Introduction
Human papillomavirus (HPV)-associated cervical
cancer is the third most commonly diagnosed cancer and the fourth
leading cause of cancer deaths in women worldwide (1). While the broad use of Papanicolaou
(Pap) screening and conventional treatment have led to a decline in
mortality from cervical cancer, many women still die of the disease
in developing countries (2). When
detected at an early stage, invasive cervical cancer is one of the
most successfully treated cancers. However, the 5-year relative
survival rate drops to 13–46% if detected at an advanced stage
(3). Invasive cervical cancer is
generally treated with a combination of surgery, radiation, and/or
chemotherapy. Cisplatin-based chemotherapy has traditionally been
reserved for metastatic or recurrent cervical cancer (4).
Cisplatin, a DNA-damaging cancer treatment that
readily induces apoptosis in vitro, is widely used in a
variety of human carcinomas, including head and neck, colorectal,
ovarian, cervical, testicular, and small cell lung cancer (5). A major limitation of cisplatin-based
chemotherapy is the development of cellular resistance, which is
implicated in its failure to treat malignant tumors (6,7). The
mechanisms of cisplatin resistance have been identified as a
reduction in drug uptake, the induction of DNA repair enzymes such
as Bcl-2, the enhancement of anti-oxidant activity with
glutathione, and the inhibition of caspase activity (7–9).
Based on the accumulated evidence, various experimental and
clinical trials are under investigation to overcome these
mechanisms of drug resistance. New cisplatin-based combination
therapies are necessary to improve outcomes in advanced cervical
cancer.
CM1 has been identified as a pro-apoptosis molecule
on B-cell lymphoma cells. It was first identified by a monoclonal
antibody developed against concanavalin-A stimulated mono-nuclear
cells in the peripheral blood. CM1 molecules are distributed across
germinal centers in the human tonsil and expressed on activated T
and B lymphocytes (10). It has
previously been reported that cross-linking CM1 with anti-CM1
antibody induces apoptosis in Burkitt’s lymphoma cells,
EBV-transformed B cells, and lung cancer cells. These effects are
primarily mediated by activation of the caspase cascade and
generation of reactive oxygen species (ROS) (11–13).
These observations encouraged us to evaluate whether HeLa cervical
cancer cells express CM1, which we found to be the case after
exposure to cisplatin. Therefore, this study aimed to investigate
the detailed signal mechanism of CM1-mediated apoptosis in
cisplatin-exposed HeLa cells.
Materials and methods
Cells and reagents
Cells from the HeLa cervical cancer cell line were
obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA). Cells were grown and maintained in RPMI-1640 medium
(Hyclone, Logan, UT, USA) containing 2 mM L-gultamine, 100 U/ml
penicillin, 100 μg/ml streptomycin and 10% heat-inactivated
fetal bovine serum (Hyclone) at 37°C in a 5% CO2
incubator. Anti-CM1 monoclonal antibody was generated using a
murine hybridoma cell line-secreting antibody, as previously
described (10). Cisplatin,
N-acetylcysteine (NAC), and phorbol myristate acetate (PMA) were
purchased from Sigma-Aldrich (St. Louis, MO, USA); 3,
3′-dihexyloxacarbocyanine iodide (DiOC6) and
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) were purchased
from Molecular Probes (Eugene, OR, USA). Fluorescein isothiocyanate
(FITC)-conjugated anti-Fas and anti-Fas ligand monoclonal
antibodies were purchased from BD Bioscience (San Jose, CA, USA).
Rabbit anti-mouse IgG antibody, secondary antibody, and MOPC21
isotype control antibody were purchased from Sigma-Aldrich. Mouse
anti-human apoptosis inducing factor (AIF) antibody and goat
anti-human endonuclease G (EndoG) antibody were purchased from
Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Z-VAD-fmk, a
pan-caspase inhibitor, was purchased from Calbiochem (La Jolla, CA,
USA).
Cell proliferation assay
HeLa cells were pre-incubated with each
concentration of cisplatin (0–100 μM) for 48 h, then
cultured in media containing 10% FBS in a 96-well flat-bottom plate
(5×104 cells/well). Cell proliferation was estimated
using an AlamarBlue assay kit (Serotec, Raleigh, NC, USA) as
previously described (13). To
analyze the effect of CM1 ligation on cell proliferation,
cisplatin-exposed HeLa cells were cross-linked with immobilized
anti-CM1 antibody (IgG1κ, 10 μg/ml) and MOPC21
(IgG1κ; Sigma-Aldrich) as isotype control for 40 min.
Next, cells were cultured in media containing 10% FBS at 37°C for
48 h in a 96-well flat-bottom plate (2×105 cells/well).
To immobilize the anti-CM1 and MOPC21 antibodies, 10 μg/ml
of each antibody in phosphate-buffered saline (PBS) were applied to
a 96-well culture plate (0.1 ml/well; wells were washed with PBS
before use) after overnight incubation at 4°C. Cell proliferation
was estimated using an AlamarBlue assay kit (Serotec). Briefly,
AlamarBlue was added (10% by total volume) to each well, and the
relative fluorescence value was tested 7 h later using a
fluorometer (Synergy HT, Bio-Tek Instruments Inc., Winnoski, VT;
excitation, 530 nm; emission, 590 nm).
Analysis of CM1 expression with flow
cytometry and confocal microscopy
Flow cytometry was employed to evaluate the
expression of surface CM1 molecules. First, HeLa cells were
incubated with or without cisplatin at various concentrations (0,
10, 20, 50 and 100 μM). Cells were washed twice with
ice-cold PBS, then incubated with FITC-conjugated mouse anti-human
CM1 antibodies (mouse IgG1) for 30 min on ice, followed
by 2 more washes with ice-cold PBS. To detect intracellular
molecules, HeLa cells were treated with a permeabilization buffer
(0.1% saponin in PBS) before staining with FITC-conjugated mouse
anti-human CM1 antibodies. All samples were assessed with a
FACSCalibur flow cytometer (BD Bioscience) and data were processed
by the program CellQuest (BD Bioscience). To confirm CM1 expression
with confocal microscopy, HeLa cells were pre-incubated with 20
μM cisplatin, with 20 ng/ml PMA, or with nothing for 48 h.
They were then incubated with FITC-conjugated anti-CM1 antibody.
Fluorescence-stained cells were examined by confocal laser-scanning
microscopy (510 META, Carl Zeiss, Jena, Germany) at x400
magnification, and images were acquired with Confocal Microscopy
Software Release 3.0 (510 META, Carl Zeiss).
Detection of CM1-mediated apoptosis
For immobilization, anti-CM1 and MOPC21 antibodies
were incubated overnight at 4°C on a 96-well culture plate (10
μg/ml, 250 μl, 2.5 μg/well). To evaluate
apoptosis by CM1 cross-linking, HeLa cells were treated with
cisplatin (20 μM) for 48 h and then 5×105
cells/well were cross-linked with immobilized anti-CM1 antibody or
MOPC21 isotype control antibody (2.5 μg/well). Next, cells
were incubated at 37°C for the indicated amount of time. Following
treatment, cells were washed twice with cold PBS then analyzed for
Annexin V expression by flow cytometry as previously described
(13).
Measurement of mitochondrial membrane
potential and ROS generation
To measure ROS levels and mitochondrial membrane
potentials (Δψ), cisplatin-exposed HeLa cells were cultured with
DCFH-DA at 37°C for 30 min or DiOC6 at 37°C for 15 min
as molecular probes. Cells were further incubated with anti-CM1
antibody as described above. Cells were harvested at the indicated
time and their ROS levels and Δψ were determined by an FACSCalibur
flow cytometer (BD Bioscience).
Apoptosis-blocking experiments
To investigate the effects of caspase inhibitors and
ROS on CM1-mediated apoptosis, cisplatin-exposed HeLa cells as
described above were pre-treated with z-VAD-fmk (20 μM in
DMSO, a broad-spectrum caspase inhibitor) or NAC for 2 h before
antibody stimulation. Cells were further incubated with anti-CM1
antibody as described above. The apoptosis-blocking effects of
z-VAD-fmk and NAC were detected using Annexin V, DCFH-DA and
DiOC6 staining as described above. To block Fas-FasL
interaction, antagonistic anti-Fas antibody ZB4 (0.5 mg/ml) was
added 2 h before treatment with anti-CM1 antibody. ZB4 was removed
from cell cultures before stimulation with anti-CM1 antibody.
Apoptosis was determined by flow cytometry after staining with
Annexin V.
Confocal microscopy to detect
apoptosis-related intracellular molecules
To detect intracellular apoptosis-related molecules,
cisplatin-exposed HeLa cells were incubated with anti-CM1 antibody
as described above. To detect the blocking effects of z-VAD-fmk nd
NAC, cells were pre-treated with each substance for 2 h before
antibody stimulation. Cells were incubated with primary antibodies
against cytochrome c (mouse IgG2b) or AIF (mouse IgG2b) and
then incubated with FITC-conjugated goat anti-mouse IgG for 30 min.
Nuclei were stained with PI for 10 min at room temperature. After
being washed 3 times with PBS, the fluorescence-stained cells were
examined under a confocal laser-scanning microscope (Carl Zeiss,
510 META) at x400 original magnification using Confocal Microscopy
Software Release 3.0 (Carl Zeiss, 510 META).
Reverse-transcription polymerase chain
reaction
Total RNA was isolated using an RNeasy Mini kit
(Qiagen, Hilden, Germany). RNA was transcribed into cDNA using
oligo (dT) primers (Bioneer, Daejeon, Korea) and reverse
transcriptase. Polymerase chain reaction (PCR) amplification was
performed using specific primer sets (Bioneer) for the Fas ligand
(upstream primer, 5′-GGT CCA TGC CTC TGG AAT GG; downstream primer,
5′-CAC ATC TGC CCA GTA GTG CA, 250-bp product), Bcl-2 (upstream
primer, 5′-GGA TTG TGG CCT TCT TTG AG; downstream primer, 5′-CAG
CCA GGA GAA ATC AAA CAT, 209-bp product), and Bad (upstream primer,
5′-CGA GTG AGC AGG AAG ACT CC; downstream primer, 5′-CTG TGC TGC
CCA GAG GTT, 299-bp product). For the control group, a specific
primer set for β-actin was used (upstream primer, 5′-ATC CAC GAA
ACT ACC TTC AA; downstream primer, 5′-ATC CAC ACG GAG TAC TTG C),
which yielded a 200-bp product. PCR (25 cycles; 20 sec at 94°C, 10
sec at 60°C, and 30 sec at 72°C) was performed using Prime Taq
Premix (GeNet Bio, Chungnam, Korea). The PCR products were
visualized on 2.5% agarose gels with ethidium bromide.
Western blot analysis for
apoptosis-related proteins
Cisplatin-exposed HeLa cells were incubated with
anti-CM1 antibody (10 μg/ml) as described above. Cells were
harvested and washed twice with PBS. Cells were lysed in RIPA
buffer (Elpis Biotech, Daejeon, Korea) containing a protease
inhibitor cocktail (AEBSF, aprotinin, bestatin hydrochloride, E-64,
EDTA, and leupeptin hemisulfate salt; Sigma-Aldrich). For western
blot analysis, an equal amount of protein (40 μg) was
separated by electrophoresis on SDS-polyacrylamide gels and
transferred to polyvinylidene difluoride membranes (Amersham
Biosciences, Braunschweig, Germany) by electroblotting.
Subsequently, membranes were incubated overnight at 4°C in a
solution of PBS supplemented with 5% non-fat dry milk. Blots were
probed with the desired antibodies [caspase-8, caspase-3, β-actin,
Bid, Bcl-2, phospho-ERK, ERK and poly-ADP-ribose polymerase (PARP)]
for 1 h, incubated with diluted enzyme-linked secondary antibody,
and then visualized by enhanced chemiluminescence as instructed by
the manufacturer (Amersham Bioscience). Protein loading equivalence
was assessed by the expression of β-actin. Each experiment was
repeated at least in triplicate.
Results
Ligation of CM1 with anti-CM1 antibody
causes apoptosis in cisplatin-exposed HeLa cells
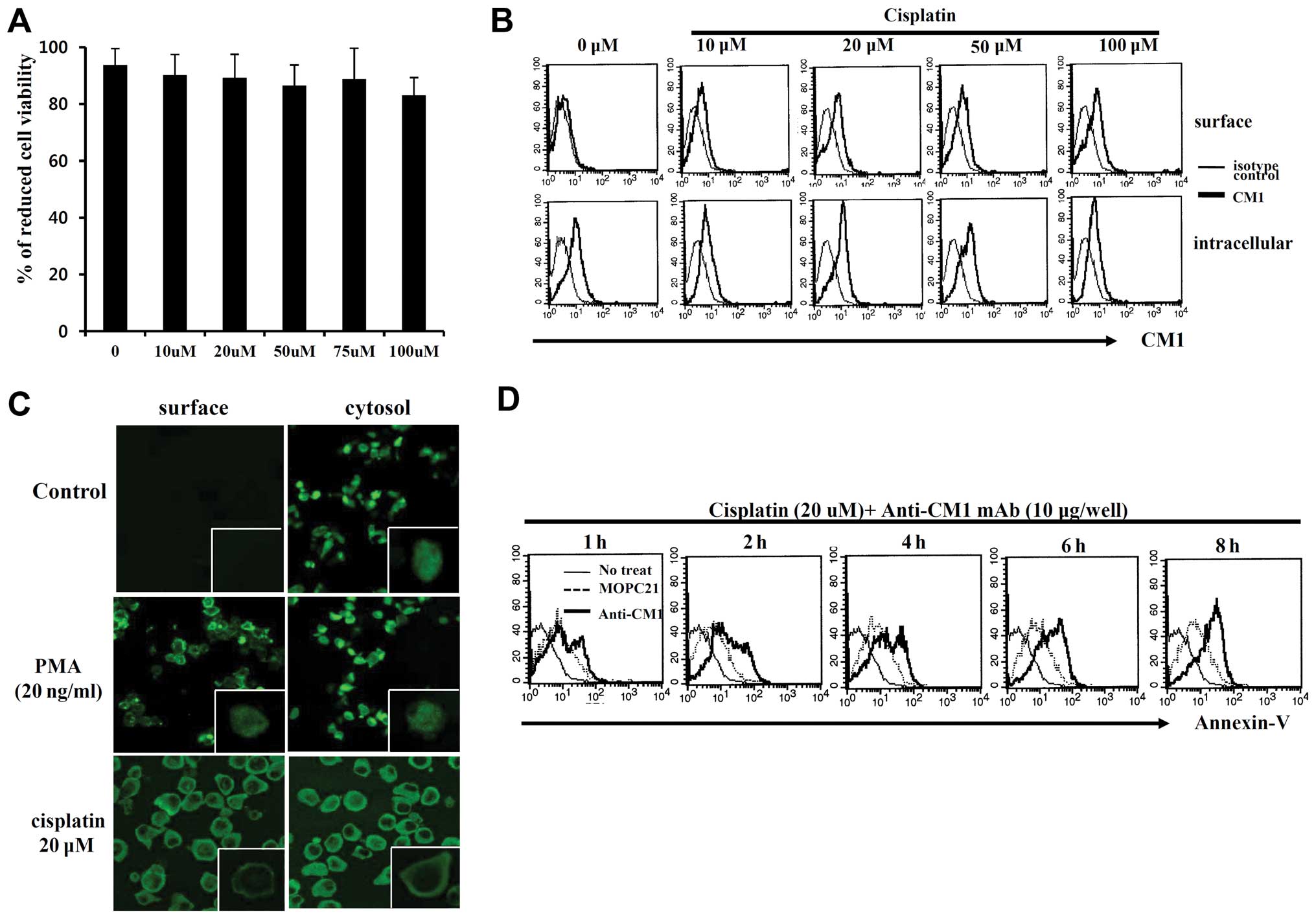
Cell viability of HeLa cells treated with cisplatin
was not significantly reduced compared to the control group based
on Alamar blue assay (Fig. 1A).
Although CM1 is present in the cytoplasm of HeLa cells, it is not
present on the cell surface until after exposure to cisplatin
(Fig. 1B), as evidenced by
confocal microscopic data (Fig.
1C). To determine the function of the surface CM1 molecule
induced by cisplatin, HeLa cells were cross-linked with immobilized
anti-CM1 antibody (10 μg/ml) or MOPC21 isotype control
antibody. Stimulation of surface CM1 with anti-CM1 antibody
increased the quantity of Annexin V-positive apoptotic cells in a
time-dependent manner (Fig. 1D).
These results suggest that surface CM1 promotes apoptosis in HeLa
cells.
Cross-linking of CM1 induces apoptosis by
ROS generation and caspase activation in cisplatin-exposed HeLa
cells
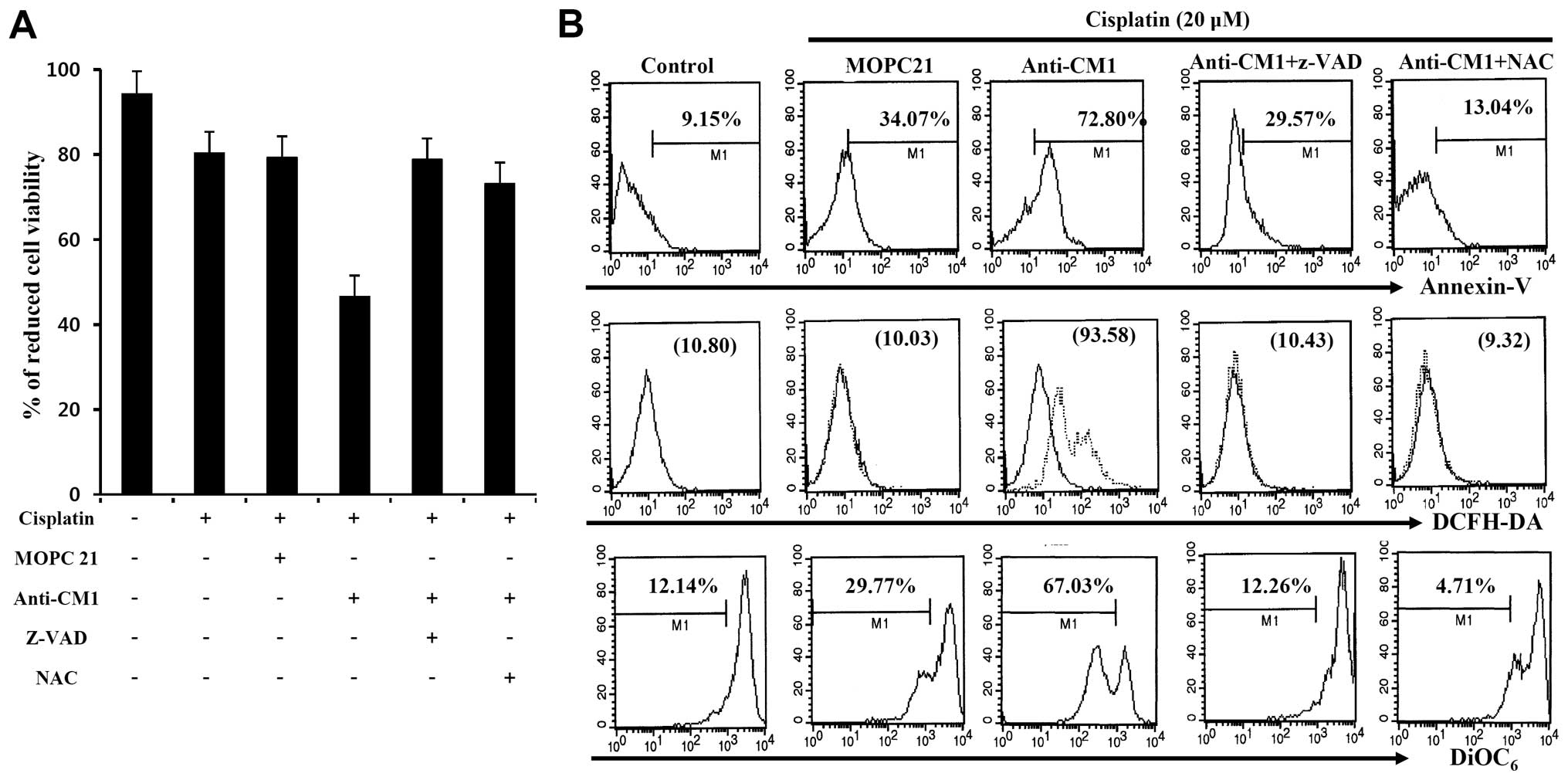
We examined whether CM1-mediated apoptosis was
related to ROS production or caspase activity following
cross-linking with anti-CM1 antibody. Cisplatin-exposed (20
μM) HeLa cells were pre-incubated with Z-VAD-fmk, a
pan-caspase inhibitor, or NAC, a ROS inhibitor for 2 h before
stimulation with anti-CM1 antibody. Cross-linking CM1 with
immobilized anti-CM1 antibody inhibited cell proliferation when
compared to cells stimulated with isotype antibody. We found that
both Z-VAD-fmk and NAC pretreatment reversed the inhibition caused
by anti-CM1 antibody stimulation (Fig.
2A). Furthermore, Z-VAD-fmk and NAC pretreatment also blocked
CM1-mediated apoptosis in cisplatin-exposed HeLa cells (Fig. 2B). These results suggest that
CM1-mediated apoptosis is associated with ROS generation and
caspase activity in cisplatin-exposed HeLa cells.
CM1 stimulation on cisplatin-exposed HeLa
cells results in mitochondrial release of AIF and EndoG
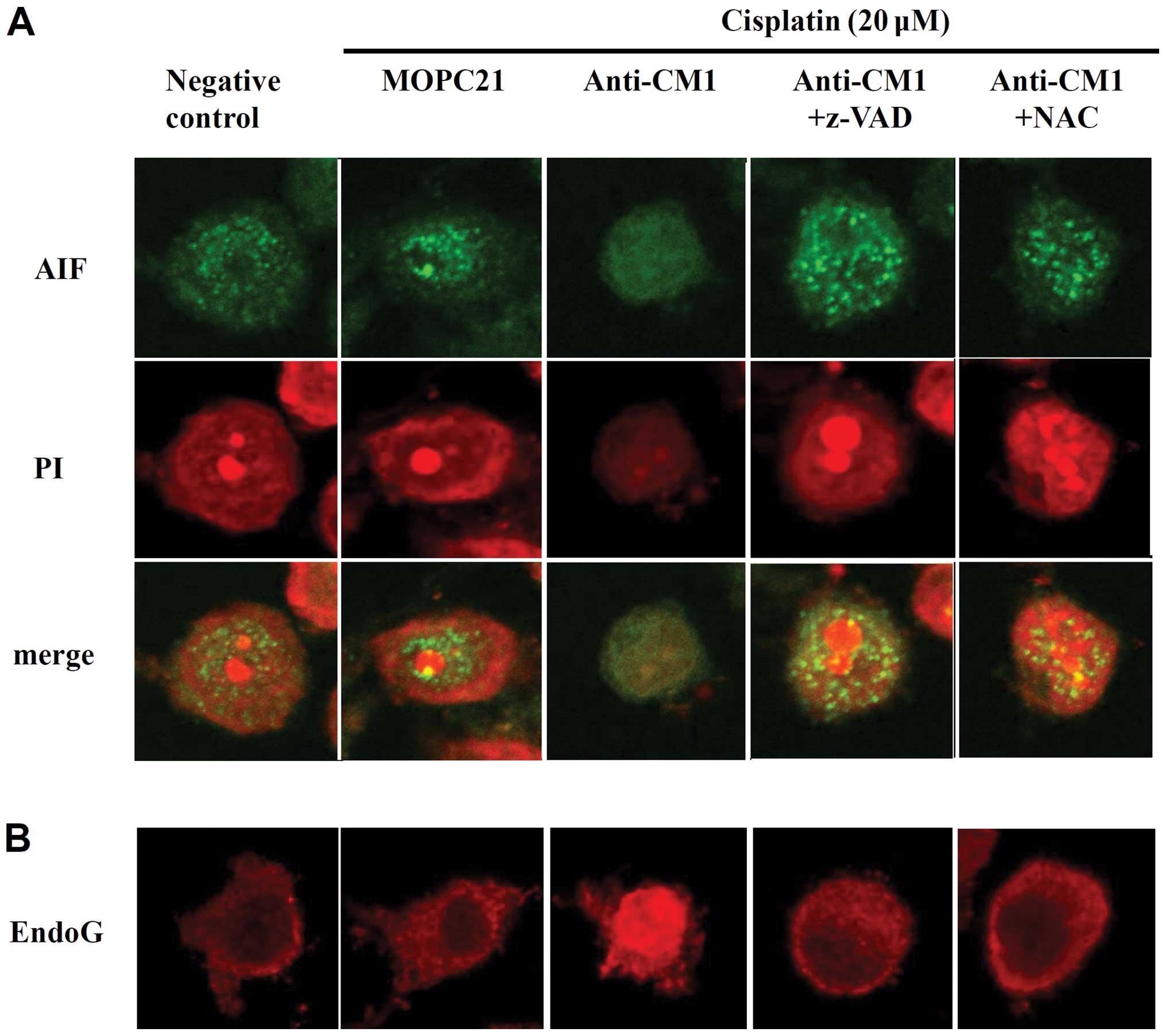
After observing that CM1 ligation disrupted
mitochondrial membrane potentials, we used confocal microscopy to
measure changes in the expression of apoptosis-inducing factor
(AIF) and EndoG, which are stored in the mitochondria.
Cross-linking CM1 with anti-CM1 antibody resulted in a significant
release of AIF and EndoG from the mitochondria into the cytoplasm.
Again, treatment with the pan-caspase inhibitor z-VAD-fmk and the
ROS quencher NAC prevented this translocation of both AIF and EndoG
(Fig. 3). These results suggest
that CM1-mediated apoptosis in cisplatin-exposed HeLa cells
involves the mitochondria.
Cross-linking of CM1 induces Fas-mediated
apoptosis in cisplatin-exposed HeLa cells
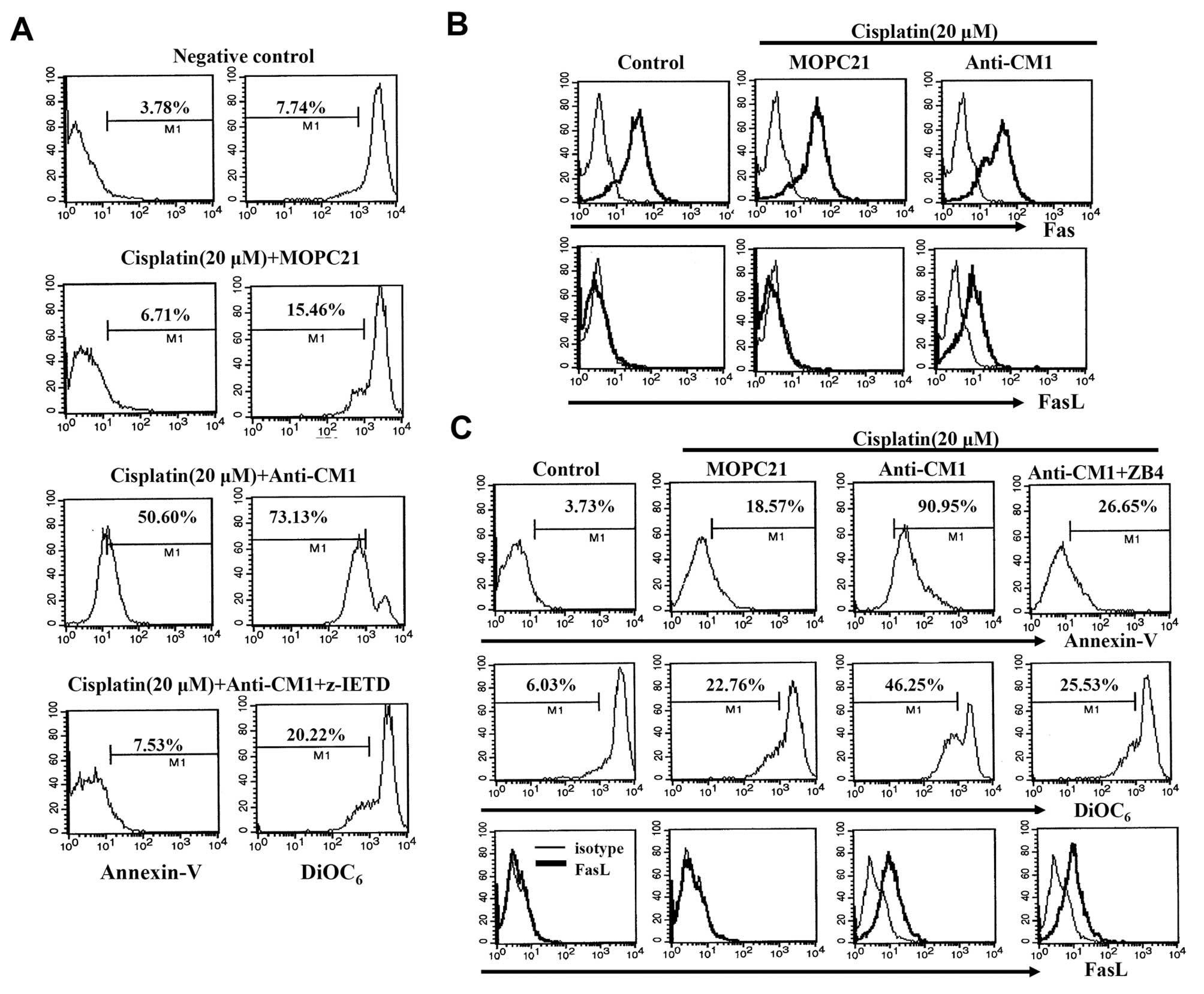
It is well-known that the death receptor and its
ligand can initiate apoptosis. As described above, we observed that
Z-VAD-fmk pretreatment prevents CM1-mediated apoptosis in
cisplatin-exposed HeLa cells. In addition, Z-IETD-fmk, a caspase-8
specific inhibitor, also blocks CM1-mediated apoptosis in
cisplatin-exposed HeLa cells (Fig.
4A). We also investigated whether the Fas/FasL interaction was
involved in CM1-mediated apoptosis in cisplatin-exposed HeLa cells.
HeLa cells expressed the Fas molecule constitutively, but did not
express FasL. Although Fas expression was not affected by CM1
cross-linking, FasL expression was dramatically increased (Fig. 4B). We also foud that pre-treatment
with ZB4, an antagonistic anti-Fas antibody, for 2 h blocked
CM1-mediated apoptosis, although FasL expression was unchanged
(Fig. 4C). These results suggest
that CM1 ligation on cisplatin-exposed HeLa cells affects FasL
expression, and that the Fas/FasL interaction is closely involved
in CM1-mediated apoptosis.
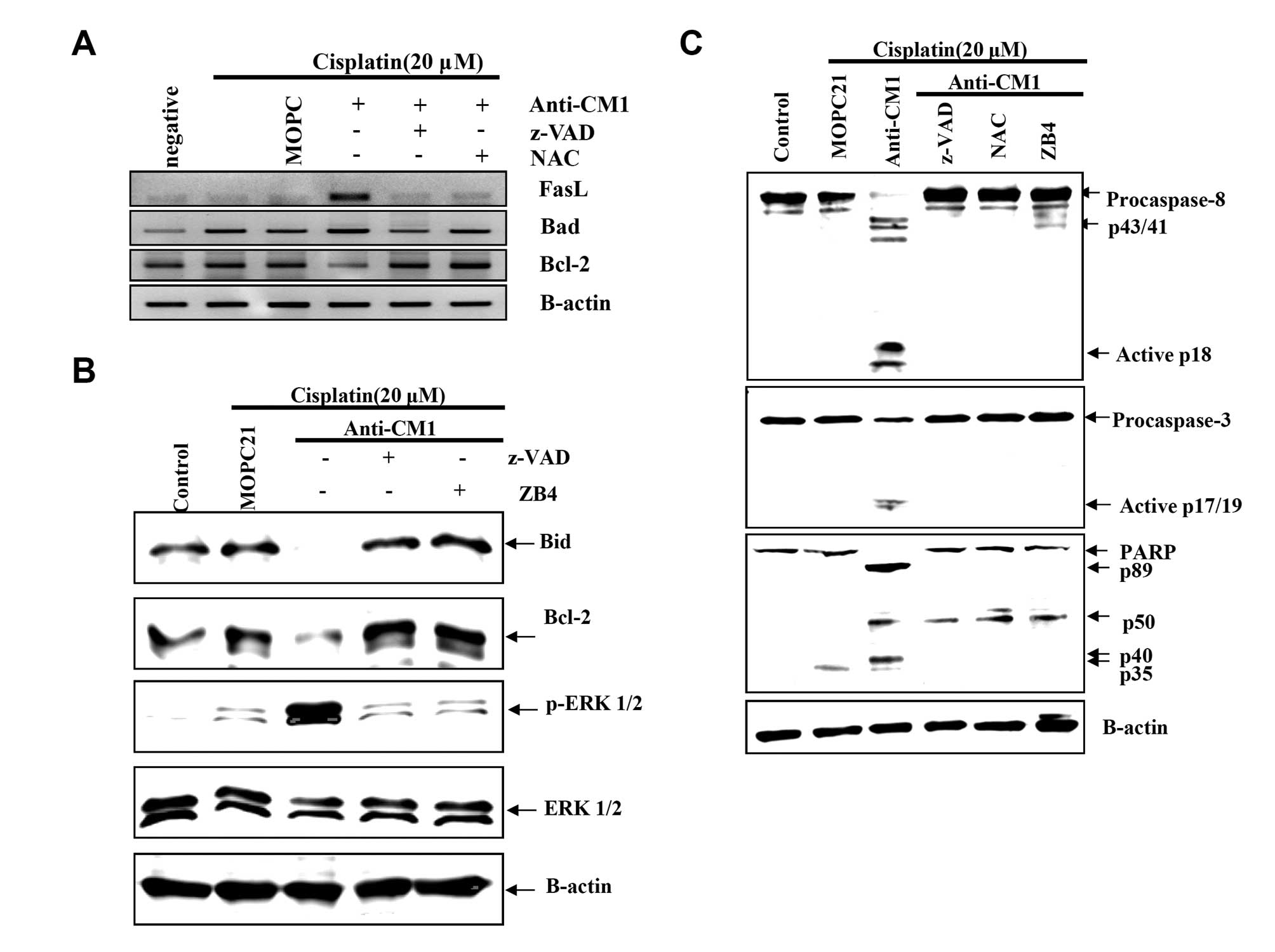
CM1 ligation induces changes in
apoptosis-related genes and activation of ERK and caspase in
cisplatin-exposed HeLa cells
To investigate the detailed signaling mechanism of
CM1-mediated apoptosis in cisplatin-exposed HeLa cells, we examined
the expression of genes related to mitochondrial membrane
disruption using RT-PCR. After CM1 ligation, FasL mRNA expression
significantly increased, Bad (a proapoptotic gene) mRNA expression
slightly increased, and Bcl-2 mRNA expression significantly
decreased (Fig. 5A). Moreover,
expression of Bcl-2 and Bid proteins, both anti-apoptotic proteins,
also decreased significantly after CM1 cross-linking (Fig. 5B). Both z-VAD-fmk and NAC inhibited
these changes in mRNA and protein expression.
Next, we examined the activation of ERK1/2, a
mitogen-activated protein kinase (MAPK), which regulates
apoptosis-related genes. CM1 ligation induced the phosphorylation
of ERK1/2 (Fig. 5B), as well as
the activation of caspase-8 and -3, and the cleavage of PARP
(Fig. 5C). Pretreatment with
z-VAD-fmk, NAC, and ZB4 blocked these changes in apoptosis-related
proteins caused by CM1 ligation in cisplatin-exposed HeLa cells
(Fig. 5B and C). These results
suggest that the CM1-mediated ROS generation leads to the
expression of FasL to generate the apoptosis signal through
cleavage of procaspase-8.
Discussion
Cisplatin is conventionally used as chemotherapy for
various cancers (9) and is an
important first-line drug in advanced cervical cancer. Despite a
consistent rate of initial response, cisplatin treatment often
results in the development of chemoresistance, which ultimately
leads to therapeutic failure (14). Recently, several studies reported
that impaired apoptosis involving the caspase cascade and other
apoptotic proteins lead to resistance to cisplatin in ovarian and
uterine cancers (15–17). Many studies have reported that
combination chemotherapy improves outcomes compared to cisplatin
monotherapy (15). In recent
years, a new paradigm of cancer therapeutics is emerging involving
molecularly-targeted therapeutics rather than conventional
cytotoxic drugs (18,19). Given the high rate of cisplatin
failure in advanced cervical cancer, new anticancer target
molecules are urgently needed. Our results suggest that the
cross-linking of CM1 with anti-CM1 antibody on cisplatin-exposed
HeLa cells represents a novel potential combinational therapy
capable of regulating cancer cell death.
It has previously been reported that CM1
cross-linking increases ROS generation and mitochondrial membrane
disruption (12). It has also been
reported that pretreatment with the pan-caspase inhibitor z-VAD and
the ROS quencher, NAC, inhibits CM1-mediated apoptosis in Burkitt’s
lymphoma cells, EBV-transformed B cells, and lung cancer cells
(12,13). Cisplatin-induced cytotoxicity is
associated with increased intracellular ROS accumulation and
apoptosis mediated by proteolytically activated caspase-3 and -9.
Several studies have found that ROS generation and therefore
apoptosis can be inhibited by upregulating the expression of
anti-oxidant proteins and by NAC exposure (20–22).
Taken together with this prior research, our results suggest that
ligation of CM1 with anti-CM1 antibody generates additional ROS,
overcoming the upregulation of anti-oxidants implicated in the
rescue mechanism against cancer cell death in cisplatin-exposed
HeLa cells.
It has been reported that cisplatin-induced
apoptosis is mediated through the Fas/FasL signal transduction
pathway in many solid tumors and hematologic malignancies (23–25).
Several studies have reported that chemoresistance is correlated
with decreased Fas expression (26–28).
HeLa cells constitutively express Fas, but not FasL. We found that
CM1 cross-linking on cisplatin-exposed HeLa cells significantly
increased FasL expression on both protein and RNA levels. In
addition, our results show that CM1-mediated apoptosis was blocked
after treatment with ZB4, an antagonistic anti-Fas antibody.
The extrinsic apoptosis pathway is triggered by the
binding of tumor necrosis factor (TNF)-family death ligands to
their appropriate cell surface receptors, followed by caspase-8
cleavage. The intrinsic apoptosis pathway involves dysregulation of
mitochondrial pro- and anti-apoptosis proteins; caspase-8 and Bid,
a pro-apoptosis BH3 family member, are then cleaved by the death
ligands. Activated caspase-8 and truncated Bid are correlated with
the production of high levels of anti-apoptosis gene products and
mitochondrial membrane disruption (29). Similarly, our results showed that
CM1 cross-linking disrupts the mitochondrial membrane potential and
dramatically increased the release of apoptotic molecules from the
mitochondria, such as AIF and EndoG, while decreasing Bcl-2 levels.
Pretreatment with Z-VAD-fmk, NAC, or ZB4 completely inhibited
apoptosis by blocking the activation of caspase-8, -3, and PARP
cleavage, as well as normalizing levels of Bcl-2 and Bid proteins,
in cisplatin-exposed HeLa cells. These results suggest that
CM1-stimulated FasL upregulation generates the apoptosis signal via
procaspase-8 cleavage and mitochondrial membrane disruption in
cisplatin-exposed HeLa cells.
The MAPK signaling cascade plays a pivotal role in
regulating cell growth and survival (23). Several studies have reported that
cisplatin-induced ERK activation is associated with increased
resistance to its cytotoxicity in cancer cells (30,31).
Our results showed that CM1 ligation induced the phosphorylation of
ERK1/2 in cisplatin-exposed HeLa cells. Treatment of z-VAD-fmk and
NAC effectively blocked ERK1/2 phosphorylation and other changes in
apoptosis-related proteins. Although the correlation between
Fas-Fas ligand upregulation and ERK1/2 phosphorylation remains
controversial, our results are consistent with a previous report
that CM1 ligation induces apoptosis and ERK1/2 phosphorylation in
A549 lung cancer cells (13). Some
studies have shown that Fas and Fas ligand proteins can be
upregulated via p38 MARK/ERK activation (32,33).
It has been also reported that ROS induces caspase activity and JNK
phosphorylation, as well as participates in ERK1/2 signaling
(34,35).
Based on these results, we provide a more detailed
understanding of the functions of the CM1 molecule in cervical
cancer cells. We propose combination chemotherapy with cisplatin
and anti-CM1 antibody as a new therapeutic strategy to overcome
chemoresistance in human cervical cancer.
Acknowledgements
This study was supported by the Korea
Research Foundation Grant funded by the Korean Government
(313-2008-2-E00023) and a grant from the Korea Healthcare
Technology R&D Project of the Ministry of Health and Welfare
Affairs, Republic of Korea (grant no. HI12C0005).
References
|
1.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2.
|
ZurHausen H: Papillomaviruses and cancer:
from basic studies to clinical application. Nat Rev Cancer.
2:342–350. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Sankaranarayanan R, Swaminathan R, Brenner
H, Chen K, Chia KS, Chen JG, Law SC, Ahn YO, Xiang YB, Yeole BB,
Shin HR, Shanta V, Woo ZH, Martin N, Sumitsawan Y, Sriplung H,
Barboza AO, Eser S, Nene BM, Suwanrungruang K, Jayalekshmi P,
Dikshit R, Wabinga H, Esteban DB, Laudico A, Bhurgri Y, Bah E and
Al-Hamdan N: Cancer sur vival in Africa, Asia, and Central America:
a population-based study. Lancet Oncol. 11:165–173. 2010.
View Article : Google Scholar
|
|
4.
|
Al-Mansour Z and Verschraegen C: Locally
advanced cervical cancer: what is the standard of care? Curr Opin
Oncol. 22:503–512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Eastman A: Alkylating and platinum-based
agents. Curr Opin Oncol. 2:1109–1114. 1990. View Article : Google Scholar
|
|
6.
|
Brabec V and Kasparkova J: Molecular
aspects of resistance to antitumor platinum drugs. Drug Resist
Updat. 5:147–161. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Djeu JY and Wei S: Clusterin and
chemoresistance. Adv Cancer Res. 105:77–92. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Siddik ZH: Cisplatin: mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Eastman A: Mechanisms of resistance to
cisplatin. Cancer Treat Res. 57:233–249. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Hur DY, Kim S, Kim YI, Min HY, Kim DJ, Lee
DS, Cho D, Hwang YI, Hwang DH, Park SH, Ahn HK, Chang KY, Kim YB
and Lee WJ: CM1, a possible novel activation molecule on
humanlymphocytes. Immunol Lett. 74:95–102. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Kim D, Hur DY, Kim YS, Lee K, Lee Y, Cho
D, Kang JS, Kim YI, Hahm E, Yang Y, Yoon S, Kim S, Lee WB, Park HY,
Kim YB, Hwang YI, Chang KY and Lee WJ: CM1 ligation initiates
apoptosis in a caspase 8-dependent manner in Ramos cells and in a
mitochondria-controlled manner in Raji cells. Hum Immunol.
63:576–587. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kim YS, Park GB, Choi YM, Kwon OS, Song
HK, Kang JS, Kim YI, Lee WJ and Hur DY: Ligation of
centrocyte/centroblast marker 1 on Epstein-Barr virus-transformed B
lymphocytes induces cell death in a reactive oxygen
species-dependent manner. Hum Immunol. 67:795–807. 2006. View Article : Google Scholar
|
|
13.
|
Lee HK, Park GB, Kim YS, Song H, Broaddus
VC and Hur DY: Ligation of CM1 enhances apoptosis of lung cancer
cells through different mechanisms in conformity with EGFR
mutation. Int J Oncol. 42:469–477. 2013.PubMed/NCBI
|
|
14.
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar
|
|
15.
|
Jin KL, Park JY, Noh EJ, Hoe KL, Lee JH,
Kim JH and Nam JH: The effect of combined treatment with cisplatin
and histone deacetylase inhibitors on HeLa cells. J Gynecol Oncol.
21:262–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Yunos NM, Beale P, Yu JQ and Huq F:
Synergism from sequenced combinations of curcumin and
epigallocatechin-3-gallate with cisplatin in the killing of human
ovarian cancer cells. Anticancer Res. 31:1131–1140. 2011.PubMed/NCBI
|
|
17.
|
Zhang Y, Wang C, Wang H, Wang K, Du Y and
Zhang J: Combination of Tetrandrine with cisplatin enhances
cytotoxicity through growth suppression and apoptosis in ovarian
cancer in vitro and in vivo. Cancer Lett. 304:21–32. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Zagouri F, Sergentanis TN, Chrysikos D,
Filipits M and Bartsch R: Molecularly targeted therapies in
cervical cancer. A systematic review. Gynecol Oncol. 126:291–303.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Soonthornthum T, Arias-Pulido H, Joste N,
Lomo L, Muller C, Rutledge T and Verschraegen C: Epidermal growth
factor receptor as a biomarker for cervical cancer. Ann Oncol.
22:2166–2178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Chen J, Adikari M, Pallai R, Parekh HK and
Simpkins H: Dihydrodiol dehydrogenases regulate the generation of
reactive oxygen species and the development of cisplatin resistance
in human ovarian carcinoma cells. Cancer Chemother Pharmacol.
61:979–987. 2008. View Article : Google Scholar
|
|
21.
|
Miyajima A, Nakashima J, Tachibana M,
Nakamura K, Hayakawa M and Murai M: N-acetylcysteine modifies
cis-dichlorodiammineplatinum-induced effects in bladder cancer
cells. Jpn J Cancer Res. 90:565–570. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Pak JH, Choi WH, Lee HM, Joo WD, Kim JH,
Kim YT, Kim YM and Nam JH: Peroxiredoxin 6 overexpression
attenuates cisplatin-induced apoptosis in human ovarian cancer
cells. Cancer Invest. 29:21–28. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Brozovic A, Fritz G, Christmann M,
Zisowsky J, Jaehde U, Osmak M and Kaina B: Long-term activation of
SAPK/JNK, p38 kinase and fas-L expression by cisplatin is
attenuated in human carcinoma cells that acquired drug resistance.
Int J Cancer. 112:974–985. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Etter AL, Bassi I, Germain S, Delaloye JF,
Tschopp J, Sordat B and Dupuis M: The combination of chemotherapy
and intraperitoneal MegaFas Ligand improves treatment of ovarian
carcinoma. Gynecol Oncol. 107:14–21. 2007. View Article : Google Scholar
|
|
25.
|
Hougardy BM, van der Zee AG, van den
Heuvel FA, Timmer T, de Vries EG and de Jong S: Sensitivity to
Fas-mediated apoptosis in high-risk HPV-positive human cervical
cancer cells: relationship with Fas, caspase-8, and Bid. Gynecol
Oncol. 97:353–364. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Baguley BC: Novel strategies for
overcoming multidrug resistance in cancer. BioDrugs. 16:97–103.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Ikuta K, Takemura K, Kihara M, Naito S,
Lee E, Shimizu E and Yamauchi A: Defects in apoptotic signal
transduction in cisplatin-resistant non-small cell lung cancer
cells. Oncol Rep. 13:1229–1234. 2005.PubMed/NCBI
|
|
28.
|
Wu W, Wang HD, Guo W, Yang K, Zhao YP,
Jiang YG and He P: Up-regulation of Fas reverses cisplatin
resistance of human small cell lung cancer cells. J Exp Clin Cancer
Res. 29:492010. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Wang J, Zhou JY and Wu GS: ERK-dependent
MKP-1-mediated cisplatin resistance in human ovarian cancer cells.
Cancer Res. 67:11933–1141. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Wu ZZ, Sun NK, Chien KY and Chao CC:
Silencing of the SNARE protein NAPA sensitizes cancer cells to
cisplatin by inducing ERK1/2 signaling, synoviolin ubiquitination
and p53 accumulation. Biochem Pharmacol. 82:1630–1640. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Liu WH and Chang LS: Piceatannol induces
Fas and FasL up-regulation in human leukemia U937 cells via
Ca2+/p38alpha MAPK-mediated activation of c-Jun and
ATF-2 pathways. Int J Biochem Cell Biol. 42:1498–1506. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Duan SG, Cheng L, Li DJ, Zhu J, Xiong Y,
Li XW and Wang SG: The role of MAPK-ERK pathway in 67-kDa laminin
receptor-induced FasL expression in human cholangiocarcinoma cells.
Dig Dis Sci. 55:2844–2852. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Wang M, Zhang L, Han X, Yang J, Qian J,
Hong S, Samaniego F, Romaguera J and Yi Q: Atiprimod inhibits the
growth of mantle cell lymphoma in vitro and in vivo and induces
apoptosis via activating the mitochondrial pathways. Blood.
109:5455–5462. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Filomeni G, Aquilano K, Rotilio G and
Ciriolo MR: Reactive oxygen species-dependent c-Jun NH2-terminal
kinase/c-Jun signaling cascade mediates neuroblastoma cell death
induced by diallyl disulfide. Cancer Res. 63:5940–5949.
2003.PubMed/NCBI
|