell lines showed that the mitofusins, Mfn1 and Mfn2, which
regulate outer mitochondrial membrane fusion, were greater in
androgen receptor-positive LNCaP and CWR-R1 cell lines as compared
to androgen receptor-negative DU145 and PC3 cell lines or the
normal prostate epithelial cell line, p69 (Fig. 1, left panel). Interestingly, Drp1,
a protein involved in mitochondrial fission, was also higher in
LNCaP and CWR-R1 cell lines (Fig.
1, right panel) compared to the other cell lines studied,
suggesting that both fusion- and fission-related proteins are
expressed at similar levels in these cell lines, presumably to
maintain an equilibrium between the proteins having opposite roles
in the regulation of mitochondrial morphology and function. The
levels of OPA1, which regulates inner mitochondrial membrane
fusion, were essentially similar in all cell lines except in the
CWR-R1 cell line which showed a markedly lower OPA1 level (Fig. 1, left panel).
Mfn1 is degraded upon CGP treatment
In previous reports we have shown that CGP
sensitizes prostate cancer cells, including the LNCaP and DU145
cell line, to TRAIL-induced apoptosis (33) and induces mitochondrial fission
(11,14). To understand the mechanism of CGP
action in the induction of mitochondrial fission, we examined the
levels of Mfn1, Mfn2, Fis1 and Drp1 in LNCaP cells treated with
CGP. The levels of Mfn1 protein decreased significantly
(approximately 50%) in CGP-treated LNCaP cells (Fig. 2A). However, the expression levels
of the other fusion protein, Mfn2, and the fission proteins Fis1
and Drp1, were unaltered after CGP treatment of LNCaP cells
(Fig. 2B). As LNCaP cells were
treated with 50 μM CGP in the earlier experiment (Fig. 2A), LNCaP cells were treated with
increasing concentrations of CGP (up to 80 μM) to obtain a
dose curve. Western blot analysis showed decreases in Mfn1 protein
only when cells were treated with 50 or 80 μM CGP (Fig. 2C). As LNCaP cells were treated for
18 h in the earlier experiments, a time course was determined by
treating these cells with the minimal effective dose of 50
μM CGP for various time periods up to 24 h. A marked
decrease in Mfn1 was observed after 8 h of CGP treatment (Fig. 2D), which was not further altered
with treatment for up to 24 h; Mfn2 levels remained largely
unaltered (data not shown), consistent with results shown in
Fig. 2B. Treatment of LNCaP cells
with an inhibitor of protein synthesis, cycloheximide, resulted in
a decrease in the levels of Mfn1 at about 18 h of treatment
(Fig. 2E), whereas CGP induced a
reduction in Mfn1 levels as early as 8 h after treatment,
suggesting that the decrease in Mfn1 levels in CGP-treated cells is
due to degradation rather than changes in
transcription/translation. CGP treatment of other cell lines (DU145
and CWR-R1) also resulted in a decrease in Mfn1 protein levels
(Fig. 2F), suggesting that the
ability of CGP to induce Mfn1 degradation may be common to prostate
cancer cells in general.
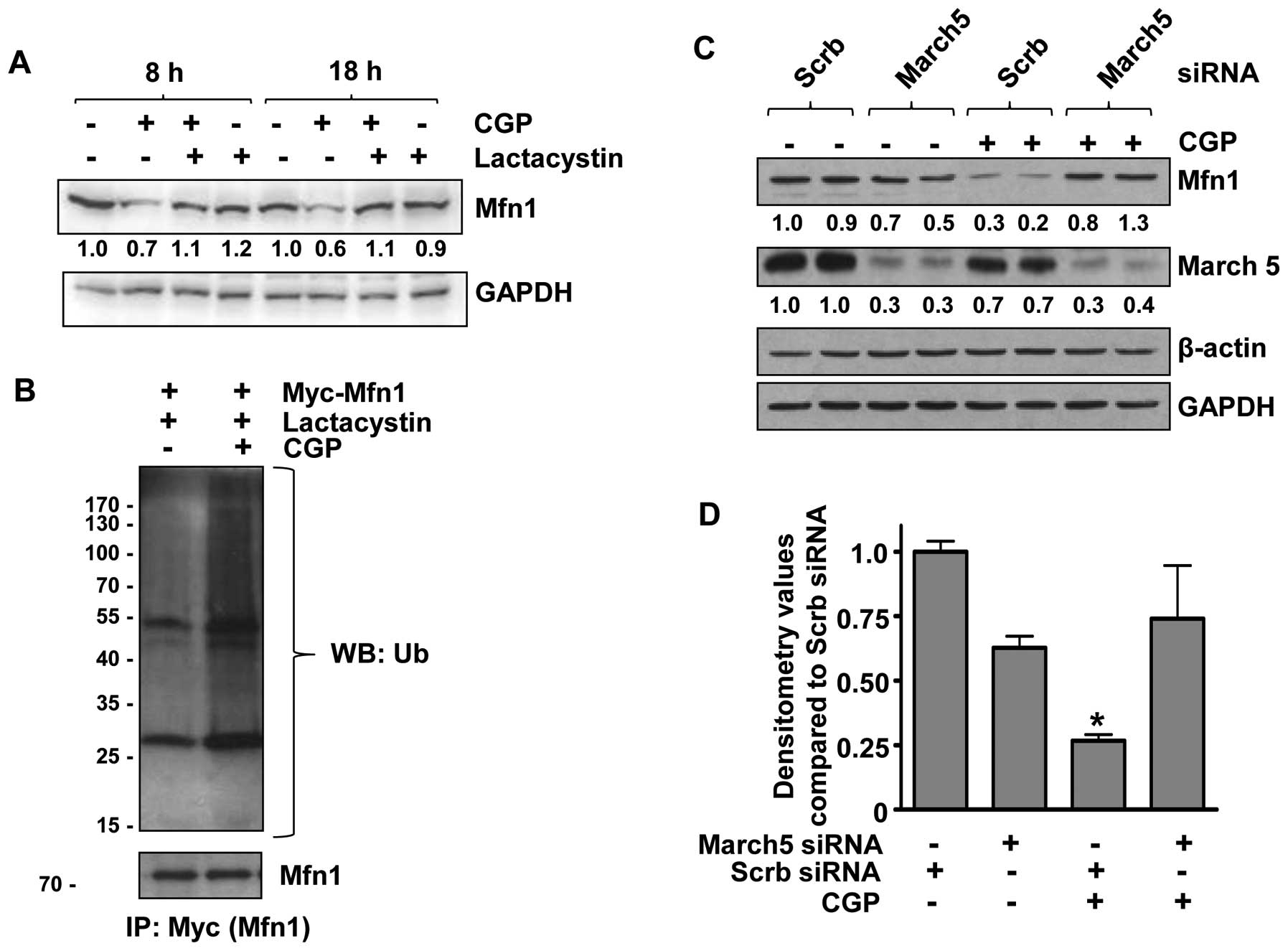
Mfn1 is degraded by a
ubiquitin-proteasome pathway involving the E3 ubiquitin ligase,
March5
To investigate whether CGP-induced Mfn1 degradation
was mediated by proteasomes, LNCaP cells were pretreated with the
proteasome inhibitor, lactacystin. Pretreatment with lactacystin
rescued Mfn1 from CGP-induced degradation (Fig. 3A), suggesting the involvement of
proteasomes in CGP-induced Mfn1 degradation. As ubiquiti-nation is
a key step in the proteasomal degradation of a protein, we
investigated whether Mfn1 is ubiquitinated upon CGP treatment.
LNCaP cells were transfected with the Myc-tagged Mfn1
overexpression plasmid and treated with CGP. Proteins were
immunoprecipitated using anti-Myc antibody and subjected to western
blot analysis with an anti-ubiquitin antibody, which revealed that
Mfn1 was ubiquitinated in CGP-treated cells (Fig. 3B). These results support our
hypothesis that treatment with CGP results in ubiquitination of
Mfn1, which is then targeted for degradation by proteasomes. As the
E3 ubiquitin ligase, March5, has been reported to be involved in
the degradation of mammalian Mfn1 protein in HeLa cervical cancer
and Chang liver cells (32), its
role in CGP-induced degradation of Mfn1 was examined. LNCaP cells
were transfected with siRNA against March5, which significantly
reduced March5 protein expression (Fig. 3C and D). Treatment of transfected
cells (March5 siRNA) with CGP resulted in Mfn1 levels that were
similar to the scrambled siRNA-transfected controls (Fig. 3C and D), indicating the involvement
of March5 E3 ubiquitin ligase in the ubiquitination and degradation
of Mfn1 upon CGP treatment.
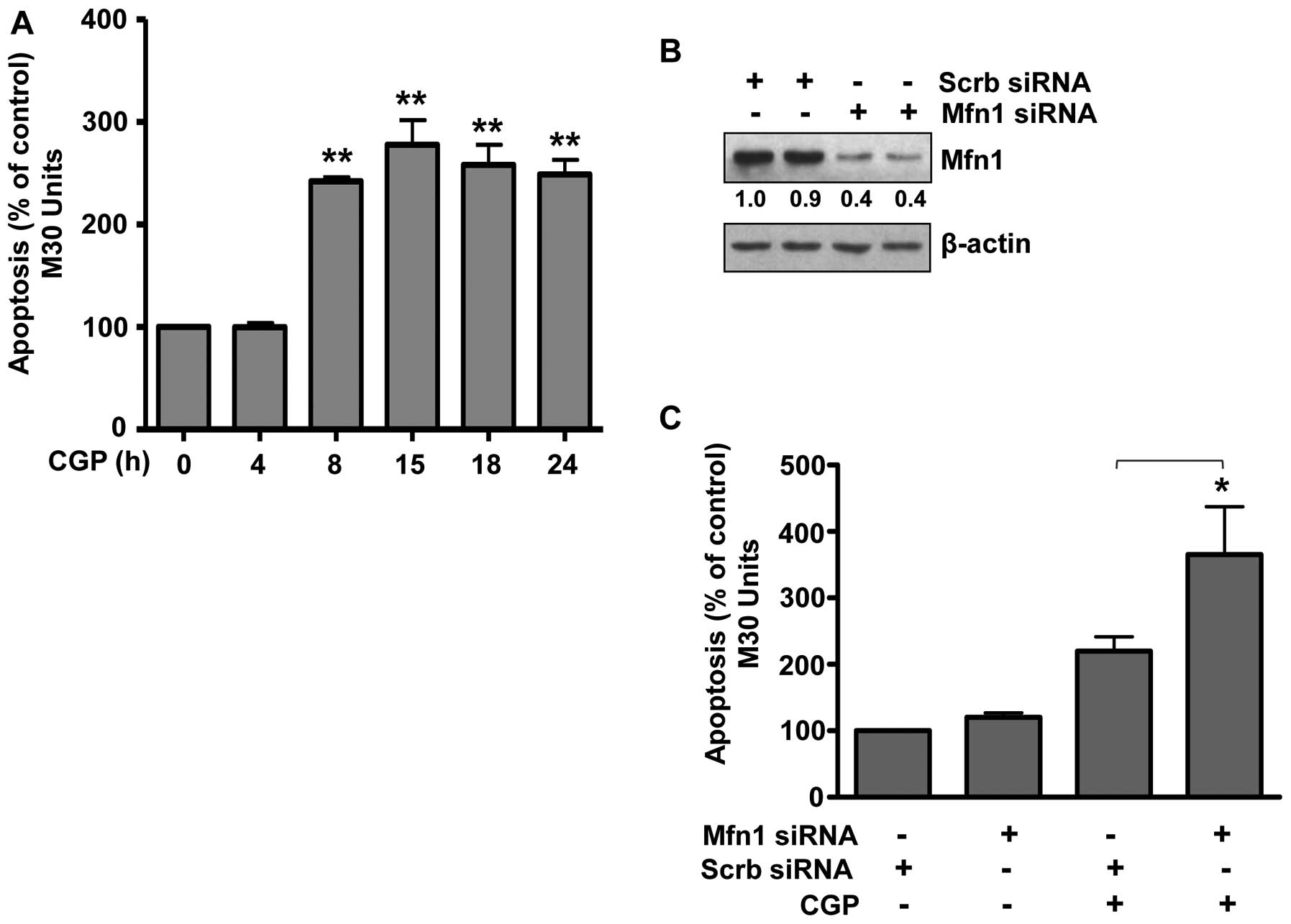
Degradation of Mfn1 sensitizes prostate
cancer LNCaP cells to CGP-induced apoptosis
Earlier, we reported that combining CGP with TRAIL
enhanced the apoptotic response to TRAIL in prostate cancer cells
(14,33). In these previous studies cells were
treated with CGP alone or in combination with TRAIL for 4 h;
however, we did not examine the apoptotic effect of CGP alone over
time. Therefore, LNCaP cells were treated with CGP for 4 to 24 h
and apoptosis was measured. Treatment with CGP for 4 h did not
induce apoptosis (Fig. 4A)
confirming our earlier findings (14,33).
However, treatment with CGP for 8 h induced significant apoptosis.
Prolonged treatment up to 24 h did not increase the apoptotic
response, which was similar to that observed after an 8 h treatment
(Fig. 4A). As Mfn1 was degraded
upon CGP treatment within this same time frame (Fig. 2A), we hypothesized that Mfn1
degradation sensitized prostate cancer cells to CGP resulting in
apoptosis. To test this hypothesis, the expression of Mfn1 was
decreased by transfecting cells with Mfn1 siRNA (Fig. 4B) and then also treating with CGP.
Results demonstrated a significant increase in CGP-induced
apoptosis in cells expressing decreased levels of Mfn1 (Fig. 4C), suggesting that low levels of
Mfn1 sensitized prostate cancer cells to CGP-induced apoptosis. In
summary, we have demonstrated that the mitofusin Mfn1 may play a
key role in the response of prostate cancer cells to
apoptosis-inducing agents such as CGP. Treatment with CGP decreased
the levels of Mfn1 through a process involving ubiquitination via
the E3 ligase March5 and proteasomal degradation. When the levels
of Mfn1 were decreased, cells were readily induced to undergo
apoptosis.
Discussion
Mitochondrial dysfunction has been linked to cancer
(1–4), and manipulation of mitochondrial
structure and function is gaining importance in the field of cancer
therapeutics (11,14,34–36).
We reported previously that alteration of mitochondrial function by
CGP significantly increased the apoptotic response to TRAIL in
TRAIL-resistant prostate cancer cells (33) and that CGP can induce mitochondrial
fission in these cells (14). We
have also shown that increased expression of Drp1, a protein that
is known to induce mitochondrial fission and alter the function of
mitochondria, induces apoptosis in these cells (14). Here, we expand our previous
findings and show that reducing the levels of the mitofusin Mfn1, a
protein that is involved in mitochondrial fusion to oppose the
action of Drp1, sensitized prostate cancer cells to apoptotic
agents, such as CGP.
The mitochondrial fusion and fission proteins play a
critical role in the normal functioning of a cell. It is being
increasingly appreciated that a delicate equilibrium is maintained
between these proteins with opposing effects. Any alteration in the
levels of these proteins may result in malfunctioning of the
mitochondria (37,38). Our previous observations and the
data presented here concur with the above hypothesis. It is
interesting to note that the expression of Mfn1 and Drp1
complemented each other. For example, cells expressing higher
levels of Mfn1 (LNCaP and CWR-R1) expressed higher levels of Drp1,
while those expressing lower levels of Mfn1 showed low levels of
Drp1 (Fig. 1). These observations
argue for the likely importance of a balance between mitochondrial
fission and fusion proteins in the survival of prostate cancer
cells.
There are several mechanisms described for
regulation of the mitochondrial fission protein Drp1 (11,39–41).
On the other hand, degradation seems to be an important mechanism
for regulating the function of mitochondrial fusion proteins.
Several studies reported Mfn1 degradation in yeast (31,42),
as well as in mammals (32,43,44).
Our results are consistent with the above observations in that
treatment with CGP resulted in degradation of the mitochondrial
fusion protein, Mfn1. Our results showed that CGP treatment
resulted in ubiquitination of Mfn1 proteins leading to degradation
by proteasomes. Subsequent experiments demonstrated the involvement
of March5, a mitochondrial E3 ubiquitin ligase (Fig. 3C). These results agree with
published data that showed that March5 is required for the
degradation of Mfn1 in other cell types (32). However, we found no change in the
apoptotic response of the cells to CGP when the expression of
March5 was knocked down using siRNA (data not shown), possibly due
to the fact that March5 is also involved in the regulation of the
mitochondrial fission protein Drp1 (45). Thus, decreased degradation and
increased levels of Mfn1 in cells expressing low levels of March5
could potentially be compensated for by elevations in Drp1. This
duality emphasizes the complex interactions that regulate the
response of the cells to apoptosis-promoting and -inhibiting
proteins. The above results clearly emphasize the potential
importance of mitochondrial fusion and fission proteins in devising
therapeutic options. Although it is intuitive to manipulate
proteins such as Drp1 involved in mitochondrial fission leading to
apoptosis, it is equally critical to pay attention to the function
of mitochondrial fusion proteins, such as Mfn1. Here, the
enhancement of the apoptotic response to CGP in cells with reduced
Mfn1 expression suggests that Mfn1 plays a pro-survival role in
prostate cancer cells, although the mechanisms involved in this
function require further investigation. Our results are supported
by the observations that under hypoxic conditions, Mfn1 protects
cancer cells from the apoptotic stimuli, staurosporine and
etoposide (46), and inhibition of
mitochondrial fission protects the heart against
ischemia/reperfusion injury (47).
Thus, manipulating mitochondrial fusion and fission machinery in
prostate cancer cells may provide a new avenue of treatment for the
disease.
Acknowledgements
We thank Dr Alexander van der Bliek,
University of California, for providing us the Myc-Mfn1 plasmid. We
also thank Dr Elizabeth Wilson and Dr Leland Chung for providing us
the CWR-R1 and P69 cell lines, respectively. This research was
funded by VA Merit Awards to M.V.K. and W.B.B. and an award from
the American Legion of Georgia to both M.V.K. and W.B.B. The
contents of this article do not represent the views of the
Department of Veterans Affairs or the United States Government.
References
|
1.
|
Chen G, Wang F, Trachootham D and Huang P:
Preferential killing of cancer cells with mitochondrial dysfunction
by natural compounds. Mitochondrion. 10:614–625. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Frezza C, Pollard PJ and Gottlieb E:
Inborn and acquired metabolic defects in cancer. J Mol Med (Berl).
89:213–220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Kuznetsov AV, Margreiter R, Amberger A,
Saks V and Grimm M: Changes in mitochondrial redox state, membrane
potential and calcium precede mitochondrial dysfunction in
doxorubicin-induced cell death. Biochim Biophys Acta.
1813:1144–1152. 2011. View Article : Google Scholar
|
|
4.
|
Willers IM and Cuezva JM:
Post-transcriptional regulation of the mitochondrial H(+)-ATP
synthase: a key regulator of the metabolic phenotype in cancer.
Biochim Biophys Acta. 1807:543–551. 2010.
|
|
5.
|
McBride HM, Neuspiel M and Wasiak S:
Mitochondria: more than just a powerhouse. Curr Biol. 16:R551–R560.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Bereiter-Hahn J and Voth M: Dynamics of
mitochondria in living cells: shape changes, dislocations, fusion,
and fission of mitochondria. Microsc Res Tech. 27:198–219. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Griparic L and van der Bliek AM: The many
shapes of mitochondrial membranes. Traffic. 2:235–244. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Bossy-Wetzel E, Barsoum MJ, Godzik A,
Schwarzenbacher R and Lipton SA: Mitochondrial fission in
apoptosis, neurodegeneration and aging. Curr Opin Cell Biol.
15:706–716. 2003. View Article : Google Scholar
|
|
9.
|
Skulachev VP: Mitochondrial filaments and
clusters as intra-cellular power-transmitting cables. Trends
Biochem Sci. 26:23–29. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Brooks C, Wei Q, Cho SG and Dong Z:
Regulation of mitochondrial dynamics in acute kidney injury in cell
culture and rodent models. J Clin Invest. 119:1275–1285. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Choudhary V, Kaddour-Djebbar I,
Lakshmikanthan V, et al: Novel role of androgens in mitochondrial
fission and apoptosis. Mol Cancer Res. 9:1067–1077. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Frank S, Gaume B, Bergmann-Leitner ES, et
al: The role of dynamin-related protein 1, a mediator of
mitochondrial fission, in apoptosis. Dev Cell. 1:515–525. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Gomez-Lazaro M, Bonekamp NA, Galindo MF,
Jordan J and Schrader M: 6-Hydroxydopamine (6-OHDA) induces
Drp1-dependent mitochondrial fragmentation in SH-SY5Y cells. Free
Radic Biol Med. 44:1960–1969. 2008. View Article : Google Scholar
|
|
14.
|
Kaddour-Djebbar I, Choudhary V, Brooks C,
et al: Specific mitochondrial calcium overload induces
mitochondrial fission in prostate cancer cells. Int J Oncol.
36:1437–1444. 2010.PubMed/NCBI
|
|
15.
|
Gandre-Babbe S and van der Bliek AM: The
novel tail-anchored membrane protein Mff controls mitochondrial and
peroxisomal fission in mammalian cells. Mol Biol Cell.
19:2402–2412. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Loson OC, Song Z, Chen H and Chan DC:
Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in
mitochondrial fission. Mol Biol Cell. 24:659–667. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Otera H, Wang C, Cleland MM, et al: Mff is
an essential factor for mitochondrial recruitment of Drp1 during
mitochondrial fission in mammalian cells. J Cell Biol.
191:1141–1158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Palmer CS, Osellame LD, Laine D,
Koutsopoulos OS, Frazier AE and Ryan MT: MiD49 and MiD51, new
components of the mitochondrial fission machinery. EMBO Rep.
12:565–573. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Zhao J, Liu T, Jin S, et al: Human MIEF1
recruits Drp1 to mitochondrial outer membranes and promotes
mitochondrial fusion rather than fission. EMBO J. 30:2762–2778.
2011. View Article : Google Scholar
|
|
20.
|
Cipolat S, Martins de Brito O, Dal Zilio B
and Scorrano L: OPA1 requires mitofusin 1 to promote mitochondrial
fusion. Proc Natl Acad Sci USA. 101:15927–15932. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Song Z, Ghochani M, McCaffery JM, Frey TG
and Chan DC: Mitofusins and OPA1 mediate sequential steps in
mitochondrial membrane fusion. Mol Biol Cell. 20:3525–3532. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Ishihara N, Eura Y and Mihara K: Mitofusin
1 and 2 play distinct roles in mitochondrial fusion reactions via
GTPase activity. J Cell Sci. 117:6535–6546. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Rojo M, Legros F, Chateau D and Lombes A:
Membrane topology and mitochondrial targeting of mitofusins,
ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J
Cell Sci. 115:1663–1674. 2002.PubMed/NCBI
|
|
24.
|
Santel A, Frank S, Gaume B, Herrler M,
Youle RJ and Fuller MT: Mitofusin-1 protein is a generally
expressed mediator of mitochondrial fusion in mammalian cells. J
Cell Sci. 116:2763–2774. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Santel A and Fuller MT: Control of
mitochondrial morphology by a human mitofusin. J Cell Sci.
114:867–874. 2001.PubMed/NCBI
|
|
26.
|
Eura Y, Ishihara N, Yokota S and Mihara K:
Two mitofusin proteins, mammalian homologues of FZO, with distinct
functions are both required for mitochondrial fusion. J Biochem.
134:333–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Honda S, Aihara T, Hontani M, Okubo K and
Hirose S: Mutational analysis of action of mitochondrial fusion
factor mitofusin-2. J Cell Sci. 118:3153–3161. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Yuan H, Gerencser AA, Liot G, et al:
Mitochondrial fission is an upstream and required event for bax
foci formation in response to nitric oxide in cortical neurons.
Cell Death Differ. 14:462–471. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Barsoum MJ, Yuan H, Gerencser AA, et al:
Nitric oxide-induced mitochondrial fission is regulated by
dynamin-related GTPases in neurons. EMBO J. 25:3900–3911. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Cohen MM, Amiott EA, Day AR, et al:
Sequential requirements for the GTPase domain of the mitofusin Fzo1
and the ubiquitin ligase SCF Mdm30 in mitochondrial outer membrane
fusion. J Cell Sci. 124:1403–1410. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Cohen MM, Leboucher GP, Livnat-Levanon N,
Glickman MH and Weissman AM: Ubiquitin-proteasome-dependent
degradation of a mitofusin, a critical regulator of mitochondrial
fusion. Mol Biol Cell. 19:2457–2464. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Park YY, Lee S, Karbowski M, Neutzner A,
Youle RJ and Cho H: Loss of MARCH5 mitochondrial E3 ubiquitin
ligase induces cellular senescence through dynamin-related protein
1 and mitofusin 1. J Cell Sci. 123:619–626. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Kaddour-Djebbar I, Lakshmikanthan V,
Shirley RB, Ma Y, Lewis RW and Kumar MV: Therapeutic advantage of
combining calcium channel blockers and TRAIL in prostate cancer.
Mol Cancer Ther. 5:1958–1966. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Jiang S, Zu Y, Wang Z, Zhang Y and Fu Y:
Involvement of mitochondrial permeability transition pore opening
in 7-xylosyl-10-deacetylpaclitaxel-induced apoptosis. Planta Med.
77:1005–1012. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Karbowski M and Youle RJ: Dynamics of
mitochondrial morphology in healthy cells and during apoptosis.
Cell Death Differ. 10:870–880. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Yingkun N, Lvsong Z and Huimin Y: Shikonin
inhibits the proliferation and induces the apoptosis of human HepG2
cells. Can J Physiol Pharmacol. 88:1138–1146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Knott AB and Bossy-Wetzel E: Impairing the
mitochondrial fission and fusion balance: a new mechanism of
neurodegeneration. Ann NY Acad Sci. 1147:283–292. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Wasilewski M and Scorrano L: The changing
shape of mitochondrial apoptosis. Trends Endocrinol Metab.
20:287–294. 2009. View Article : Google Scholar
|
|
39.
|
Chang CR and Blackstone C: Cyclic
AMP-dependent protein kinase phosphorylation of Drp1 regulates its
GTPase activity and mitochondrial morphology. J Biol Chem.
282:21583–21587. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Cribbs JT and Strack S: Reversible
phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and
calcineurin regulates mitochondrial fission and cell death. EMBO
Rep. 8:939–944. 2007. View Article : Google Scholar
|
|
41.
|
Taguchi N, Ishihara N, Jofuku A, Oka T and
Mihara K: Mitotic phosphorylation of dynamin-related GTPase Drp1
participates in mitochondrial fission. J Biol Chem.
282:11521–11529. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Escobar-Henriques M, Westermann B and
Langer T: Regulation of mitochondrial fusion by the F-box protein
Mdm30 involves proteasome-independent turnover of Fzo1. J Cell
Biol. 173:645–650. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Chan NC, Salazar AM, Pham AH, et al: Broad
activation of the ubiquitin-proteasome system by Parkin is critical
for mitophagy. Hum Mol Genet. 20:1726–1737. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Rakovic A, Grunewald A, Kottwitz J, et al:
Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins
in human fibroblasts. PLoS One. 6:e167462011. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Nakamura N, Kimura Y, Tokuda M, Honda S
and Hirose S: MARCH-V is a novel mitofusin 2- and Drp1-binding
protein able to change mitochondrial morphology. EMBO Rep.
7:1019–1022. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Chiche J, Rouleau M, Gounon P,
Brahimi-Horn MC, Pouyssegur J and Mazure NM: Hypoxic enlarged
mitochondria protect cancer cells from apoptotic stimuli. J Cell
Physiol. 222:648–657. 2010.PubMed/NCBI
|
|
47.
|
Ong SB, Subrayan S, Lim SY, Yellon DM,
Davidson SM and Hausenloy DJ: Inhibiting mitochondrial fission
protects the heart against ischemia/reperfusion injury.
Circulation. 121:2012–2022. 2010. View Article : Google Scholar : PubMed/NCBI
|