Introduction
Hepatocellular carcinoma (HCC) is an endemic disease
in Asia and majority of patients are not suitable for curative
treatment strategies including surgery, liver transplantation or
local ablative therapy, because their symptoms are often at
advanced stage (1). Sorafenib is
the only effective drug approved by USA Food and Drug
Administration to increase overall survival in patients with
advanced HCC that are not amenable to transarterial
chemoembolization (TACE). However, the survival benefit is far from
satisfactory, and more effective new treatment strategy for
advanced HCC is needed (2).
Vorinostat or suberoylanilide hydroxamic acid (SAHA)
is a histone deacetylase (HDAC) inhibitor, which detaches chromatin
from core histone and activates gene transcription. HDAC is
overexpressed in HCC cell line and tumor tissues of HCC patients
and may be associated with increased invasiveness (3,4).
Among HCC patients who received surgical resection or liver
transplantation, HDAC overexpression is a biomarker for
aggressiveness and also a prognostic factor (5,6).
Inhibition of HDAC by gene knockdown or inhibitor has been shown
with anti-HCC effect in preclinical studies (3,4,7,8).
Poor survival in patients with HCC also has been shown with
overexpression of NF-κB (9,10).
It has been observed that low dose SAHA may potentiate NF-κB
activity (11,12), and may results in HCC progression
through NF-κB-regulated effector proteins, such as cyclin D1,
B-cell lymphoma 2 (BCL-2), X-linked inhibitor of apoptosis protein
(XIAP) and vascular endothelial growth factor (VEGF) (13).
Therefore, it is reasonable to combine HDAC
inhibitor, such as SAHA, with NF-κB inhibitor and the combination
effect has been unraveled in many preclinical studies. SAHA-induced
apoptosis can be enhanced by NF-κB inhibition in hematologic
malignancy and breast cancer (14–17).
Moreover, bortezomib, which is an anti-NF-κB drug in clinical use,
and HDAC inhibitor together have synergistic effect against cell
lines of pancreatic cancer and HCC (12). Combination of SAHA and bortezomib
has been tested in phase I clinical trials for advanced solid
tumors (18,19).
Sorafenib, a targeted drug for HCC, is effective in
inhibiting NF-κB activation (20).
In our previous studies, both constitutive and tumor
promoter-induced NF-κB activity can be suppressed by sorafenib
in vitro and in vivo (13,21).
Notably, whether sorafenib enhances the therapeutic efficacy of
SAHA through the suppression of NF-κB activity remains unknown. The
goal of this study was to investigate the effect of sorafenib plus
SAHA on NF-κB activation and tumor growth of HCC in vitro
and in vivo by using multimodalities of molecular imaging
including bioluminescent imaging (BLI), which can reveal the
real-time change of intracellular NF-κB signal with times; red
fluorescent protein imaging (RFPI), the signal represents the
living cells; and whole-body autoradiography to demonstrate the
tumor volume (13). In order to
study the effect of NF-κB inhibition on anti-HCC efficacy of SAHA,
HCC cells transfected with IκBα mutant vector was used as the
positive control for effect of NF-κB inhibition.
Materials and methods
Construction of plasmid vector with NF-κB
response element carrying reporter genes herpes simplex virus (HSV)
thymidine kinase (tk) and firefly luciferase (luc2)
The construction protocol has been published in our
previous study (13). In short,
CMV-IRES-dsred2 vector (Clontech, Mountain View, CA) was
digested by Aquaspirillum serpens (Ase) I and
Bacillus megaterium (Bmt) I then blunted by Klenow
enzyme to form pIres-dsred2. The NF-κB responsive element isolated
from pNF-κB-luc vector (Clontech) by Micrococcus
luteus (Mlu) I and Haemophilus influenzae Rd
(Hind) III was blunted by Klenow enzyme and then ligated
into pIres-dsred2 to form the pNF-κB-ires-dsred2
vector. The luc2, which was isolated from pGL4-luc2
(Promega, Madison, WI) by Bacillus stearothermophilus
(Bst) XI and Nocardia otitidis-caviarum (Not)
I and blunted by Klenow enzyme, was inserted downstream of
pNF-κB-ires, which was derived from pNF-κB-ires-dsred2 after
being digested by NotI and Xanthomonas badrii
(Xba) I, to form pNF-κB-ires-luc2. The
HSV1-tk, which was isolated from pORF-HSV1-tk
(InvivoGen, San Diego, CA) by HindIII and Escherichia
coli RY13 (EcoR) I then blunted Klenow enzyme, was
inserted into pNF-κB-ires-luc2 to form pNF-κB-tk-luc2
vector.
Hepatocellular carcinoma (HCC) cell
culture
Two human HCC cell lines, Huh7 and Hep3B, were used
in this study. Huh7 was kindly provided by Dr Jason Chia-Hsien
Cheng at the Department of Radiation Oncology, National Taiwan
University Hospital, Taipei, Taiwan. Hep3B was obtained from the
American Type Culture Collection (ATCC, Gaithersburg, MD). Both
cell lines were maintained in Dulbecco’s modified Eagle’s medium
(DMEM) with supplemental 10% fetal bovine serum (FBS) and cultured
at 37°C in a humidified incubator containing 5% CO2. The
Huh7/NF-κB-tk-luc2/rfp stable clone (transfection procedure
as described) was maintained in the same medium as Huh7 with
additional 500 μg/ml of G418 (Calbiochem, Darmstadt, Hesse,
Germany).
Establishment of Huh7/NF-κB-tk-luc2/rfp
stable clone from HCC cell line
Huh7 cells (2×106) were seeded in a 10-cm
dia meter dish for 24 h prior to transfection.
pNF-κB-tk-luc2 vector (8 μg) and 16 μl of
jetPEI™ solution (Polyplus Transfection, Strasbourg, Alsace,
France), diluted with 500 and 484 μl of 145 mM NaCl,
respectively, were mixed and incubated for 30 min at room
temperature to make the 1000 μl jetPEI™/DNA mixture. The
mixture was then added to Huh7 cells and incubated at 37°C for 24 h
followed by trypsinization, then cultured with DMEM containing 1
mg/ml G418 supplemented with 10% FBS for two weeks. The surviving
clones were isolated and transferred to 96-well plates for cell
growth. The expression of luc2 protein in each clone was
assayed using BLI, and re-named as Huh7/NF-κB-tk-luc2 cell
line.
CAG promoter (composed of CMV enhancer and β-actin
promoter)-driven red fluorescent protein (RFP) vector (National
RNAi Core Facility Platform, Academic Sinica, Taipei, Taiwan) was
used to transfect Huh7/NF-κB-tk-luc2 cell line with the same
strategy as the afore-mentioned protocol. Two weeks after RFP
vector transfection, cells with red fluorescence were sorted and
isolated by flow cytometry. The isolated cells were transferred to
96-well plates for growth. The RFP expression in each clone was
assayed using an IVIS50 Imaging System (Xenogen, Alameda, CA). This
bioluminescent cell clone with simultaneous red fluorescence was
renamed as Huh7/NF-κB-tk-luc2/rfp cell line.
Transfection of Huh7/NF-κB-tk-luc2 with
IκBα mutant vector
Huh7/NF-κB-tk-luc2 cells transfected with
IκBα mutant vector (pI-κBαM, Clontech) was used as the positive
control for the inhibition of NF-κB as compared with that
suppressed by sorafenib treatment. In addition,
Huh7/NF-κB-tk-luc2 cell line transfected with empty vector
was used as the negative control for NF-κB inhibition.
Extraction of sorafenib
The method for extraction of sorafenib was described
in our previous study (21).
Briefly, sorafenib was extracted from commercial
Nexavar® tablets (Bayer, Leverkusen, North
Rhine-Westphalia, Germany) composed of 200 mg sorafenib. The tablet
was ground to fine powder and transferred to a 100-ml conical
flask. The powder was washed with 15 ml deionized distilled water
three times to remove water-soluble components; 15 ml ethyl acetate
was then used to extract the precipitate three times in order to
recover the sorafenib. The organic phases were combined and dried
over anhydrous sodium sulfate, followed by evaporation under
reduced pressure. The residue was recrystallized with acetone and
hexane to yield a white solid extract weighing about 122 mg (60%
recovery). Nuclear magnetic resonance (NMR) spectra were recorded
with a spectrometer (Varian Gemini 200; Oxford Instruments,
Abingdon, Oxfordshire, UK) to determine the chemical structure of
the sorafenib extract. High-performance liquid chromatography
(HPLC) was conducted using a PU-2089 plus quaternary gradient pump,
equipped with a UV-2075 Plus intelligent UV/VIS detector (Jasco,
Tokyo, Kanto, Japan). The 1H-NMR spectrum of the recovered
sorafenib was the same as the one reported before (22). More than 98% chemical purity was
achieved for the recovered sorafenib (retention time, 16.2 min) as
determined with HPLC.
Huh7/NF-κB-tk-luc2/rfp tumor bearing
animal model
Male nude mice (n=6 per group and repeated 3 times,
total 72 mice, 4–6 weeks old, purchased from National Laboratory
Animal Center, Taipei, Taiwan) were injected subcutaneously with
1×107 Huh7/NF-κB-tk-luc2/rfp cells in the right
hind flank. Mice were randomly divided into four groups (Fig. 5): control [vehicle treated with 100
μl phosphate-buffered saline (PBS) in 1% dimethyl sulfoxide
(DMSO) daily by gavage], SAHA alone (25 mg/kg/day for 21 days by
gavage), sorafenib alone (20 mg/kg/day for 21 days by gavage) and
combination (SAHA plus sorafenib, 25 mg/kg/day plus 20 mg/kg/day
for 21 days by gavage). All treatments were administered by gavage.
Treatment was initiated when tumor volume reached about 50
mm3. Tumor volume was assayed with digital caliper twice
a week and BLI once a week. Mice were sacrificed on day 21
post-treatment for ex vivo EMSA, ex vivo western
blotting, and whole-body autoradiography. The animal study
protocols complied with institutional animal care and use guideline
of National Yang-Ming University, Taipei, Taiwan.
Cell viability assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT, Sigma-Aldrich, St. Louis, MO) was dissolved in PBS
(145 mM NaCl, 1.4 mM KH2PO4, 4.3 mM
Na2HPO4 and 2.7 mM KCl, pH 7.2).
Huh7/NF-κB-tk-luc2/rfp cells were seeded into 96-well plates
with 3×104 cells/well for 24 h, then treated with
different concentrations of SAHA (0–10 μM in 0.1% DMSO) or
SAHA combined with 10 μM sorafenib for additional 48 h.
After washing with fresh medium, 100 μl of 5 mg/ml MTT
solution was added to each well followed by incubation at 37°C for
2 h. Then 100 μl DMSO was added to dissolve the MTT
formazan, and the absorbance was determined with an enzyme-linked
immunosorbent assay (ELISA) reader (Power Wave X340, Bio-Tek,
Winooski, VT) using a wavelength of 570 nm for excitation.
DNA fragmentation assay
Huh7/NF-κB-tk-luc2/rfp cells
(1×106) were seeded in 6-well plates and treated with
vehicle (0.1% DMSO), 10 μM SAHA, 10 μM sorafenib or
combination of both for 48 h. After treatment a genomic DNA
purification kit (Axygen, Tewksbury, MA) was used to extract DNA
following the instruction provided by the manufacturer. DNA
laddering was analyzed with 1.5% agarose gel electrophoresis.
Electrophoretic mobility shift assay
(EMSA)
Forty-eight hours post-treatment with vehicle, 10
μM SAHA, 10 μM sorafenib and combination of SAHA and
sorafenib, respectively, nuclear fractions of
Huh7/NF-κB-tk-luc2/rfp cells were obtained using Nuclear
Extraction kit (Chemicon, Temecula, CA). The NF-κB/DNA binding
activity was evaluated using LightShift Chemiluminescent EMSA kit
(Thermo Scientific, Rockford, IL). The procedure followed the
protocol provided with the kit. In brief, DNA sequences were
synthesized for NF-κB binding. Sense: AGTTGAGGGGACTTTCCCAGGC and
antisense: GCCTGGGAAAGTCCCCTCAACT. Nuclear extracts were incubated
with the biotin-labeled DNA probe for 20 min at room temperature.
The protein-DNA complex was separated from free oligonucleotides on
a 5% polyacrylamide gel, then transferred to a nylon membrane and
cross-linked with UV light. The membrane was incubated with
streptavidin-horseradish peroxidase, and detected by enhanced
chemiluminescence (ECL, Thermo Scientific).
Ex vivo EMSA
Mice were sacrificed on day 21 and tumors were
removed for nuclear protein extraction by using Nuclear Extraction
kit (Chemicon). The NF-κB/DNA binding activity was evaluated using
LightShift Chemiluminescent EMSA kit (Thermo Scientific).
Western blotting
Huh7/NF-κB-tk-luc2/rfp cells
(2×106) were seeded into a 10-cm diameter dish (5
dishes/group) for 24 h prior to the treatment with vehicle, 10
μM SAHA, 10 μM sorafenib or combination of 10
μM SAHA and 10 μM sorafenib, respectively, for
another 48 h. Cells were then lysed with 100 μl lysis buffer
[50 mM Tris-HCl (pH 8.0), 120 mM NaCl, 0.5% NP-40, 1 mM
phenylmethanesulfonylfluoride]. Total proteins (40 μg) were
separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE), then transferred to a polyvinylidene
difluoride membrane (Millipore, Billerica, MA), blocked with 5%
non-fat milk in TBS-Tween buffer (0.12 M Tris-base, 0.15 M NaCl and
0.1% Tween-20) for 1 h at room temperature, and then incubated with
primary antibody (Millipore) for XIAP, BCL-2, VEGF, cyclin D1,
c-FLIP, caspase-3, caspase-8, cytochrome-C, extracellular
signal-regulated kinase (ERK) and β-actin, respectively, overnight
at 4°C, followed by incubation with secondary peroxidase-conjugated
anti-rabbit antibody for 30 min at room temperature. The
expressions of proteins were determined by ECL (Millipore). ImageJ
software (National Institutes of Health, Bethesda, MD) was used for
the quantitative analysis. A cytosol/nuclear extraction kit
(Chemicon) was used to extract cytosolic cytochrome-C following the
manufacturer’s protocol.
Ex vivo western blotting
Mice were sacrificed on day 21 after treatments.
Tumors were removed for protein extraction using T-PER kit (Thermo
Scientific). Equal amounts of protein (40 μg) were loaded
for SDS-PAGE, then transferred to nitrocellulose membranes. The
membranes were incubated with primary antibodies specific for VEGF,
XIAP, BCL-2, cyclin D1, c-FLIP, caspase-3 and caspase-8,
cytochrome-C, ERK and β-actin, followed by incubation with
horseradish peroxidase-conjugated secondary antibodies. The protein
expression was determined by ECL. A cytosol/nuclear extraction kit
(Chemicon) was used to extract cytosolic cytochrome-C following the
manufacturer’s protocol.
Therapeutic efficacy evaluation by
molecular imaging in vitro and in vivo
Procedure for each modality of molecular imaging was
described in our previous study (13). In brief, BLI was used for
monitoring the NF-κB activation in Huh7/NF-κB-tk-luc2/rfp
cells. A total of 3×104 Huh7/NF-κB-tk-luc2/rfp
cells were cultured in a 96-well plate for 48 h, then treated with
various concentrations of SAHA (0–10 μM), 10 μM
sorafenib, combination of SAHA and sorafenib, and 10 μM of
various signaling molecule inhibitors for 48 h, respectively.
D-luciferin (100 μl of 500 μM, Xenogen) was added to
each well for imaging. Relative NF-κB activity was calculated as
follows: ROI value of treatment group/ROI value of control group,
and both ROIs were corrected by cell viability which was determined
by MTT assay. BLI was also used in monitoring temporal change of
NF-κB activation in the Huh7/NF-κB-tk-luc2/rfp tumor-bearing
mice. Mice were anesthetized using 1–3% isoflurane and received 150
mg/kg D-luciferin via intraperitoneal injection 15 min before
imaging. RFPI was used for monitoring viable tumor cell growth in
the Huh7/NF-κB-tk-luc2/rfp tumor bearing mice. The photons
emitted from tumors obtained by BLI and RFPI were detected with an
IVIS50 Imaging System (Xenogen). The image acquiring periods for
BLI and RFPI were 5 min and 30 sec, respectively. Regions of
interest (ROIs) of the images were drawn conformally to the tumor
contour and quantified as photons/sec using Living Image software
(Version 2.20, Xenogen).
Whole-body autoradiography
Mice were injected intravenously with
3.7×106 Bq/0.2 ml
131I-1-(2-deoxy-2-fluoro-1-D-arabinofuranosyl)-5-iodouracil
(FIAU) on day 21 post-treatments, and sacrificed 24 h later for
whole-body autoradiography. Frontal sectioning was performed with
thickness of 30 μm at −20°C with a cryostat microtome
(Bright Instrument, Huntingdon, Cambridgeshire, UK). Sections were
placed on the imaging plate (BAS-SR2040, Fuji Photo Film, Tokyo,
Kanto, Japan) in the cassette (2040, Fuji Photo Film). After
exposure, the imaging plate was assayed with a FLA5000 reader (Fuji
Photo Film) to acquire the phosphor image. The parameters of the
reader were: resolution of 10 μm, gradation of 16 bits,
635-nm laser light and 800 V of photomultiplier tube.
Statistical analysis
All data were shown as the mean ± standard error.
Student’s t-test was used for the comparison between two groups.
Differences between the means were considered significant at
p<0.05 or less.
Results
Sorafenib enhances SAHA cytotoxicity
through extrinsic and intrinsic apoptotic pathways in HCC
The viabilities of both
Huh7/NF-κB-tk-luc2/rfp and Hep3B cells were decreased
linearly (Fig. 1A and B) with
increasing SAHA concentrations (2–10 μM) with or without 10
μM sorafenib concurrently. Either SAHA or sorafenib alone
induced apoptosis through extrinsic (cleaved caspase-8) and
intrinsic (cytochrome-C) pathways as unraveled by increased
expressions of apoptosis-related proteins (Fig. 1E and F). Combination of SAHA and 10
μM sorafenib not only enhanced HCC cytotoxicity, but
accompanied by obvious DNA fragmentation and significantly
increased expression of pro-apoptotic proteins (Fig. 1).
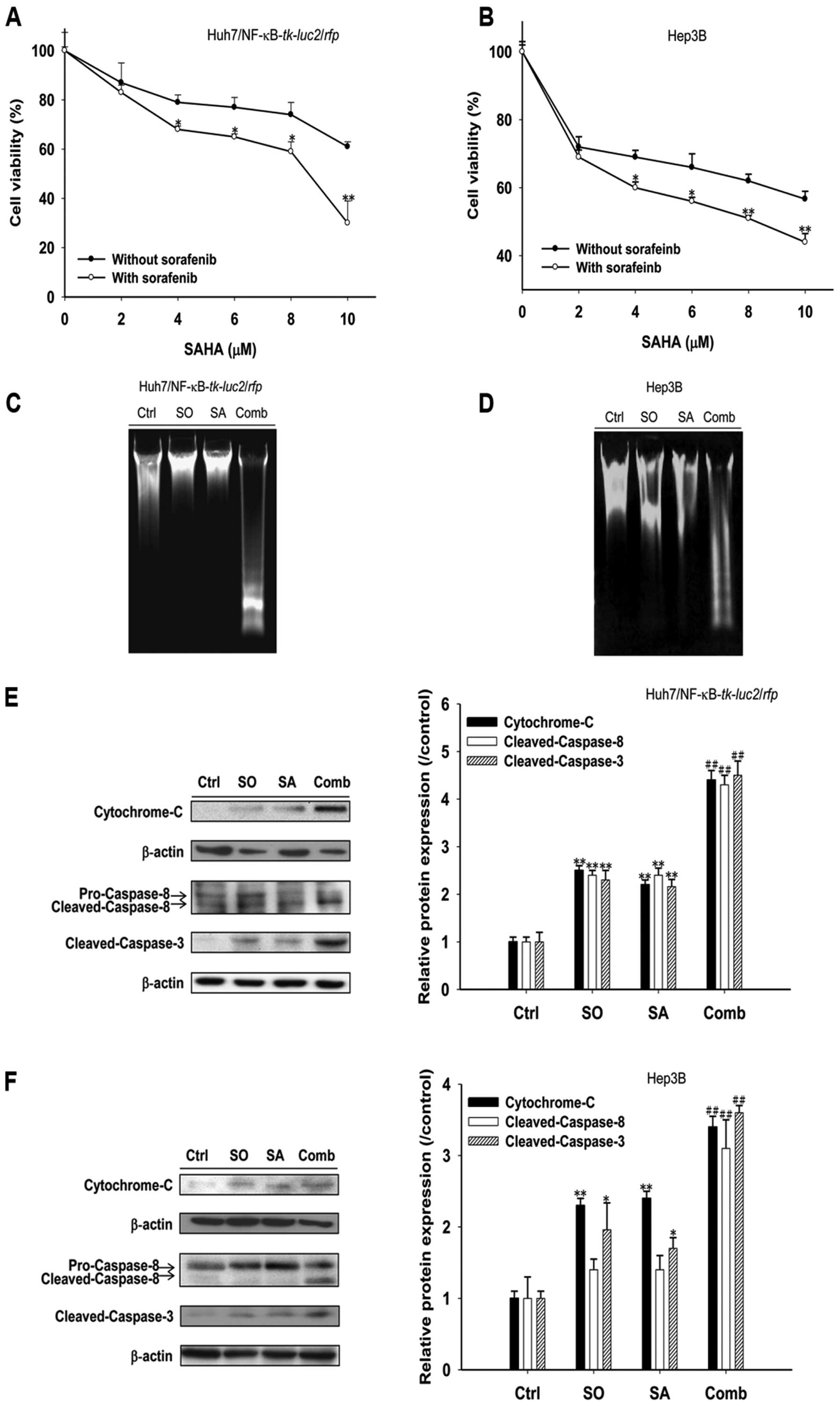 | Figure 1.Sorafenib enhances cytotoxicity of
SAHA against human HCC (Huh7/NF-κB-tk-luc2/rfp and Hep3B)
cells. (A and B) HCC cells were treated with 0–10 μM SAHA
alone or combined with 10 μM sorafeinb for 48 h in
Huh7/NF-κB-tk-luc2/rfp (A) and Hep3B (B) cells,
respectively. MTT assay was used for cytotoxicity. Enhanced
cytotoxicity was observed after treatment with 4 μM or
higher concentrations of SAHA combined with sorafenib. (C and D)
HCC cells were treated with SAHA (10 μM), sorafenib (10
μM) or combination of both for 48 h. DNA laddering,
charateristic of apoptosis, was most obvious in combination group
both in Huh7/NF-κB-tk-luc2/rfp (C) and Hep3B (D) cells. (E
and F) Huh7/NF-κB-tk-luc2/rfp (E) and Hep3B (F) cells were
treated with SAHA (10 μM), sorafenib (10 μM) or
combination of both for 48 h. Western blotting was used to evaluate
protein expression. Expression of pro-apoptotic proteins were
increased after SAHA or sorafenib treatment and most prominent in
the combination group. (Ctrl, control; SO, sorafenib; SA, SAHA;
Comb, combination; *p<0.05, **p<0.01 as
compared with that of the control, ##p<0.01 as
compared with that of sorafenib- or SAHA-treated group). |
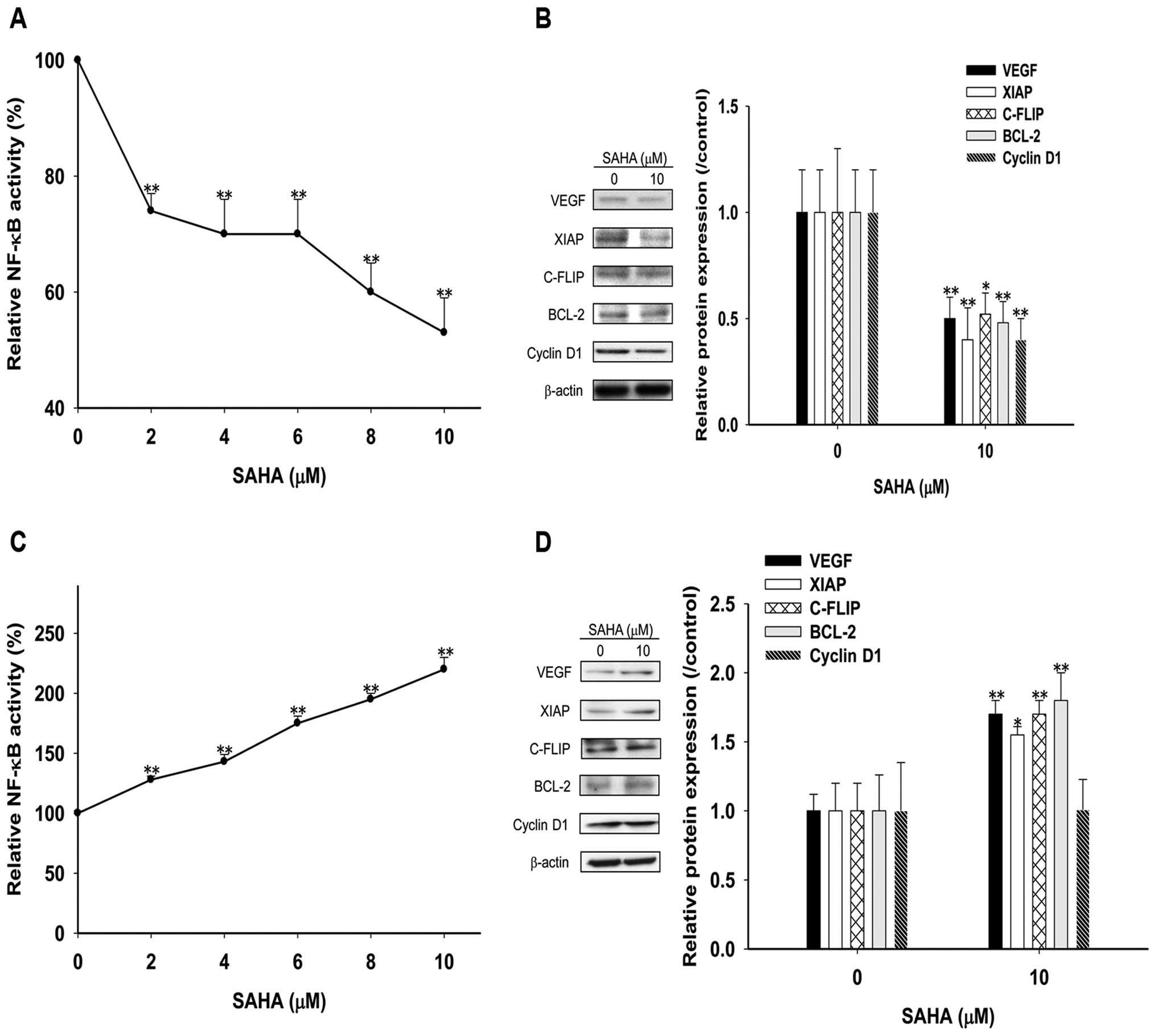
Contradictory effects of short (24 h) and
long (48 h) treatment time with SAHA on NF-κB activity and
expressions of NF-κB-regulated effector proteins in
Huh7/NF-κB-tk-luc2/rfp cells
After treatments with various concentrations of SAHA
for 24 h, the NF-κB activity was inhibited significantly in HCC
cells in a dose-dependent manner (Fig.
2A), and 10 μM SAHA reduced effector proteins
expressions (Fig. 2B) to 50% of
those of the controls. Notably, SAHA significantly induced NF-κB
activity in a dose-dependent manner after 48 h treatments (Fig. 2C), and increased the expressions of
NF-κB-regulated effector proteins by 1.5- to 2-fold except cyclin
D1 as compared to those of the controls (Fig. 2D).
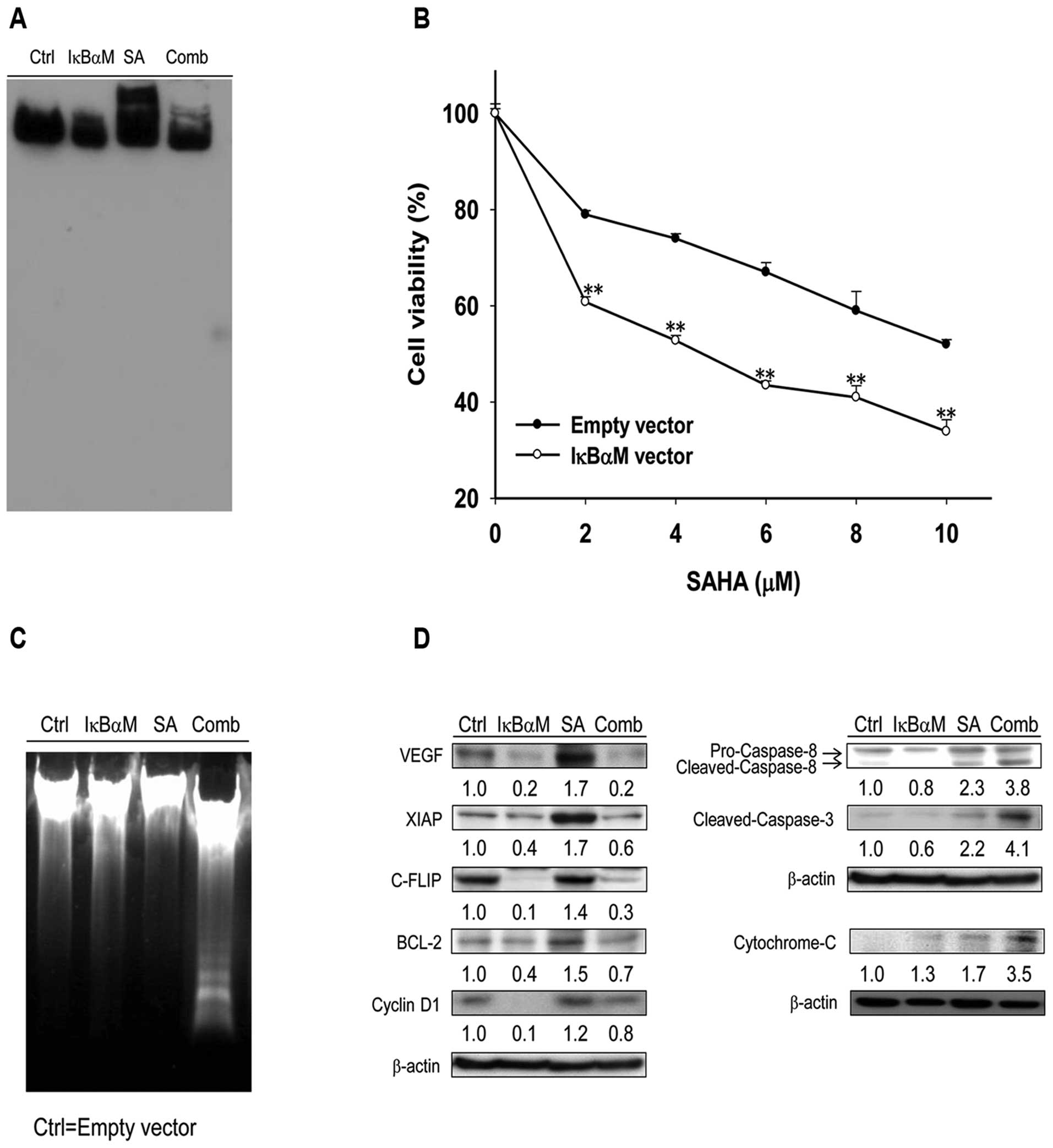
NF-κB inhibition enhances anticancer
effects of SAHA against Huh7/NF-κB-tk-luc2 cells
SAHA-induced NF-κB/DNA binding activity was
inhibited with the transfection of pI-κBαM in
Huh7/NF-κB-tk-luc2 cells (Fig.
3A). Cell viability after SAHA treatment was significantly
decreased in Huh7/NF-κB-tk-luc2 cells transfected with
pI-κBαM as compared with that transfected with empty vector
(Fig. 3B). Apoptosis based on the
DNA fragmentation assay was significantly increased in the
combination group as compared with those of others groups (Fig. 3C). The transfection of pI-κBαM
inhibited SAHA-induced expression of NF-κB-regulated downstream
effector proteins and increased SAHA-induced apoptotic-related
proteins (Fig. 3C and D).
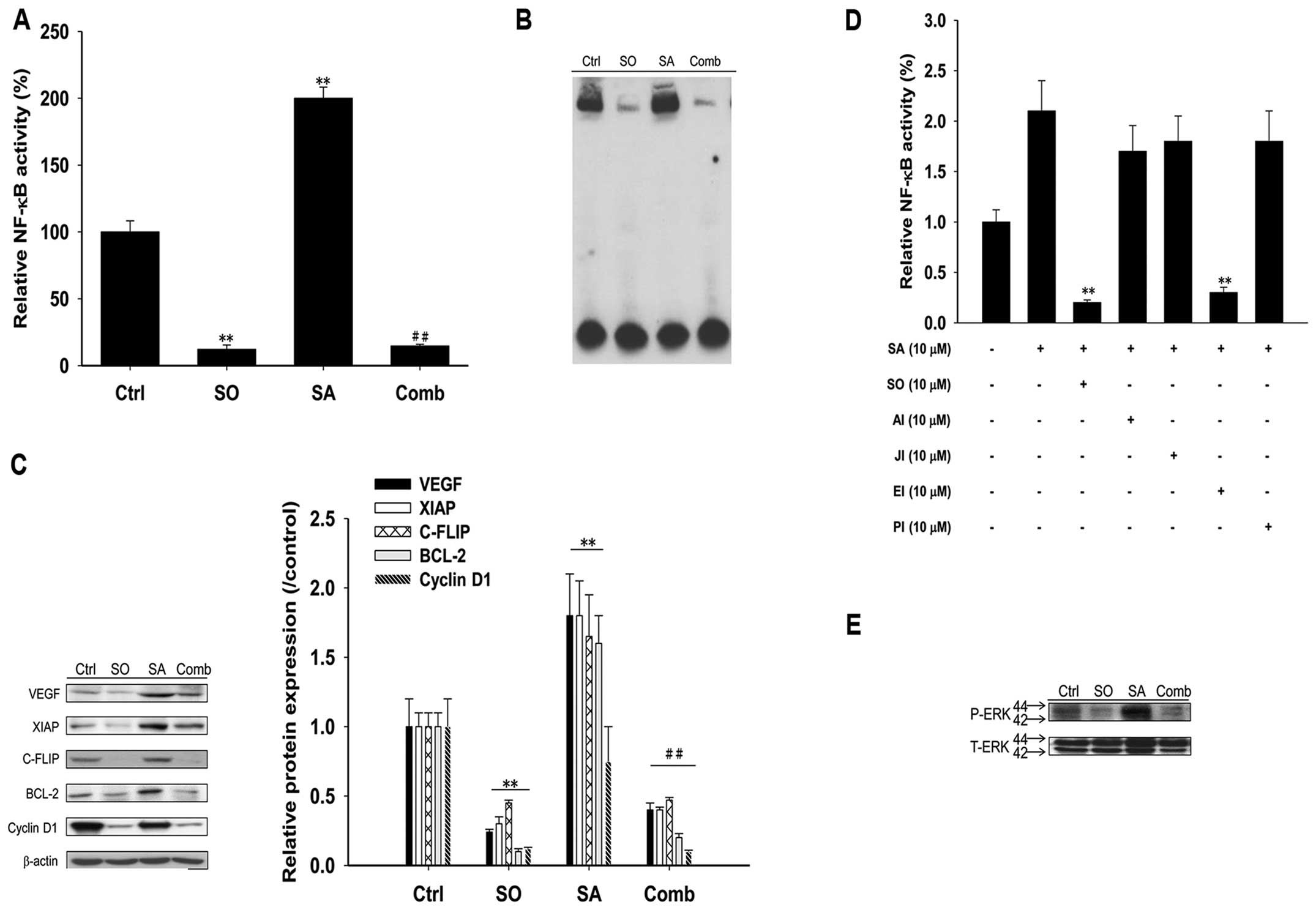
Sorafenib inhibits SAHA-induced NF-κB
activity and NF-κB-regulated effector proteins expressions via
suppression of ERK activation in Huh7/NF-κB-tk-luc2/rfp cells
Intrinsic NF-κB activation, SAHA-induced NF-κB
activity and expressions of downstream effector proteins were
suppressed significantly by sorafenib as shown in Fig. 4A–C. Among the inhibitors for signal
transduction, i.e., AKT inhibitor, c-Jun N-terminal kinase (JNK)
inhibitor, ERK inhibitor and P38 inhibitor, only ERK inhibitor
shows similar effect as sorafenib in suppressing SAHA-induced NF-κB
activation (Fig. 4D). Furthermore,
SAHA-induced ERK phosphorylation in Huh7/NF-κB-tk-luc2/rfp
cells was suppressed by sorafenib as shown in Fig. 4E.
Sorafenib enhances therapeutic efficacy
of SAHA in Huh7/NF-κB-tk-luc2/rfp tumor-bearing mice
Experimental design for the study in vivo is
shown in Fig. 5. Though the tumor
volumes of SAHA-treated mice were smaller as compared with those of
the control, no significant difference was found. Both
sorafenib-treated and combination groups had significantly smaller
tumor volumes throughout the experimental period until day 21 as
compared with those of the control. Notably, the highest tumor
suppression was found in the combination group (Fig. 6A). The viable tumor cell growth was
evaluated with RFPI once a week. The emitted photon flux correlates
well with the number of viable tumor cells (13). The results demonstrated in Fig. 6B were similar to those shown in
Fig. 6A. Since the NF-κB level
could be increased by SAHA treatment for 48 h, the NF-κB activation
inside the tumor was observed by BLI once a week. The higher photon
flux represents the more intense NF-κB activity (20). SAHA significantly increased the
NF-κB activity as compared with that of the control, but the
increase of NF-κB activity could be suppressed by sorafenib as
shown in the combination group (Fig.
6C). Simultaneous imaging of living tumor cells and NF-κB
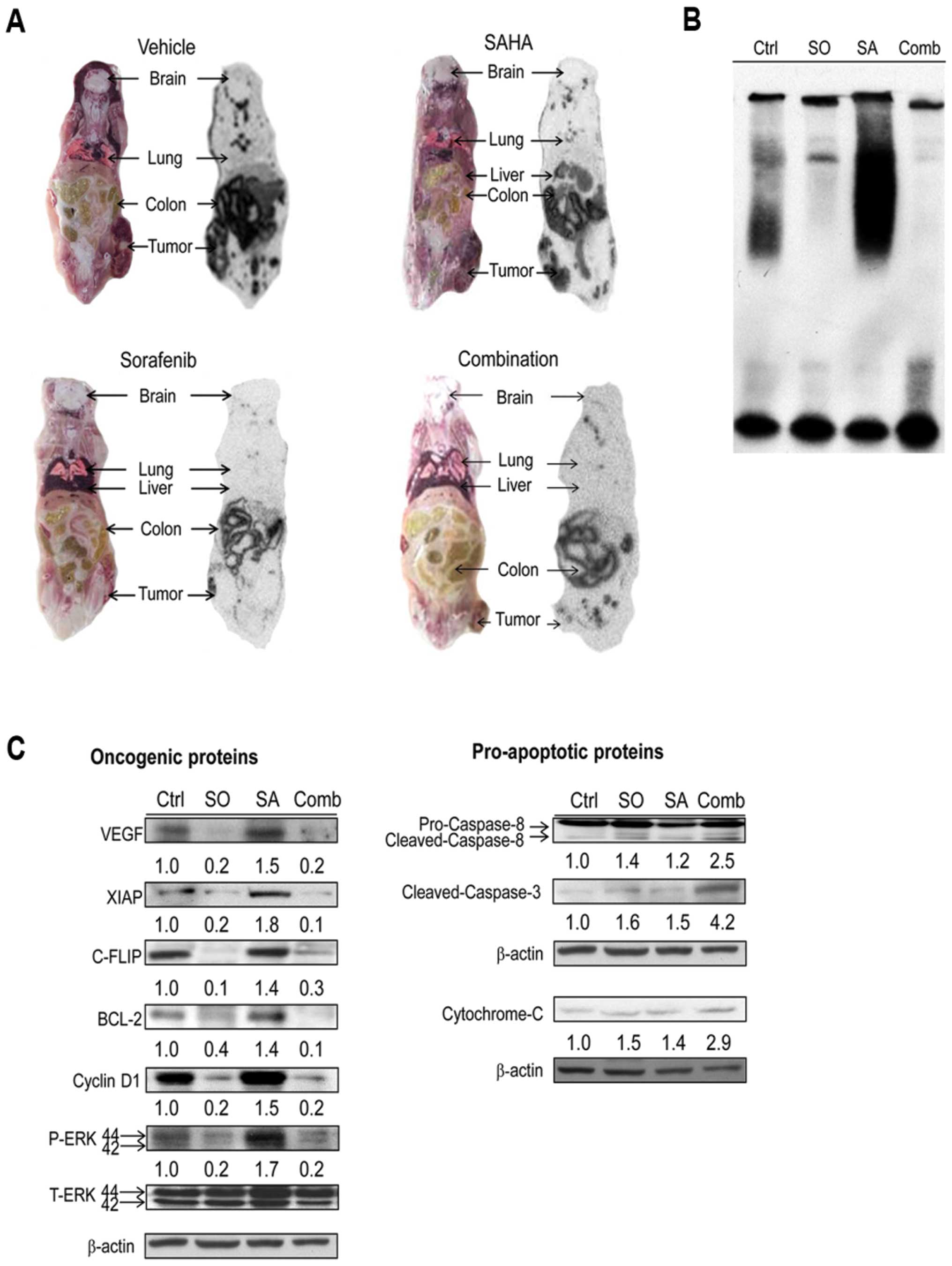
activity by RFPI and BLI, respectively, was shown in Fig. 6D. In addition, the uptake of
131I-FIAU in tumors, which represented the NF-κB
activity, assayed by whole-body autoradiography (13) were also shown to be the most
prominent in SAHA-treated mice. Lower uptake of
131I-FIAU in sorafenib-treated and combination groups
than that of the control was found (Fig. 7A). Furthermore, sorafenib not only
inhibited SAHA-induced NF-κB/DNA binding activity, but expression
of oncogenic proteins as well, while enhanced expression of
SAHA-induced pro-apoptotic proteins by ex vivo western
blotting (Fig. 7C).
 | Figure 6.Sorafenib enhances therapeutic
efficacy of SAHA in mice with Huh7/NF-κB-tk-luc2/rfp
xenografts. (A) Left panel: the photograph of representative tumors
obtained from different groups on day 21. Right panel: tumors
obtained from sorafenib-treated and combination groups showed
smaller and smallest, respectively, by caliper measurement
(ap<0.01 as compared with that of the control,
bp<0.01 as compared with that of SAHA-treated group,
cp<0.01 as compared with that of sorafenib-treated
group). (B) Fewer viable tumor cells in sorafenib-treated mice and
the fewest in combination group were demonstrated by red
fluorescent protein imaging (RFPI) (ap<0.01 as
compared with that of the control, bp<0.01 as
compared with that of SAHA-treated group, cp<0.01 as
compared with that of sorafenib-treated group). (C) The highest
NF-κB activation induced by SAHA, and inhibition of SAHA-induced
NF-κB activation by sorafenib were revealed by BLI
(ap<0.05, cp<0.01 as compared with that
of the control; bp<0.01 as compared with that of
SAHA-treated group). (D) Normalization of RFPI and BLI signal
intensities. Left panel: the signals of living cells after
treatments were decreased with times as compared with that of the
control (ap<0.01 as compared with that of the
control, bp<0.01 and cp<0.01 as
compared with those of SAHA- and sorafenib-treated groups,
respectively). Right panel: the signals of NF-κB were increased and
decreased with times in SAHA-treated and sorafenib-treated groups,
respectively (ap<0.05, bp<0.01 as
compared with that of the control; cp<0.01 as
compared with that of SAHA-treated group). BLI, bioluminescent
imaging; RFPI, red fluorescent protein imaging. |
Discussion
HDACs including HDAC 1, 3, 8 (class I) and 6 (class
IIB) have been shown to correlate with HCC progression, and all of
these enzymes can be inhibited by SAHA (23). NF-κB activity in the renal cell
carcinoma treated with low concentration (500 nM) of SAHA for 24–36
h could be induced by 1.35- to 1.8-fold above the control (11). NF-κB has also been reported to be
related to chemoresistance through enhancing tumor proliferation,
anti-apoptosis, and invasiveness (24). Here we used high SAHA concentration
(10 μM) instead of 2.5–5 μM as used in most
literature (23,25,26),
and longer treatment time (48 h) to achieve higher cytotoxicity for
HCC cells (Fig. 1A and B). This
strategy resulted in 2-fold increase in NF-κB activity as compared
to that of the control (Figs. 2C,
4A and 4D). As a result, the subsequent increased
expressions of NF-κB-regulated oncogenic proteins (Figs. 2D, 4C and 7C), which may hamper treatment efficacy,
made it a concern when adopting high dose SAHA to treat HCC in
clinic. Notably, cancer cells treated with short-term (24 h) SAHA
suppressed NF-κB activity in a dose-dependent manner as shown in
Fig. 2A. This was also observed
with other cancer cell lines (11,26).
Though the preclinical and clinical phase I evidence indicate that
HDAC inhibition is effective against various cancer cell lines
(8), the potential menace of NF-κB
activation after heavily pre-treated solid or hematologic
malignancy should be addressed. In our previous studies, we have
shown that sorafenib could be used as an NF-κB inhibitor to
suppress constitutive and induced NF-κB activity (13,21).
Here we demonstrated that NF-κB activity and expressions of its
regulated oncogenic proteins were significantly induced in
Huh7/NF-κB-tk-luc2/rfp cells treated with 10 μM SAHA,
which was equivalent to the maximal clinical dosage (27), for 48 h but could be suppressed by
10 μM sorafenib (Fig. 4).
Since combination of sorafenib and SAHA results in the better
therapeutic efficacy both in vitro and in vivo
(Figs. 1A and 6A), this approach may be a potential
strategy for the treatment of HCC in clinic. Combination of low
dose (500 nM) SAHA and sorafenib (3 μM) has been reported to
kill HCC synergistically through the inhibition of cellular
FLICE-like inhibitory protein (c-FLIP) and induction of cluster of
differentiation 95 (CD95), the latter is ceramide-PP2A-reactive
oxygen species-(ROS) dependent (11,28,29).
c-FLIP expression in HCC cells could be suppressed by the treatment
of high dose (2–10 μM) short-term (24 h) SAHA (Fig. 2B), but increased after 48 h
treatment (Fig. 2D). c-FLIP
expression was also suppressed in HCC cells transfected with
pI-κBαM (Fig. 3D) or treated with
the regular clinical dose (10 μM) of sorafenib (Fig. 4C), the latter effectively inhibited
constitutive or induced NF-κB activation in Huh7 cells via
suppression of ERK/NF-κB pathway (13,21).
Since sorafenib has been reported to eradicate HCC through multiple
kinase inhibition (2), we used
multiple signal transduction inhibitors to suppress SAHA-induced
NF-κB activity (Fig. 4D). Notably,
only ERK inhibitor showed the same effect as sorafenib to suppress
SAHA-induced NF-κB activity. Herein we propose that sorafenib
inhibits SAHA-induced NF-κB activity is through the
dephosphorylation of ERK both in vitro and in vivo
(Figs. 4E and 7C). It is reasonable to deduce that
sorafenib enhances anticancer effect of SAHA through the
suppression of ERK/NF-κB pathway.
Paradoxical increase of c-FLIP and induced NF-κB
activation after high dose, long-term (48 h) SAHA treatment is the
niche for sorafenib to be complementary in the combination regimen
for cancer treatment. Here we demonstrated the combination strategy
worked well in vivo (Figs.
6 and 7). Though the HDAC
inhibitor is not yet tested in clinical trial for patients with
HCC, phase I study for sorafenib combined with SAHA in patients
with solid tumors including renal cell carcinoma (RCC) and
non-small cell lung cancer has been completed (30). Among 8 patients with RCC, there
were 1 partial response and 5 minor responses. The promising result
may make future phase II trial more likely. Another small phase II
trial exhibited the efficacy of SAHA plus bortezomib in refractory
hematologic malignancy (31).
However, unfavorable phase II clinical trial results for solid
tumors (32,33) and SAHA in combination with NF-κB
inhibition may be accompanied with intolerable toxicity (30,31).
In our previous study, we demonstrated the
visualization of temporal change of intracellular NF-κB signal
activation in Huh7/NF-κB-tk-luc2/rfp tumor-bearing animal
model (13). Here the constitutive
NF-κB signals were found to be increased with the progression of
tumor growth in both the control and SAHA treated groups. In
contrast, lower NF-κB activation and decreased expressions of
NF-κB-regulated oncogenic proteins were observed in
sorafenib-treated (with or without SAHA) mice, and the tumor growth
was inhibited throughout the experiment, particularly in mice
treated with SAHA plus sorafenib (Figs. 6C, 6D and 7C). Although tumor growth in mice treated
with SAHA alone was found to be slower as compared with that of the
control, the NF-κB activity induced by SAHA allowed tumors to grow
more rapidly than mice treated with sorafenib. Therefore,
suppression of NF-κB activation during HCC treatment with SAHA may
be critical for the improvement of therapeutic efficacy.
In conclusion, we found that NF-κB signal can be
activated by SAHA with long treatment time (48 h), and results in
increased expressions of NF-κB-regulated oncogenic proteins.
Sorafenib inhibits SAHA-induced NF-κB activity and expressions of
NF-κB-regulated oncogenic proteins while enhances therapeutic
efficacy of SAHA against HCC both in vitro and in
vivo through suppression of ERK/NF-κB signaling pathway.
Sorafenib combined with SAHA may have potential as a new treatment
strategy for the treatment of advanced HCC.
Abbreviations:
|
BCL-2
|
B-cell lymphoma 2;
|
|
BLI
|
bioluminescent imaging;
|
|
ERK
|
extracellular signal-regulated
kinases;
|
|
HCC
|
hepatocellular carcinoma;
|
|
HDAC
|
histone deacetylase;
|
|
luc
|
luciferase;
|
|
NF-κB
|
nuclear factor κ-light-chain-enhancer
of activated B cells;
|
|
rfp
|
red fluorescent protein;
|
|
RFPI
|
red fluorescent protein imaging;
|
|
SAHA
|
suberoylanilide hydroxamic acid;
|
|
TACE
|
transarterial chemo-embolization;
|
|
tk
|
thymidine kinase;
|
|
VEGF
|
vascular endothelial growth
factor;
|
|
XIAP
|
X-linked inhibitor of apoptosis
protein
|
Acknowledgements
This study was supported by grants
NSC 101-2314-B-010-045-MY3, NSC 102-2321-B-010-005 from National
Science Council, Taipei, Taiwan; and RD2014-012 from National
Yang-Ming University Hospital, Yilan, Taiwan. The imaging facility
was supported by Taiwan Mouse Clinic.
References
|
1.
|
Feng M and Ben-Josef E: Radiation therapy
for hepatocellular carcinoma. Semin Radiat Oncol. 21:271–277. 2011.
View Article : Google Scholar
|
|
2.
|
Zhang X, Yang XR, Huang XW, et al:
Sorafenib in treatment of patients with advanced hepatocellular
carcinoma: a systematic review. Hepatobiliary Pancreat Dis Int.
11:458–466. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Kanno K, Kanno S, Nitta H, et al:
Overexpression of histone deacetylase 6 contributes to accelerated
migration and invasion activity of hepatocellular carcinoma cells.
Oncol Rep. 28:867–873. 2012.PubMed/NCBI
|
|
4.
|
Wu J, Du C, Lv Z, et al: The up-regulation
of histone deacetylase 8 promotes proliferation and inhibits
apoptosis in hepatocellular carcinoma. Dig Dis Sci. 58:3545–3553.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Rikimaru T, Taketomi A, Yamashita Y, et
al: Clinical significance of histone deacetylase 1 expression in
patients with hepatocellular carcinoma. Oncology. 72:69–74. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Wu LM, Yang Z, Zhou L, et al:
Identification of histone deacetylase 3 as a biomarker for tumor
recurrence following liver transplantation in HBV-associated
hepatocellular carcinoma. PLoS One. 5:e144602010. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Machado MC, Bellodi-Privato M, Kubrusly
MS, et al: Valproic acid inhibits human hepatocellular cancer cells
growth in vitro and in vivo. J Exp Ther Oncol. 9:85–92.
2011.PubMed/NCBI
|
|
8.
|
Coradini D and Speranza A: Histone
deacetylase inhibitors for treatment of hepatocellular carcinoma.
Acta Pharmacol Sin. 26:1025–1033. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Yoshida K, Sasaki R, Nishimura H, et al:
Nuclear factor-kappaB expression as a novel marker of
radioresistance in early-stage laryngeal cancer. Head Neck.
32:646–655. 2010.PubMed/NCBI
|
|
10.
|
Ni W, Chen B, Zhou G, et al: Overexpressed
nuclear BAG-1 in human hepatocellular carcinoma is associated with
poor prognosis and resistance to doxorubicin. J Cell Biochem.
114:2120–2130. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Zhang G, Park MA, Mitchell C, et al:
Vorinostat and sorafenib synergistically kill tumor cells via FLIP
suppression and CD95 activation. Clin Cancer Res. 14:5385–5399.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Spratlin JL, Pitts TM, Kulikowski GN, et
al: Synergistic activity of histone deacetylase and proteasome
inhibition against pancreatic and hepatocellular cancer cell lines.
Anticancer Res. 31:1093–1103. 2011.PubMed/NCBI
|
|
13.
|
Wang WH, Chiang IT, Liu YC, et al:
Simultaneous imaging of temporal changes of NF-kappaB activity and
viable tumor cells in Huh7/NF-kappaB-tk-luc2/rfp tumor-bearing
mice. In vivo. 27:339–350. 2013.PubMed/NCBI
|
|
14.
|
Dai Y, Rahmani M, Dent P and Grant S:
Blockade of histone deacetylase inhibitor-induced RelA/p65
acetylation and NF-kappaB activation potentiates apoptosis in
leukemia cells through a process mediated by oxidative damage, XIAP
downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell
Biol. 25:5429–5444. 2005. View Article : Google Scholar
|
|
15.
|
Domingo-Domenech J, Pippa R, Tapia M,
Gascon P, Bachs O and Bosch M: Inactivation of NF-kappaB by
proteasome inhibition contributes to increased apoptosis induced by
histone deacetylase inhibitors in human breast cancer cells. Breast
Cancer Res Treat. 112:53–62. 2008. View Article : Google Scholar
|
|
16.
|
Dai Y, Guzman ML, Chen S, et al: The NF
(nuclear factor)-kappaB inhibitor parthenolide interacts with
histone deacetylase inhibitors to induce MKK7/JNK1-dependent
apoptosis in human acute myeloid leukaemia cells. Br J Haematol.
151:70–83. 2010. View Article : Google Scholar
|
|
17.
|
Dai Y, Chen S, Wang L, et al: Disruption
of IkappaB kinase (IKK)-mediated RelA serine 536 phosphorylation
sensitizes human multiple myeloma cells to histone deacetylase
(HDAC) inhibitors. J Biol Chem. 286:34036–34050. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Schelman WR, Traynor AM, Holen KD, et al:
A phase I study of vorinostat in combination with bortezomib in
patients with advanced malignancies. Invest New Drugs.
31:1539–1546. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Deming DA, Ninan J, Bailey HH, et al: A
Phase I study of intermittently dosed vorinostat in combination
with bortezomib in patients with advanced solid tumors. Invest New
Drugs. 32:323–329. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Liu YC, Chiang IT, Hsu FT and Hwang JJ:
Using NF-kappaB as a molecular target for theranostics in radiation
oncology research. Expert Rev Mol Diagn. 12:139–146. 2012.
View Article : Google Scholar
|
|
21.
|
Chiang IT, Liu YC, Wang WH, et al:
Sorafenib inhibits TPA-induced MMP-9 and VEGF expression via
suppression of ERK/NF-kappaB pathway in hepatocellular carcinoma
cells. In vivo. 26:671–681. 2012.
|
|
22.
|
Bankston D, Dumas J, Natero R, et al: A
scaleable synthesis of BAY 43-9006 a potent Raf kinase inhibitor
for the treatment of cancer. Org Process Res Dev. 6:777–781. 2002.
View Article : Google Scholar
|
|
23.
|
Marks PA and Breslow R: Dimethyl sulfoxide
to vorinostat: development of this histone deacetylase inhibitor as
an anticancer drug. Nat Biotechnol. 25:84–90. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Li F and Sethi G: Targeting transcription
factor NF-kappaB to overcome chemoresistance and radioresistance in
cancer therapy. Biochim Biophys Acta. 1805:167–180. 2010.PubMed/NCBI
|
|
25.
|
Galimberti S, Canestraro M, Khan R, et al:
Vorinostat and bortezomib significantly inhibit WT1 gene expression
in MO7-e and P39 cell lines. Leukemia. 22:628–631. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Takada Y, Gillenwater A, Ichikawa H and
Aggarwal BB: Suberoylanilide hydroxamic acid potentiates apoptosis,
inhibits invasion, and abolishes osteoclastogenesis by suppressing
nuclear factor-kappaB activation. J Biol Chem. 281:5612–5622. 2006.
View Article : Google Scholar
|
|
27.
|
Chen KF, Chen HL, Tai WT, et al:
Activation of phosphatidylinositol 3-kinase/Akt signaling pathway
mediates acquired resistance to sorafenib in hepatocellular
carcinoma cells. J Pharm Exp Ther. 337:155–161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Park MA, Zhang G, Martin AP, et al:
Vorinostat and sorafenib increase ER stress, autophagy and
apoptosis via ceramide-dependent CD95 and PERK activation. Cancer
Biol Ther. 7:1648–1662. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Park MA, Mitchell C, Zhang G, et al:
Vorinostat and sorafenib increase CD95 activation in
gastrointestinal tumor cells through a Ca(2+)-de novo
ceramide-PP2A-reactive oxygen species-dependent signaling pathway.
Cancer Res. 70:6313–6324. 2010.PubMed/NCBI
|
|
30.
|
Dasari A, Gore L, Messersmith WA, et al: A
phase I study of sorafenib and vorinostat in patients with advanced
solid tumors with expanded cohorts in renal cell carcinoma and
non-small cell lung cancer. Invest New Drugs. 31:115–125. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Warlick ED, Cao Q and Miller J: Bortezomib
and vorinostat in refractory acute myelogenous leukemia and
high-risk myelodys-plastic syndromes: produces stable disease but
at the cost of high toxicity. Leukemia. 27:1789–1791. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Hoang T, Campbell TC, Zhang C, et al:
Vorinostat and bortezomib as third-line therapy in patients with
advanced non-small cell lung cancer: a Wisconsin Oncology Network
Phase II study. Invest New Drugs. 32:195–199. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Friday BB, Anderson SK, Buckner J, et al:
Phase II trial of vorinostat in combination with bortezomib in
recurrent glioblastoma: a north central cancer treatment group
study. Neuro Oncol. 14:215–221. 2012. View Article : Google Scholar : PubMed/NCBI
|