Introduction
Pancreatic cancer is the fourth leading cause of
cancer-related death, with a 5-year survival rate below 5%
(1). The main reasons for this
poor prognosis include invasive behavior and multifactorial
chemoresistance of the cancer. Gemcitabine is considered as the
standard first-line chemotherapeutic agent for the treatment of
advanced pancreatic cancer and has offered some relief to affected
patients over the past two decades (2). However, its efficacy remains moderate
and the median overall survival times range from 5 to 8 months.
Furthermore, pancreatic cancer has been reported to easily acquire
resistance to gemcitabine after a few cycles of administration. For
this reason, numerous studies have sought to increase the efficacy
of chemotherapy by combining gemcitabine with other agents.
However, administration of other chemotherapeutics, monoclonal
antibodies, or radiation in addition to gemcitabine has not
resulted in any meaningful improvement in the survival of
pancreatic cancer patients (3–5).
Thus, pancreatic cancer remains a refractory cancer and one of the
most challenging problems in oncology (6). These disappointing results
necessitate novel combination therapies to improve the survival of
pancreatic cancer patients and to identify novel molecular targets
in this advanced disease (6).
The phosphatidylinositol-3-kinase (PI3K)/Akt
signaling pathway plays an important role in biological processes
including cell proliferation, differentiation and survival
(7,8). The PI3K pathway is activated by
various extracellular signals and leads to the phosphorylation of
Akt and its downstream effectors (9). When Akt is phosphorylated, it in
turn, phosphorylates proteins such as mTOR and 4EBP1 (9). Dysregulation of PI3K/Akt pathway is a
prominent feature of pancreatic cancers, because of the high
prevalence of abnormalities that regulate this pathway, including
K-ras mutations that occur in approximately 90% of cases, and
increased expression of EGFR, which is an important regulator in
PI3K/Akt signaling (10–13). Previous studies have also shown
that the activation of the PI3K/Akt signaling pathway is a result
of aberrant expression of PTEN in pancreatic cancer cell lines
(14,15). In addition, Akt is reported to be
constitutively overexpressed in various pancreatic cancer cell
lines (16). Importantly, the
PI3K/Akt pathway has been reported as a crucial factor in the
conferring of chemoresistance to gemcitabine in pancreatic cancer
(17–20). Furthermore, inhibition of the
PI3K/Akt pathway by LY294002 or wortmannin enhances
gemcitabine-induced apoptosis in human pancreatic cancer cells
(17). Considering these findings,
it is likely that the activation of the PI3K/Akt pathway has an
important role in the aggressive clinical features of pancreatic
cancer. Although PI3K/Akt inhibitors have therapeutic potential
when used either alone or in combination with gemcitabine in the
treatment of pancreatic cancer, severe cytotoxicity has been
observed in preclinical animal studies, leading to their limited
use in clinical trials. Thus, the PI3K/Akt pathway is still a
promising target for therapeutic intervention in human pancreatic
cancer, and the identification of effective novel PI3K inhibitors
to enhance clinical efficacy is important.
HS-104 is a synthetic PI3K inhibitor belonging to
the imidazopyridine derivative class (21). In our previous study, we reported
that HS-104 suppressed tumor proliferation and angiogenesis in
various cancers (22,23). On the basis of our previous
findings, we decided to combine gemcitabine and HS-104, which may
represent a major advantage over traditional chemotherapies. The
effect of these combinations on cancer cell growth, death and
angiogenesis is unknown. In this study, we tested the hypothesis
that co-treatment of the new PI3K inhibitor HS-104 could enhance
the anticancer effect of gemcitabine by blocking multiple pathways,
including the PI3K/Akt signaling pathway, thereby halting the
progression of pancreatic cancer.
Gemcitabine and HS-104 acted synergistically to
induce apoptosis and inhibit cell growth or angiogenesis in
pancreatic cancer. This combination treatment led to a stronger
inhibitory effect on cell viability than did treatment with either
drug alone. Our findings suggest that a combination treatment
comprising gemcitabine and HS-104 may be a promising therapeutic
strategy for pancreatic cancer.
Materials and methods
Cells and materials
Human pancreatic cancer cell lines (AsPC-1 and
PANC-1) used in this study were purchased from the American Type
Culture Collection (ATCC, Manassas, VA). AsPC-1 cells were grown in
Roswell Park Memorial Institute-1640 (RPMI-1640) medium and PANC-1
cells were grown in Dulbecco’s modified Eagle’s medium (DMEM). Both
types of media were supplemented with 10% fetal bovine serum (FBS)
and 1% penicillin/streptomycin. Cell cultures were maintained at
37°C in a CO2 incubator with a controlled humidified
atmosphere composed of 95% air and 5% CO2. Gemcitabine
[4-amino-1-(2-deoxy-2,2-difluoro-β-D-erythro-pentofuranosyl)
pyrimidin-2(1H)-on] was purchased from LTK Corporation
(Wilmington, DE), and HS-104
[N-(5-(3-(3-methyl-1,2,4-oxadiazol-3-yl)imidazo[1,2-a]pyridin-6-yl)pyridin-3-yl)
benzenesulfonamide], a new PI3K inhibitor, was synthesized
according to our previous methods (21).
Cell growth assay
The growth rate of gemcitabine- or HS-104-treated
cells was determined using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)
assay. Exponentially dividing cells (PANC-1, 6,000 cells; AsPC-1,
8,000 cells) were plated in 96-well plates. The following day, the
medium was removed and the cells were treated with either dimethyl
sulfoxide (DMSO) and/or DDW as a control or gemcitabine (1
μM) and/or HS-104 (1, 5 and 10 μM). After the cells
were incubated for 72 h, 20 μl of MTT solution (2 mg/ml) was
added to each well and the plate was incubated for another 4 h at
37°C. The formazan crystals formed were dissolved in DMSO (100
μl/well) with constant shaking for 30 min. The plate was
then read using a microplate reader at a wavelength of 540 nm. The
median inhibitory concentration for cell growth (IC50,
the drug concentration at which cell growth was inhibited by 50%)
was assessed from the dose-response curves. The combination index
(CI), used to measure drug interaction between gemcitabine and
HS-104, was calculated according to the Chou-Talalay method
(24). Data were analyzed using
the CalcuSyn software (Biosoft, Ferguson, MO). The CI index has
been used for data analysis of two-drug combinations. Index values
of CI<1, CI=1 and CI>1 indicate synergism, additive effect
and antagonism, respectively.
Western blot analysis
Total cellular protein content was extracted using a
lysis buffer containing 1% Triton X-100, 1% Nonidet P-40, and the
following protease and phosphatase inhibitors: aprotinin (10
mg/ml), leupeptin (10 mg/ml; Icn Biomedicals, Asse-Relegem,
Belgium), phenylmethylsulfonyl fluoride (1.72 mM), NaF (100 mM),
NaVO3 (500 mM), and
Na4P2O7 (500 mg/ml; Sigma-Aldrich,
St. Louis, MO). The proteins were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto nitrocellulose membranes. The blots were
immunostained with the appropriate primary antibodies followed by
secondary antibodies conjugated to horseradish peroxidase. Antibody
binding was detected with an enhanced chemiluminescence reagent
(Amersham Biosciences, Cambridge, MA). Antibodies against p-Mek
(Thr286), p-Erk (Thr202/Tyr204),
Erk, p-mTOR (Ser2448), mTOR, p-Akt (Ser473),
Akt, p-4EBP1, 4EBP1, PARP, cleaved caspase-3, Bcl-2, Bax and
β-actin were purchased from Cell Signaling Technology (Danvers,
MA).
Immunofluorescence
AsPC-1 cells were plated on 18-mm glass cover slips
with RPMI-1640 medium and grown to approximately 70% confluence for
24 h. The cells were treated with gemcitabine (1 μM) and/or
HS-104 (5 μM) for 6 h, washed twice with PBS, and fixed in
an acetic acid: 70% ethanol (1:2) solution for 10 min at −20°C.
Following fixation, the cells were washed with 1% Triton X-100 in
PBS (pH 7.4). Next, the cells were blocked in 1.5% horse serum in
PBS for 30 min at room temperature, and then incubated overnight at
4°C with primary antibodies (1:50) for p-Mek, p-4EBP1, p-Erk (Cell
Signaling Technology), p-Akt (Abcam, Cambridge, MA) and p-mTOR
(eBioscience, San Diego, CA), in a humidified chamber. After
washing twice with PBS, the cells were incubated with rabbit
tetramethylrhodamine isothiocyanate (TRITC)-labeled secondary
antibody (1:100, Dianova, Hamburg, Germany) for 1 h at room
temperature. The cells were also stained with
4,6-diamidino-2-phenylindole (DAPI) to visualize the nuclei. The
slides were then washed twice with PBS, and covered with Dabco
(Sigma-Aldrich) before confocal laser scanning microscopy was
performed (Olympus, Tokyo, Japan).
Analysis of cytochrome c
localization
AsPC-1 cells were plated on 18-mm glass coverslips
with RPMI-1640 medium and grown to approximately 70% confluence for
24 h. The cells were treated with gemcitabine (1 μM) alone
or in combination with HS-104 (5 μM) for 6 h, and then
incubated with 500 nM MitoTracker Red probe (Molecular Probes Inc.,
Eugene, OR) for 30 min at 37°C. Next, the cells were washed twice
with PBS, fixed in an acetic acid: 70% ethanol (1:2) solution for
10 min at −20°C, and then incubated overnight at 4°C with
cytochrome c antibody (1:50; Santa Cruz Biotechnologies,
Santa Cruz, CA). On the following day, the cells were washed twice
with PBS and incubated with mouse fluorescently labeled secondary
antibody (1:100, Dianova) for 1 h at room temperature. The cells
were also stained with DAPI to visualize the nuclei. The slides
were then washed twice with PBS, and covered with Dabco
(Sigma-Aldrich) before confocal laser scanning microscopy was
performed (Olympus).
DAPI staining and TUNEL assay
AsPC-1 cells were plated on 18-mm glass coverslips
with RPMI-1640 medium and grown to approximately 70% confluence for
24 h. The cells were treated with gemcitabine (1 μM) and/or
HS-104 (5 μM) for 24 h, washed twice with PBS, and fixed in
an acetic acid: 70% ethanol (1:2) solution for 10 min at −20°C.
Following fixation, the cells were washed with 1% Triton X-100 in
PBS (pH 7.4). Next, the cells were blocked in 1.5% horse serum in
PBS for 30 min at room temperature, and then stained with 2
μg/ml of DAPI (Sigma-Aldrich) for 20 min at 37°C. The
stained cells were examined under a fluorescence microscope for
evidence of nuclear fragmentation. TUNEL assay was performed using
the In Situ Cell Death Detection Kit (Roche Co. Ltd, Mannheim,
Germany).
Matrigel plug assay
All animal manipulations were performed in
accordance with the animal care guidelines of the Guide for Animal
Experiments by the Korean Academy of Medical Sciences. Male BALB/c
6-week-old mice were obtained from Orient-Bio Laboratory Animal
Research Center Co., Ltd. (Kapyung, South Korea). The animals were
fed with standard mice chow with free access to tap water in an
animal house with controlled temperature (21°C) and humidity and
alternating 12 h light-dark cycles. Liquid Matrigel was mixed at
4°C with concentrated VEGF (100 ng/ml), and 1 μM gemcitabine
and/or 5 μM HS-104 and/or PBS (10 μl) were injected
subcutaneously into the mice. Matrigel plugs were surgically
harvested on day 7 and photographed. The plugs were fixed in 4%
buffered formaldehyde, embedded in paraffin and sectioned. The
8-μm-thick sections were stained with hematoxylin and eosin
(H&E). H&E staining was performed to identify the formation
and infiltration of new, functional microvessels. Functional
vessels with intact RBCs were quantified manually by using a
microscope [high-power field (HPF) ×200 magnification].
Animal experiments
A total of 20 BALB/c nu/nu mice were
intraperitoneally injected with a ketamine:Rumpun mixture (9:1, 25
μl/mice) to induce anesthesia and were then arranged in the
decubitus position. Then, 100 μl of an AsPC-1 single-cell
suspension (1×106 ml−1) prepared using a 1-ml
injector was surgically implanted into the pancreas, after which
the needle was promptly pulled out. After injection, the mice were
maintained in an animal house with controlled temperature (21°C)
and humidity and alternating 12 h light-dark cycles, and fed with
standard mouse chow and had free access to tap water. One week
after tumor implantation, the mice were randomly divided into 4
groups to receive gemcitabine and HS-104 formulations. One group of
mice was left untreated, the second group received gemcitabine, the
third group received HS-104, and the fourth group received both
gemcitabine and HS-104. HS-104 was administered orally (20
mg/kg/day) as a 5% w/v solution in DMSO:PEG 400:water (1:5:4, v/v),
and gemcitabine was diluted in sterile saline and injected
intraperitoneally (25 mg/kg/day) 5 times per week for 4 weeks. Body
weight of the mice and tumor growth were monitored.
Immunohistochemistry
Immunostaining was performed using 8-μm-thick
sections of the tumor samples after deparaffinization. Microwave
antigen retrieval was performed in citrate buffer (pH 6.0) for 10
min prior to peroxidase quenching with 3% hydrogen peroxide
(H2O2) in PBS for 10 min. The sections were
then washed in water and preblocked with normal goat or horse serum
for 10 min. Next, the tissue sections were incubated overnight at
4°C in 1:50 dilutions of primary antibodies against PCNA, cleaved
caspase-3, CD34 (Santa Cruz Biotechnology), p-Akt and p-Mek (Cell
Signaling Technologies). The sections were then incubated with
biotinylated secondary antibodies (1:100) for 1 h. After washing
with PBS, streptavidin-HRP was applied. Finally, the sections were
developed with diaminobenzidine tetrahydrochloride substrate for 10
min, and counterstained with hematoxylin. At least three random
fields of each section were examined at a ×400 magnification.
Pimonidazole staining
Hypoxic regions in mouse tissues were visualized
using the Hydroxyprobe-1 kit (Chemicon International, Temecula,
CA). These experiments were approved by the animal experimentation
ethics committee, according to local and governmental regulations.
Briefly, mice were injected intraperitoneally with pimonidazole (60
mg/kg body weight), which forms adducts when reductively activated
in hypoxic cells. Mice were sacrificed 90 min later by asphyxiation
in CO2. The hydroxyprobe-1 monoclonal antibody was
biotinylated using d-biotinyl-ɛ-aminocaproic acid
N-hydroxysuccinimide ester (Boehringer, Mannheim, Germany)
and was used to stain pimonidazole adducts in formalin-fixed
paraffin-embedded tissues according to the manufacturer’s
instructions (25).
Statistical analysis
Data are expressed as the mean ± SD, and were
analyzed with an ANOVA and unpaired Student’s t-test. A p-value
≤0.05 was considered statistically significant. Statistical
calculations were performed using SPSS software for the Windows
operating system (version 10.0; SPSS, Chicago, IL).
Results
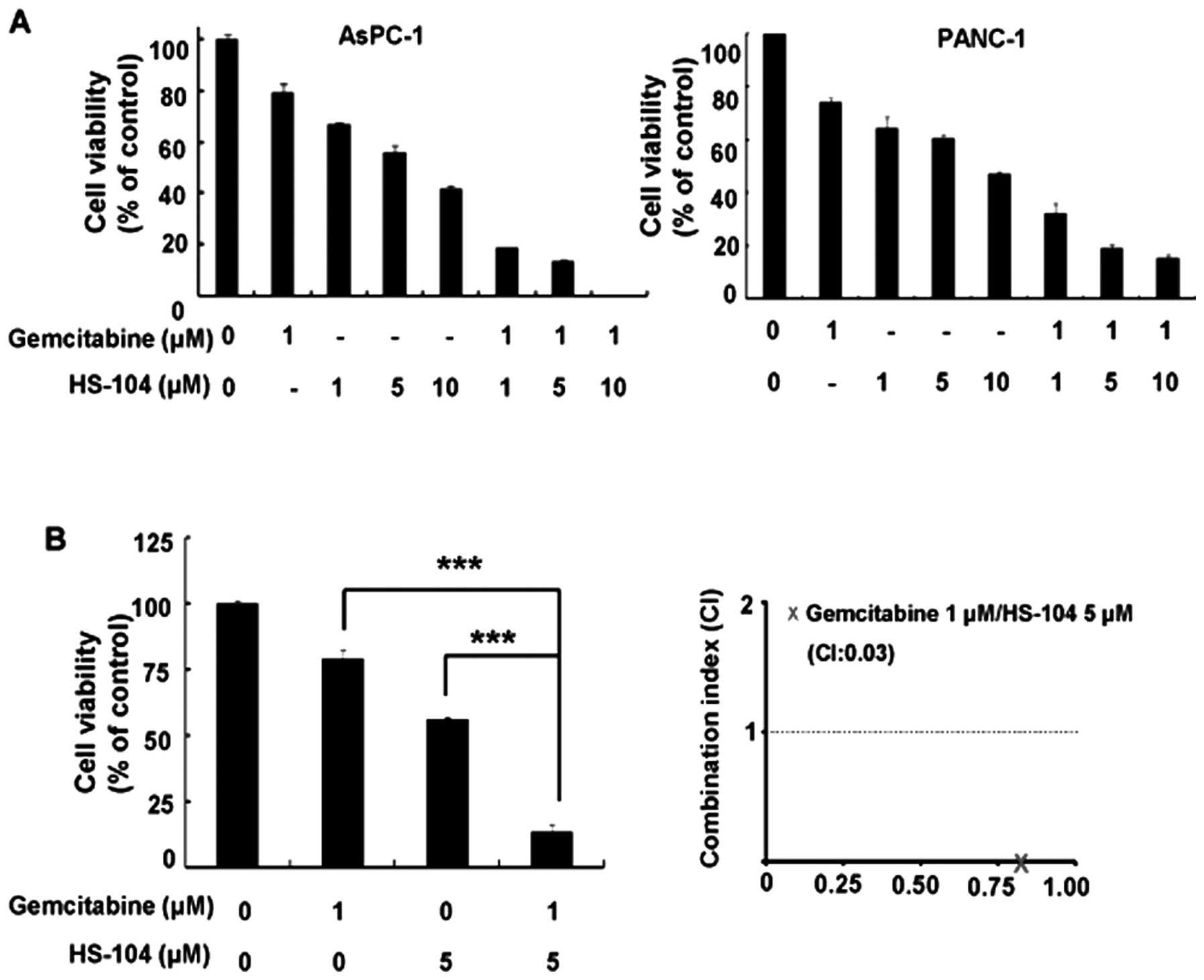
Gemcitabine and HS-104 synergistically
inhibit the proliferation of pancreatic cancer cells
We evaluated the effect of the gemcitabine and
HS-104 combination treatment on PANC-1 and AsPC-1 pancreatic cancer
cells by using a cell viability assay. The cells were treated with
a fixed concentration of gemcitabine (1 μM) alone or with
various concentrations of HS-104 (0, 1, 5 or 10 μM) for 72
h. Compared to treatment with either agent alone, the combination
of gemcitabine and HS-104 significantly inhibited growth in the two
human pancreatic cancer cell lines (Fig. 1A). To identify the synergistic
effect between gemcitabine and HS-104, we further examined the
combination index (CI) values using CalcuSyn software. Indeed, a
synergistic effect of gemcitabine and HS-104 was more significantly
observed in the AsPC-1 cells (Fig.
1B). The CI values were <1 for the combination of 1
μM gemcitabine and 5 μM HS-104 in AsPC-1 cells
(CI=0.03).
Gemcitabine and HS-104 synergistically
induce apoptosis
Since the combination treatment of gemcitabine and
HS-104 caused a significant reduction in cell proliferation, we
next investigated whether the treatment with gemcitabine and HS-104
induced apoptosis by performing DAPI and TUNEL staining in AsPC-1
cells. As shown in Fig. 2A, among
the cells simultaneously treated with the two drugs, the number of
TUNEL-positive cells was higher than that among the cells treated
with each agent alone. We next performed cytochrome c
staining to identify the involvement of the combined treatment of
gemcitabine and HS-104 in changes in mitochondrial membrane
potential (MMP), which induces the release of mitochondrial
cytochrome c into the cytosol, a hallmark of intrinsic
pathway-mediated apoptosis. As shown in Fig. 2B, we observed that the combined
treatment synergistically increased cytochrome c release
with a concomitant decrease in the colocalization of cytochrome
c and mitochondria. In addition, the combination treatment
significantly increased the expression levels of cleaved caspase-3
and Bax as well as decreased Bcl-2 expression in AsPC-1 cells
(Fig. 2C). Collectively, these
results indicate that the combination of gemcitabine and HS-104
synergistically induced apoptosis of pancreatic cancer cells.
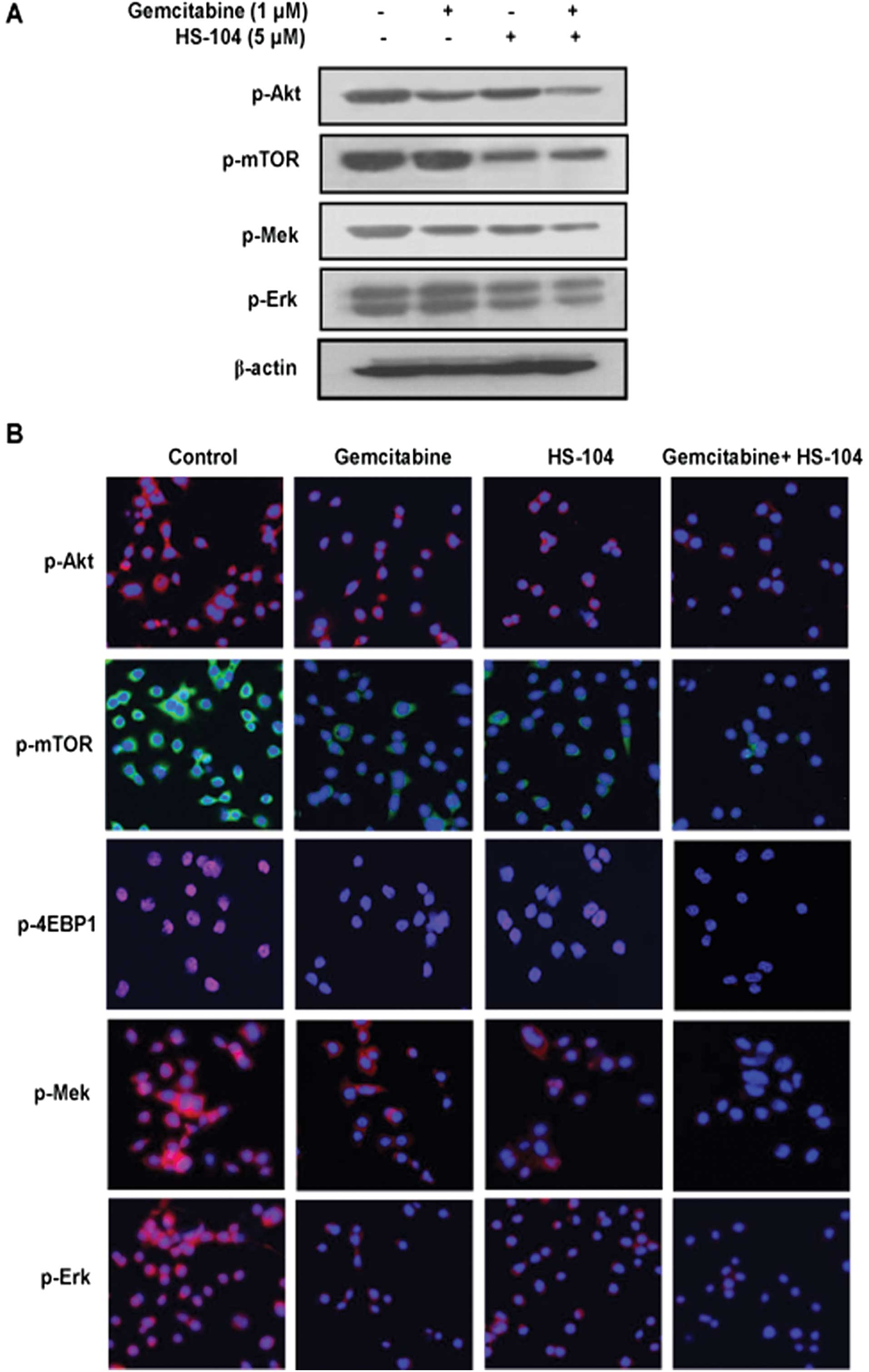
The combination of gemcitabine and HS-104
inhibit key enzymes in both PI3K/Akt and RAF/Mek signaling
pathways
The PI3K/Akt pathway is highly activated in
pancreatic cancer, consequently promoting cell proliferation,
survival and tumorigenesis (26,27).
Additionally, the Ras kinase-mediated cascade is the most widely
known pathway (28), and
upregulation of this pathway has been thought to play an important
role in pancreatic cancer cell growth (29). Hence, we examined the combination
effect of HS-104 (a PI3K inhibitor) and gemcitabine (a clinically
approved first-line anticancer drug) on the two given pathways in
pancreatic cancer cells. HS-104 clearly inhibited the expression of
p-Akt, p-mTOR and p-4EBP1, main mediators of the PI3K/Akt pathway,
while gemcitabine weakly inhibited the expressions of p-Akt,
p-mTOR, p-Mek and p-Erk in AsPC-1 cells. However, compared to
treatment with each agent alone, the combination treatment with
both agents decreased the expression of p-Akt, p-mTOR, p-Mek and
p-Erk, indicating synergistic suppression of both PI3K/Akt and
RAF/Mek pathways (Fig. 3A). These
results were confirmed using confocal fluorescent microscopy
(Fig. 3B).
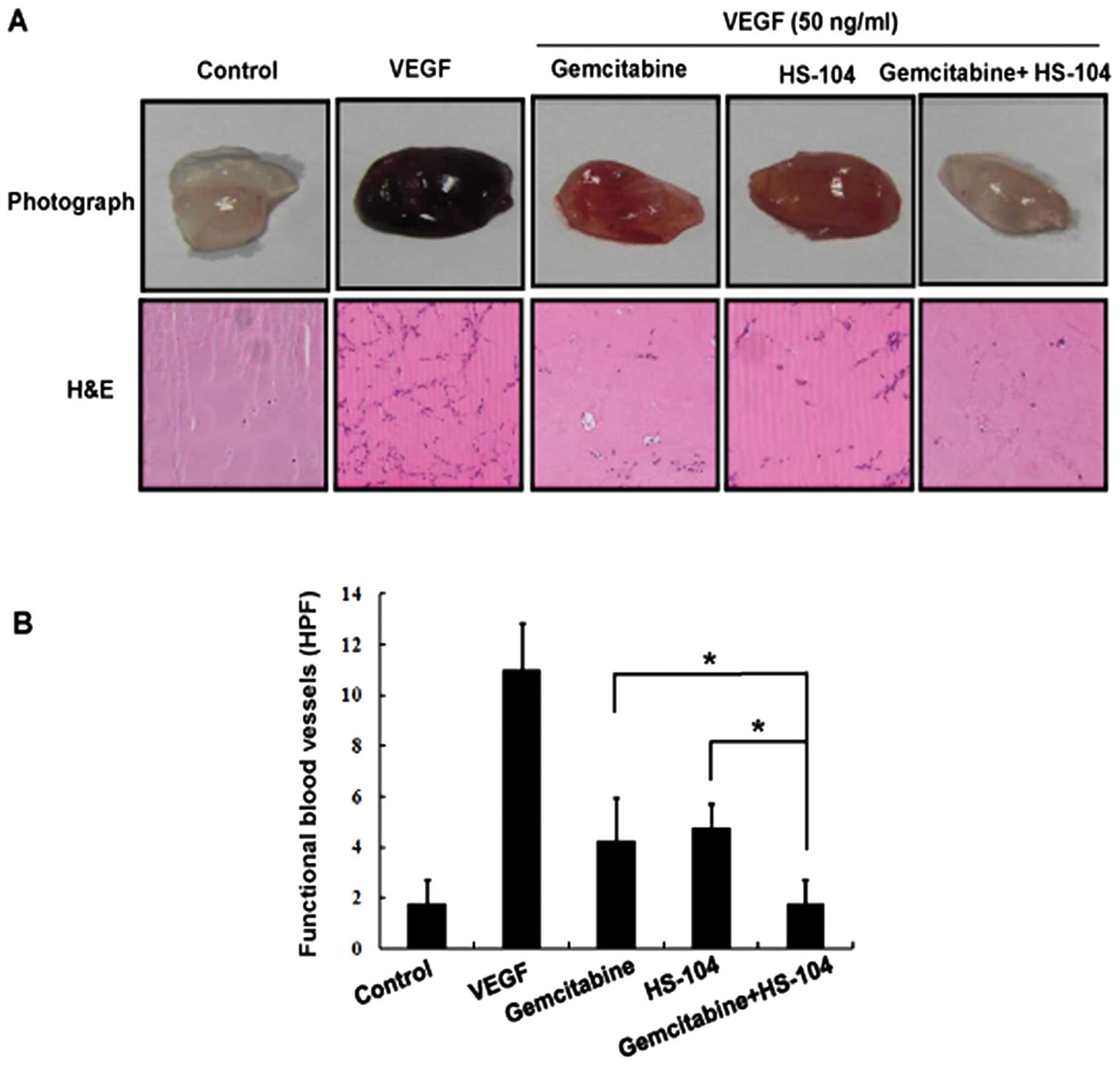
The combination of gemcitabine and HS-104
synergistically suppresses angiogenesis in an in vivo Matrigel plug
assay
We further explored the anti-angiogenic activity of
combined treatment of gemcitabine and HS-104 by using an in
vivo Matrigel plug assay. Matrigel containing VEGF alone or
with gemcitabine and/or HS-104 was subcutaneously injected into
male BALB/c mice and removed 7 days after implantation. As shown in
Fig. 4A, blood vessels were rarely
observed in Matrigel plugs without VEGF. The Matrigel plugs
containing VEGF alone appeared deep red in color because of the
presence of red blood cells (RBCs), indicating that new blood
vessels were formed inside the Matrigel via angiogenesis initiated
by VEGF. However, compared to treatment with a single agent alone,
the combination treatment of gemcitabine and HS-104 considerably
inhibited vascular formation (Fig.
4A). We performed H&E staining to determine functional
vasculature in the Matrigel plugs. As expected, the combination of
gemcitabine and HS-104 suppressed VEGF-induced functional blood
vessels formation to a greater degree than treatment with a single
agent alone (Fig. 4B). These
results suggest that the combination of gemcitabine with HS-104
showed a more potent inhibitory effect against VEGF-induced vessel
formation in vivo than that shown by treatment with either
agent alone.
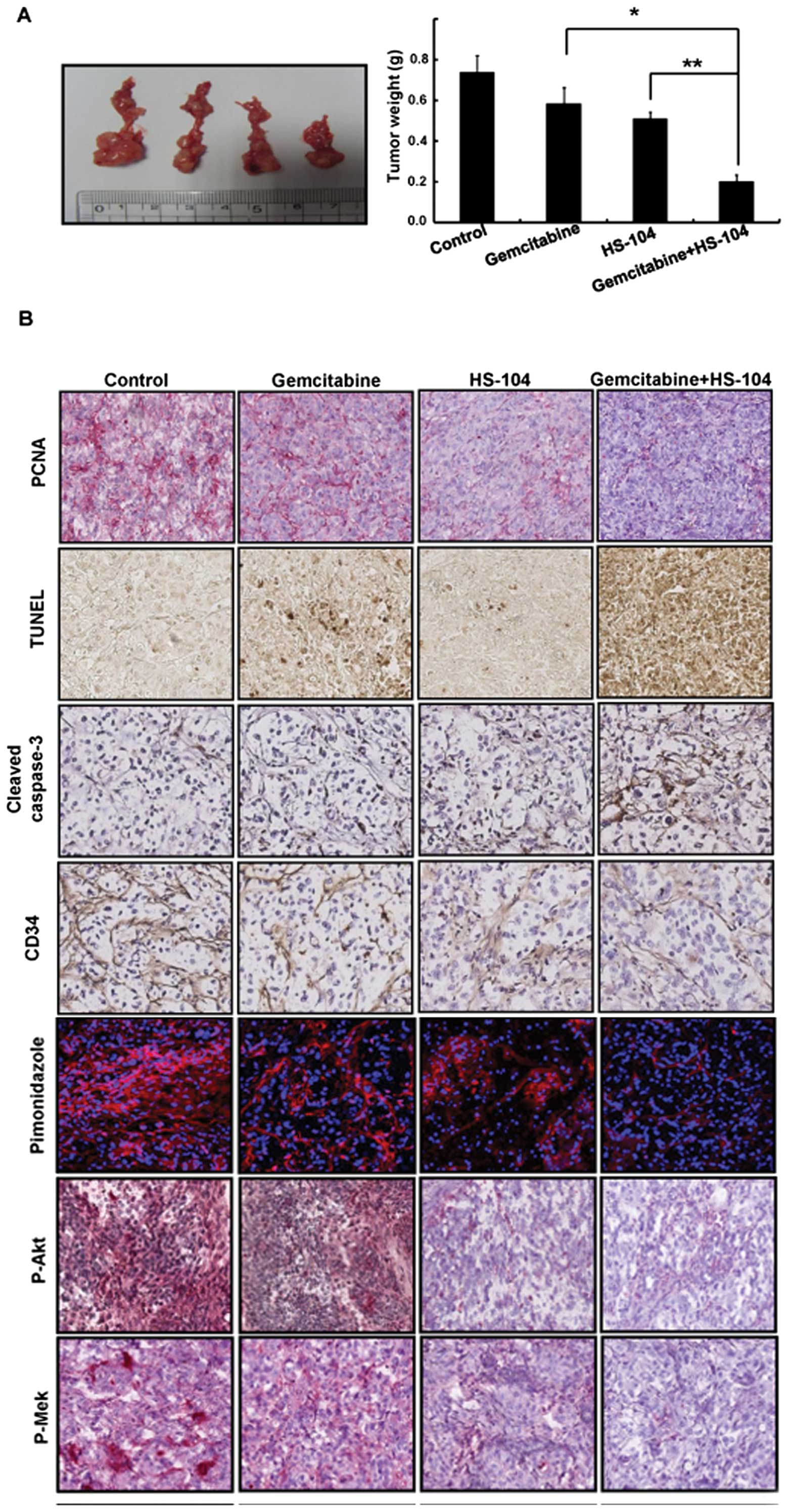
The combination effects of gemcitabine
with HS-104 in orthotopic pancreatic cancer animal models
To evaluate the antitumor efficacy of HS-104, alone
and in combination with gemcitabine, we used orthotopic models of
pancreatic cancer with AsPC-1 cells in BALB/c nu/nu mice. All
treatments were well tolerated, with no significant differences in
body weight among the treatment groups. The incidence of tumor
formation was more than 95% after implantation in the pancreas.
Treatment with HS-104 (20 mg/kg/day, p.o.) or gemcitabine (25
mg/kg/day, i.p.) inhibited the growth of the primary pancreatic
tumor by 33 or 23%, respectively, compared with the control
treatment. In particular, the antitumor effect was substantially
enhanced when the two agents were combined, with 73% growth
inhibition observed, compared to that observed with the control
treatment (Fig. 5A).
The combination of gemcitabine with
HS-104 inhibits proliferation and angiogenesis with induction of
apoptosis in the orthotopic pancreatic cancer models
In a histopathological analysis, we observed that
the combination treatment of HS-104 with gemcitabine
synergistically decreased the expression of PCNA, a cell
proliferation marker and CD34, an angiogenesis marker, compared
with the control treatment. In addition, the combination treatment
increased apoptotic cells in tumor tissues, as identified by
expression of cleaved caspase-3 and DNA fragments by performing the
TUNEL assay (Fig. 5B). Owing to
the antitumor effects of the combination treatment, we further
examined the hypoxic regions in the tumors, which were visualized
using a hypoxic probe (pimonidazole) that has been widely used to
detect cellular and tissue hypoxia (30). Interestingly, the pimonidazole
levels expectedly decreased because of the combination treatment
compared to treatment with a single agent alone. Moreover, the
combination treatment decreased the phosphorylation levels of Akt
and Mek for cell survival and proliferation.
Discussion
Pancreatic cancer has one of the poorest prognoses
among all cancers because of its tendency for late detection and
its peculiar resistance to chemotherapy. Chemotherapeutic options
for pancreatic cancer are limited, and the standard of care,
gemcitabine, improves survival only minimally (31). Combination therapy is typically
employed to achieve a better response rate than that achieved with
monotherapy and is generally designed empirically by using drugs
that act through different cytotoxic mechanisms with less
overlapping toxicity. Clinical studies indicate that the
combination of gemcitabine and various anticancer drugs is needed
for survival prolongation, although previous uses of combination
therapies have not resulted in meaningful improvement. Currently, a
major challenge is to identify new targets and more effective
combination therapies with gemcitabine, a first-line drug in the
treatment of pancreatic cancer.
Pancreatic cancers are associated with high
incidence of K-ras mutations with increased signaling through the
PI3K/Akt pathway and the cancer cells show high Akt activation
(16), suggesting PI3K/Akt as a
target for gemcitabine combinations. In addition, we previously
reported that HS-104, a novel PI3K inhibitor, inhibited tumor
growth by inhibiting PI3K/Akt signaling in hepatocellular carcinoma
and breast cancer (22,23). These findings have led us to
investigate the combination effect of gemcitabine and HS-104 in
pancreatic cancer. Our study showed that i) the combination of
gemcitabine and HS-104 could decrease the concentrations of
gemcitabine that were needed to inhibit pancreatic cancer cell
growth and induce apoptosis; ii) apoptosis by the combination of
gemcitabine and HS-104 was mediated by mitochondrial potential;
iii) the combination of gemcitabine and HS-104 enhanced cell death
by inhibiting PI3K/Akt signaling along with the Mek/Erk signaling
pathway; and iv) the combination of gemcitabine and HS-104
synergistically inhibited tumor growth in orthotopic pancreatic
cancer animal models.
The use of combinations of various agents, including
gemcitabine, for treating pancreatic cancer has been reported
previously. For instance, the combination of gemcitabine and
5-fluorouracil (5-FU) (32), as
well as gemcitabine with cetuximab (33) or bevacizumab (34), failed to improve survival rates in
pancreatic cancer patients. In this respect, our combination
experiment of gemcitabine and HS-104 is a meaningful trial. Indeed,
the present study showed that the combination of gemcitabine with
HS-104 significantly inhibited the growth of pancreatic cancer
cells compared to treatment with either agent alone. When we
calculated CI values to further characterize the synergistic effect
between gemcitabine and HS-104, the combination of 1 μM
gemcitabine with 5 μM HS-104 was the most effective in
AsPC-1 and PANC-1 cells. Since the PI3K/Akt signaling pathway is
considered to be a viable and effective target for pancreatic
cancer therapy (35,36) and the inhibition of Akt can enhance
the efficacy of gemcitabine chemotherapy in pancreatic cancer
(37–39), we identified signaling pathways via
which the combination of gemcitabine and HS-104 accelerated
inhibition of cell growth in pancreatic cancer cells. We found that
HS-104 inhibited substantially more the expression of p-Akt and
p-mTOR than that of p-Mek and p-Erk, whereas gemcitabine weakly
inhibited the expression of p-Akt, p-mTOR, p-Mek and p-Erk compared
to inhibition by HS-104. However, the combination treatment
decreased the expression of p-Mek, p-Erk, p-Akt, p-mTOR and p-4EBP1
more effectively than did treatment with either agent alone,
indicating synergistic suppression of both the PI3K/Akt and RAF/Mek
pathways. Considering these results, this synergistic effect by
both agents is likely to be mediated by the ability of gemcitabine
and HS-104 to inhibit both the PI3K/Akt and Raf/signaling
pathways.
Apoptosis is induced by two alternative pathways: an
extrinsic pathway mediated by the death receptor and the intrinsic
pathway mediated by mitochondria. The extrinsic pathway involves
cell surface death receptors, such as tumor necrosis factor or Fas,
which upon binding of their ligands, initiate signaling to
activated caspase-8, which cleaves caspase-3 directly to induce
apoptosis. The intrinsic pathway is initiated by mitochondrial
dysfunction and is regulated by members of the Bcl-2 family such as
Bcl-2, Bax, Bak and Bid. In particular, a mitochondrial change
activates the expression of members of the pro-apoptotic family and
triggers the release of cytochrome c, which in turn
activates caspase-9 and then caspase-3 in the cytosol (40). Our results showed that the
combination of gemcitabine and HS-104 induced apoptosis of
pancreatic cancer cells, which was observed as increased nuclear
fragmentation upon TUNEL and DAPI staining. We identified the
involvement of the combination treatment in mitochondrial membrane
potential: the combination treatment induced marked changes in
mitochondrial membrane potential and synergistically increased
cytochrome c release. In addition, it significantly
increased the expression levels of Bax and cleaved caspase-3 and
decreased that of Bcl-2 in pancreatic cancer cells. The synergistic
effect of the two agents on apoptosis was in line with the findings
reported by Wei et al (41)
and Ng et al (17), who
showed that a combination of gemcitabine and PI3K inhibitors
enhanced the induction of apoptosis in pancreatic cancer cells.
Collectively, our data showed that the combination of gemcitabine
with HS-104 synergistically induced mitochondria-mediated apoptosis
in pancreatic cancer cells.
Most importantly, anticancer efficacy of the
combination of gemcitabine and HS-104 in vitro was also
observed in vivo in pancreatic cancer orthotopic animal
models. Our study revealed that the combination of gemcitabine and
HS-104 inhibited tumor growth without loss of body weight. These
data are in accordance with induction of apoptosis due to increased
expression of cleaved caspase-3 and DNA fragmentation, as observed
in TUNEL staining, in parallel with a decrease in cell
proliferation, as observed upon PCNA immunostaining in tumor
tissues. Consequently, we considered that targeting PI3K/Akt
signaling pathways with HS-104 may enhance the antitumor activity
of gemcitabine, compared to treatment with either agent alone.
In conclusion, our results showed that a combination
treatment of gemcitabine and HS-104 synergistically enhanced
anticancer activity by inhibiting cell growth/proliferation and
inducing apoptosis in vitro and in vivo, thereby
indicating that induction of apoptosis and inhibition of cell
proliferation by two agents may be a contributing factor for the
suppression of tumor growth. In addition, our results showed that
HS-104 may augment the therapeutic effect of gemcitabine in
pancreatic cancer by direct or indirect inhibition of the PI3K/Akt
signaling pathway, thereby leading to sensitization of pancreatic
cancer cells to gemcitabine. Taken together, our findings suggest
that the combination treatment with these two agents might be a
candidate for future clinical application.
Acknowledgements
This research was supported by Inha
University Grant, the Korean Health Technology R&D Project
(A120266, A110944), Ministry of Health and Welfare, and the
National Research Foundation of Korea (NRF) funded by the Ministry
of Education, Science and Technology (NRF-2012-0002988,
2012R1A2A2A01045602).
References
|
1.
|
Brothers HM II and Kostic NM: Catalytic
activity of the serine proteases alpha-chymotrypsin and alpha-lytic
protease tagged at the active site with a (terpyridine)platinum(II)
chromophore. Biochemistry. 29:7468–7474. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD and Von Hoff DD:
Improvements in survival and clinical benefit with gemcitabine as
first-line therapy for patients with advanced pancreas cancer: a
randomized trial. J Clin Oncol. 15:2403–2413. 1997.PubMed/NCBI
|
|
3.
|
Moore MJ, Goldstein D, Hamm J, Figer A,
Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos
D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M and
Parulekar W: Erlotinib plus gemcitabine compared with gemcitabine
alone in patients with advanced pancreatic cancer: a phase III
trial of the National Cancer Institute of Canada Clinical Trials
Group. J Clin Oncol. 25:1960–1966. 2007. View Article : Google Scholar
|
|
4.
|
Van Cutsem E, Vervenne WL, Bennouna J,
Humblet Y, Gill S, Van Laethem JL, Verslype C, Scheithauer W, Shang
A, Cosaert J and Moore MJ: Phase III trial of bevacizumab in
combination with gemcitabine and erlotinib in patients with
metastatic pancreatic cancer. J Clin Oncol. 27:2231–2237. 2009.
|
|
5.
|
McGinn CJ, Zalupski MM, Shureiqi I,
Robertson JM, Eckhauser FE, Smith DC, Brown D, Hejna G, Strawderman
M, Normolle D and Lawrence TS: Phase I trial of radiation dose
escalation with concurrent weekly full-dose gemcitabine in patients
with advanced pancreatic cancer. J Clin Oncol. 19:4202–4208.
2001.PubMed/NCBI
|
|
6.
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar
|
|
7.
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Cheng JQ, Lindsley CW, Cheng GZ, Yang H
and Nicosia SV: The Akt/PKB pathway: molecular target for cancer
drug discovery. Oncogene. 24:7482–7492. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Martelli AM, Faenza I, Billi AM, Manzoli
L, Evangelisti C, Fala F and Cocco L: Intranuclear
3′-phosphoinositide metabolism and Akt signaling: new mechanisms
for tumorigenesis and protection against apoptosis? Cell Signal.
18:1101–1107. 2006.
|
|
10.
|
Agbunag C and Bar-Sagi D: Oncogenic K-ras
drives cell cycle progression and phenotypic conversion of primary
pancreatic duct epithelial cells. Cancer Res. 64:5659–5663. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Korc M: Role of growth factors in
pancreatic cancer. Surg Oncol Clin N Am. 7:25–41. 1998.
|
|
12.
|
Vincent F, de Boer J, Pfohl-Leszkowicz A,
Cherrel Y and Galgani F: Two cases of ras mutation associated with
liver hyperplasia in dragonets (Callionymus lyra) exposed to
polychlorinated biphenyls and polycyclic aromatic hydrocarbons. Mol
Carcinog. 21:121–127. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Bardeesy N and DePinho RA: Pancreatic
cancer biology and genetics. Nat Rev Cancer. 2:897–909. 2002.
View Article : Google Scholar
|
|
14.
|
Tong Z, Fan Y, Zhang W, Xu J, Cheng J,
Ding M and Deng H: Pancreas-specific Pten deficiency causes partial
resistance to diabetes and elevated hepatic AKT signaling. Cell
Res. 19:710–719. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Ali S, Banerjee S, Ahmad A, El-Rayes BF,
Philip PA and Sarkar FH: Apoptosis-inducing effect of erlotinib is
potentiated by 3,3′-diindolylmethane in vitro and in vivo using an
orthotopic model of pancreatic cancer. Mol Cancer Ther.
7:1708–1719. 2008.PubMed/NCBI
|
|
16.
|
Izuishi K, Kato K, Ogura T, Kinoshita T
and Esumi H: Remarkable tolerance of tumor cells to nutrient
deprivation: possible new biochemical target for cancer therapy.
Cancer Res. 60:6201–6207. 2000.PubMed/NCBI
|
|
17.
|
Ng SSW, Tsao MS, Chow S and Hedley DW:
Inhibition of phosphatidylinositide 3-kinase enhances
gemcitabine-induced apoptosis in human pancreatic cancer cells.
Cancer Res. 60:5451–5455. 2000.PubMed/NCBI
|
|
18.
|
Yokoi K and Fidler IJ: Hypoxia increases
resistance of human pancreatic cancer cells to apoptosis induced by
gemcitabine. Clin Cancer Res. 10:2299–2306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Bondar VM, Sweeney-Gotsch B, Andreeff M,
Mills GB and McConkey DJ: Inhibition of the phosphatidylinositol
3′-kinase-AKT pathway induces apoptosis in pancreatic carcinoma
cells in vitro and in vivo. Mol Cancer Ther. 1:989–997. 2002.
|
|
20.
|
Venkannagari S, Fiskus W, Peth K, Atadja
P, Hidalgo M, Maitra A and Bhalla KN: Superior efficacy of
co-treatment with dual PI3K/mTOR inhibitor NVP-BEZ235 and
pan-histone deacetylase inhibitor against human pancreatic cancer.
Oncotarget. 3:1416–1427. 2012.PubMed/NCBI
|
|
21.
|
Kim O, Jeong Y, Lee H, Hong SS and Hong S:
Design and synthesis of imidazopyridine analogues as inhibitors of
phosphoinositide 3-kinase signaling and angiogenesis. J Med Chem.
54:2455–2466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Jung KH, Zheng HM, Jeong Y, Choi MJ, Lee
H, Hong SW, Lee HS, Son MK, Lee S, Hong S and Hong SS: Suppression
of tumor proliferation and angiogenesis of hepatocellular carcinoma
by HS-104, a novel phosphoinositide 3-kinase inhibitor. Cancer
Lett. 328:176–187. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Lee H, Li GY, Jeong Y, Jung KH, Lee JH,
Ham K, Hong S and Hong SS: A novel imidazopyridine analogue as a
phosphatidylinositol 3-kinase inhibitor against human breast
cancer. Cancer Lett. 318:68–75. 2012. View Article : Google Scholar
|
|
24.
|
Lee KH, Lee JH, Han SW, Im SA, Kim TY, Oh
DY and Bang YJ: Antitumor activity of NVP-AUY922, a novel heat
shock protein 90 inhibitor, in human gastric cancer cells is
mediated through proteasomal degradation of client proteins. Cancer
Sci. 102:1388–1395. 2011. View Article : Google Scholar
|
|
25.
|
Greijer AE, Delis-van Diemen PM, Fijneman
RJ, Giles RH, Voest EE, van Hinsbergh VW and Meijer GA: Presence of
HIF-1 and related genes in normal mucosa, adenomas and carcinomas
of the colorectum. Virchows Arch. 452:535–544. 2008. View Article : Google Scholar
|
|
26.
|
Campbell PM, Groehler AL, Lee KM,
Ouellette MM, Khazak V and Der CJ: K-Ras promotes growth
transformation and invasion of immortalized human pancreatic cells
by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res.
67:2098–2106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Ruggeri BA, Huang L, Wood M, Cheng JQ and
Testa JR: Amplification and overexpression of the AKT2 oncogene in
a subset of human pancreatic ductal adenocarcinomas. Mol Carcinog.
21:81–86. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Sridhar SS, Hedley D and Siu LL: Raf
kinase as a target for anticancer therapeutics. Mol Cancer Ther.
4:677–685. 2005. View Article : Google Scholar
|
|
29.
|
Siu LL, Awada A, Takimoto CH, Piccart M,
Schwartz B, Giannaris T, Lathia C, Petrenciuc O and Moore MJ: Phase
I trial of sorafenib and gemcitabine in advanced solid tumors with
an expanded cohort in advanced pancreatic cancer. Clin Cancer Res.
12:144–151. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Doege K, Heine S, Jensen I, Jelkmann W and
Metzen E: Inhibition of mitochondrial respiration elevates oxygen
concentration but leaves regulation of hypoxia-inducible factor
(HIF) intact. Blood. 106:2311–2317. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Fujioka S, Niu J, Schmidt C, Sclabas GM,
Peng B, Uwagawa T, Li Z, Evans DB, Abbruzzese JL and Chiao PJ:
NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent
regulation of AP-1 activity. Mol Cell Biol. 24:7806–7819. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Reni M, Balzano G, Aprile G, Cereda S,
Passoni P, Zerbi A, Tronconi MC, Milandri C, Saletti P, Rognone A,
Fugazza C, Magli A, Di Muzio N, Di Carlo V and Villa E: Adjuvant
PEFG (cisplatin, epirubicin, 5-fluorouracil, gemcitabine) or
gemcitabine followed by chemoradiation in pancreatic cancer: a
randomized phase II trial. Ann Surg Oncol. 19:2256–2263. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Philip PA, Benedetti J, Corless CL, Wong
R, O’Reilly EM, Flynn PJ, Rowland KM, Atkins JN, Mirtsching BC,
Rivkin SE, Khorana AA, Goldman B, Fenoglio-Preiser CM, Abbruzzese
JL and Blanke CD: Phase III study comparing gemcitabine plus
cetuximab versus gemcitabine in patients with advanced pancreatic
adenocarcinoma: Southwest Oncology Group-directed intergroup trial
S0205. J Clin Oncol. 28:3605–3610. 2010. View Article : Google Scholar
|
|
34.
|
Kindler HL, Niedzwiecki D, Hollis D,
Sutherland S, Schrag D, Hurwitz H, Innocenti F, Mulcahy MF,
O’Reilly E, Wozniak TF, Picus J, Bhargava P, Mayer RJ, Schilsky RL
and Goldberg RM: Gemcitabine plus bevacizumab compared with
gemcitabine plus placebo in patients with advanced pancreatic
cancer: phase III trial of the Cancer and Leukemia Group B (CALGB
80303). J Clin Oncol. 28:3617–3622. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Yotsumoto F, Fukami T, Yagi H, Funakoshi
A, Yoshizato T, Kuroki M and Miyamoto S: Amphiregulin regulates the
activation of ERK and Akt through epidermal growth factor receptor
and HER3 signals involved in the progression of pancreatic cancer.
Cancer Sci. 101:2351–2360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
He L, Wu Y, Lin L, Wang J, Chen Y, Yi Z,
Liu M and Pang X: Hispidulin, a small flavonoid molecule,
suppresses the angiogenesis and growth of human pancreatic cancer
by targeting vascular endothelial growth factor receptor 2-mediated
PI3K/Akt/mTOR signaling pathway. Cancer Sci. 102:219–225. 2011.
View Article : Google Scholar
|
|
37.
|
Liu D, Zhang Y, Dang C, Ma Q, Lee W and
Chen W: siRNA directed against TrkA sensitizes human pancreatic
cancer cells to apoptosis induced by gemcitabine through an
inactivation of PI3K/Akt-dependent pathway. Oncol Rep. 18:673–677.
2007.PubMed/NCBI
|
|
38.
|
Yao J and Qian C: Inhibition of Notch3
enhances sensitivity to gemcitabine in pancreatic cancer through an
inactivation of PI3K/Akt-dependent pathway. Med Oncol.
27:1017–1022. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Zhang B, Shi ZL, Liu B, Yan XB, Feng J and
Tao HM: Enhanced anticancer effect of gemcitabine by genistein in
osteosarcoma: the role of Akt and nuclear factor-kappaB. Anticancer
Drugs. 21:288–296. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Soengas MS, Capodieci P, Polsky D, Mora J,
Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL,
Lazebnik YA, Cordon-Cardo C and Lowe SW: Inactivation of the
apoptosis effector Apaf-1 in malignant melanoma. Nature.
409:207–211. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Wei WT, Chen H, Wang ZH, Ni ZL, Liu HB,
Tong HF, Guo HC, Liu DL and Lin SZ: Enhanced antitumor efficacy of
gemcitabine by evodiamine on pancreatic cancer via regulating
PI3K/Akt pathway. Int J Biol Sci. 8:1–14. 2012. View Article : Google Scholar : PubMed/NCBI
|