Introduction
Numerous papers exist regarding the effects of
flavonoids aiding in the prevention of cancer growth through their
ability to act as anti-oxidants (1), enzyme inhibitors (2) and growth regulators (3). However, many of these papers limit
their conclusions to findings on cancer cells and fail to test the
impact flavonoids have on normal cells (4–7).
Studies, dating back to 1977, report a connection between high
flavonoid concentrations and potential mutagenic tendencies
(8–13). Skibola and Smith (8), in 2000, addressed the potential
danger of consuming high concentrations of flavonoids through
supplements, which are not regulated by the FDA.
The flavonol, quercetin, is a common supplement
promoted as having the ability to aid in cancer prevention, treat
chronic infections of the prostate and other common ailments
(14–16). Humans typically consume between 4
to 68 mg of flavonols daily through the consumption of fruits and
vegetables (8). Flavonoid
supplements, which range from 500 to 1000 mg per tablet, can
provide approximately 15 times more of the flavonol than can be
obtained through dietary consumption. Therefore, it is important to
determine flavonoid toxicity in normal cells in order to establish
if flavonoid supplements are truly safe for human consumption.
Additionally, one must consider the rate of flavonoid absorption
and bioavailability when designing a study to examine flavonoid
activity in vitro (14,17–20).
Flavonoids with concentrations above the absorption and
bioavailability limit are not relevant to real world applications,
since they will not be absorbed by the body.
Flavonoids have previously been studied as potential
therapeutic agents for breast (21,22),
prostate (23), lung (24,25),
colon (26) and skin (27) cancers. To be a good potential
therapeutic agent, flavonoids must be able to reduce cell viability
in the cancerous cells, while having a minimum effect on the normal
cells. In prostate cancer, polar natural flavonols (fisetin,
galangin, kaempferol, morin, myricetin and quercetin) have commonly
been found to exhibit these characteristics (3,28–30).
However, there is limited information about the effects of
hydrophobic and lipophilic flavonols on prostate cancer. The more
hydrophobic (alkoxyl, geranyl > dimethylallyl > halogen >
monolignol > methoxy > hydroxyl > glycosyl) and lipophilic
flavonols (I > Br > Cl > F) are the most potent inhibitors
of P-glycoprotein (P-gp), which is an important protein involved in
drug sensitivity and resistance (31). Halogenated flavonols could also
interact with Lewis bases, such as amines or alcohols of amino
acids, to potentially form non-covalent halogen bonds within the
cancer cells (32). Based on this
limited information, a series of more hydrophobic and lipophilic
analogs were synthesized with the assumption that they would
decrease cell viability of DU-145 and PC-3 prostate cancer more
effectively than the polar natural flavonols. The effects of
flavonols on the viability of normal human infant foreskin
fibroblasts (HIFF) were also studied.
Flavonoid cell viability has most commonly been
measured through colorimetric assays. However, in 2002, Bruggisser
et al (33) found MTT to be
an unreliable technique for studying flavonols due to the ability
of flavonols to reduce MTT in the absence of cells. Since then, at
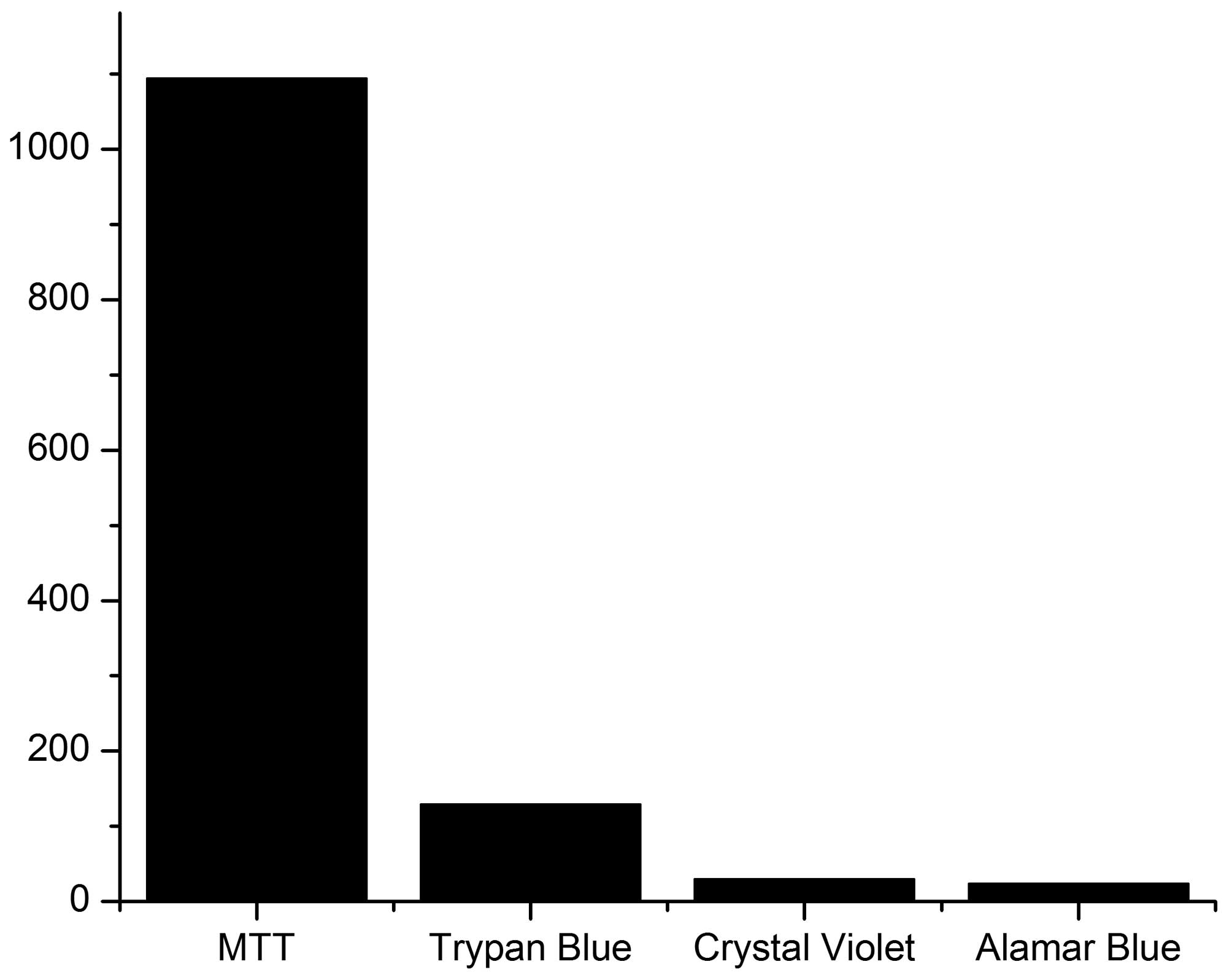
least 1019 flavonoid articles have been published using the MTT
assay. (A SciFinder search was conducted on 01/22/14 and consisted
of keyword: flavonoids; refine: MTT; sort by: publication year) A
recent review of the literature indicated that Alamar Blue, crystal
violet and trypan blue had also been used to determine flavonoid
cytotoxicity (Fig. 1). Thus, we
evaluated the reliability of these lesser utilized methods in the
presence of hydrophilic, hydrophobic and lipophilic flavonols to
find a more accurate method of measuring flavonoid
cytotoxicity.
Materials and methods
Chemistry reagents
All chemicals and solvents were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Spectra were obtained on a
Perkin-Elmer Paragon 1000 FT-IR spectrometer. Proton and carbon NMR
spectra were recorded on a Varian Mercury Vx 300 or 500 MHz
spectrometer with (CD3)2CO and
CD3OD as the solvents. High resolution mass spectral
(HRMS) data were obtained on an Applied Biosystems/MDS SCIEX 4800
Plus MALDI TOF/TOF mass spectrometer. Melting points were
determined on a Thomas Hoover Uni-melt and are uncorrected. The
purity of the compounds was determined to be >95% by elemental
analysis (Galbraith Laboratories, Inc., Knoxville, TN, USA).
General procedure for the synthesis of
the flavonol analogs.Synthesis of the flavonol methyl ethers
Methoxyphloroacetophenone (50 mg, 2.53 mmol) was
placed into a round bottom flask, dissolved in 1,2-dichloroethane
(5 ml) and 4 ml were distilled to remove water. The carboxylic acid
(3.54 mmol) and the acid chloride (8.33 mmol) were then added and
dissolved in triethylamine (5 ml) and dimethylformamide (DMF) (15
ml). The solution was stirred at room temperature under argon for 3
h before being heated to reflux for an additional 12 h. The
triethylamine and DMF were then removed under vacuum. The residue
was dissolved in 3.8 ml of ethanol and heated to reflux. A solution
of KOH (1.0 g) in H2O (1.4 ml) was then added and the
mixture was refluxed for an additional 60 min. The solvents were
removed under vacuum. The solid was dissolved in water (30 ml) and
the crude product was precipitated out of solution by the addition
of 10% HCl until the solution reached a pH of 5.0. The crude
precipitate was extracted into chloroform (5 ml) and rinsed with
sodium bicarbonate (10 ml × 2). Since the desired product was only
partially soluble in chloroform, these last two steps must be
repeated several times (6–10 x: until the chloroform extractions
are no longer yellow). The chloroform was removed under vacuum. The
crude product was then dissolved in a minimal amount of acetone
(~0.5 ml per 10 mg of flavonol). The solution becomes cloudy upon
the addition of hexanes (10 ml) and was filtered through a pipet
packed with glass wool. The filtrate contains the desired yellow
product. The filtrate was evaporated under vacuum and set aside.
Since some of the product remained in the precipitate, the
precipitate on the glass wool in the pipet was dissolved in
acetone, the acetone was evaporated, and trituration with
acetone/hexanes was repeated (2–4 x: until the filtrate is no
longer yellow). The NMR spectrum confirmed that the entire product
was removed from the crude precipitate.
Deprotection
The methyl ether flavonol (20 mg, 0.055 mmol) was
placed into a round bottom flask and dissolved in acetic acid (16
ml). HBr (48% aqueous solution, 2 ml) was added via a syringe. The
solution was heated to reflux for 24 h. Water (150 ml) was then
added, resulting in a cloudy solution. The precipitate was isolated
by filtration. This crude product was dissolved in chloroform (10
ml) and rinsed with sodium bicarbonate (10 ml × 3). The chloroform
was evaporated under vacuum. The crude product was dissolved in a
minimal amount of acetone (0.5 ml) and an excess of hexanes (10 ml)
was added. The filtrate was collected and the process was repeated
several times. The filtrate was dried under vacuum to give a yellow
precipitate.
2-(3-Benzoylphenyl)-5,7-dihydroxy-3-methoxy-4H-chromen-4-one
(
Fig. 2, 4a)
55% yield (108 mg, 0.278 mmol); 1H NMR
(300 MHz, CDCl3) δ 8.47 (dd, 1H, J=1.7, 3.3 Hz), 8.28
(ddd, 1H, J=1.4, 2.9, 8.1 Hz), 7.95 (ddd, 1H, J=1.4, 2.8, 7.8 Hz),
7.86 (m, 1H), 7.64 (m, 1H), 7.54 (m, 1H), 6.42 (d, 1H, J=2.0 Hz),
6.30 (d, 1H, J=2.0 Hz), 3.88 (s, 3H); 13C NMR (75 MHz,
acetone) δ 195.7 (C), 178.3 (C), 167.8 (C), 162.1 (C), 157.4 (C),
153.8 (C), 139.6 (C), 138.0 (C), 137.4 (C), 132.9 (CH), 131.9 (CH),
131.6 (CH), 131.2 (CH), 130.00 (CH), 129.97 (CH), 129.1 (C), 128.7
(CH), 104.35 (C), 99.8 (CH), 94.4 (CH), 60.04 (CH3).
2-(3-Benzoylphenyl)-3,5,7-trihydroxy-4H-chromen-4-one (3′-BPK)
(
Fig. 2, 5a)
84% yield (16 mg, 0.043 mmol); 1H NMR
(300 MHz, acetone) δ 12.10 (s, 1H), 8.70 (dd, 1H, J=1.5, 3.0 Hz),
8.52 (ddd, 1H, J=1.3, 1.8, 8.1 Hz), 7.90 (m, 1H), 7.76 (m, 1H),
7.62 (m, 1H), 6.58 (d, 1H, J=2.1 Hz), 6.40 (d, 1H, J=1.8 Hz);
1H NMR (300 MHz, CDCl3) δ 8.47 (dd, 1H,
J=1.7, 3.3 Hz), 8.28 (ddd, 1H, J=1.4, 2.9, 8.1 Hz), 7.95 (ddd, 1H,
J=1.4, 2.8, 7.8 Hz), 7.86 (m, 1H), 7.64 (m, 1H), 7.54 (m, 1H), 6.42
(d, 1H), 6.30 (d, 1H); 13C NMR (125 MHz, MeOH) δ 197.93
(C), 177.68 (C), 166.23 (C), 162.68 (C), 158.42 (C), 145.47 (C),
139.16 (C), 138.96 (C), 138.53 (C), 134.03 (CH), 133.04 (C), 132.41
(CH), 131.92 (CH), 131.15 (CH), 130.28 (CH), 129.80 (CH), 129.62
(CH), 104.72 (C), 99.55 (CH), 94.57 (CH); Anal. Calcd. for
C22H14O6 · H2O: C,
67.35%; H, 4.11%. Found: C, 66.94%; H, 4.08%.
2-(4-Benzoylphenyl)-5,7-dihydroxy-3-methoxy-4H-chromen-4-one
(
Fig. 2, 4b)
62% yield (121 mg, 0.312 mmol); 1H NMR
(300 MHz, acetone) δ 12.10 (s, 1H), 8.28 (ddd, 2H, J=1.8, 3.6, 8.4
Hz), 7.96 (ddd, 2H, J=1.8, 3.6, 8.7 Hz), 7.85 (m, 2H), 7.71 (dddd,
1H, J=1.7, 3.4, 7.4, 14.7 Hz), 7.60 (dddd, 2H, J=1.6, 3.2, 7.4,
14.7 Hz), 6.56 (d, 1H, J=1.8 Hz), 6.31 (d, 1H, J=1.8 Hz), 3.96 (s,
3H); 13C NMR (75 MHz, acetone) δ 195.09 (C), 178.84 (C),
164.77 (C), 162. 46 (C), 157.29 (C), 154.27 (C), 140.27 (C), 139.30
(C), 137.38 (C), 134.31 (C), 132.95 (CH), 129.98 (CH), 129.95 (CH),
128.72 (CH), 128.52 (CH), 105.47 (C), 99.03 (CH), 94.04 (CH), 60.14
(CH3).
2-(4-Benzoylphenyl)-3,5,7-trihydroxy-4H-chromen-4-one (4′-BPK)
(
Fig. 2, 5b)
84% yield (98 mg, 0.262 mmol), dec. 221°C; IR (KBr,
cm−1) 3283.9 (b), 1655.3 (s), 1636.8 (s), 1598.1 (s),
1501.2 (m), 1376.7 (m), 1319.5 (s), 1169.0 (s); 1H NMR
(500 MHz, acetone) δ 12.02 (s), 8.43 (d, 2H, J=8.5 Hz), 7.95 (d,
2H, J=8.5 Hz), 7.85 (m, 2H), 7.69 (m, 2H), 7.59 (ddd, 1H, J=1.5,
7.5, 13.5 Hz), 6.61 (d, 1H, J=2.5 Hz), 6.3 (d, 1H, J=2.0 Hz);
13C NMR (125 MHz, acetone) δ 195.90 (C), 176.94 (C),
165.53 (C), 162.32 (C), 158.06 (C), 144.66 (C), 139.11 (C), 138.96
(C), 138.26 (C), 135.64 (C), 133.51 (CH), 130.67 (CH), 130.65 (CH),
129.36 (CH), 128.30 (CH), 104.38 (C), 99.32 (CH), 94.67 (CH); HRMS
(TOF) m/z calcd. for
C22H14O6 (M+) 375.0868,
found 375.0803. Anal. Calcd for
C22H14O6 · H2O: C,
67.35%; H, 4.11%. Found: C, 65.55; H, 4.10%.
5,7-Dihydroxy-2-(3-iodophenyl)-3-methoxy-4H-chromen-4-one (
Fig. 2, 4c)
64% yield (34 mg, 0.083 mmol); 1H NMR
(500 MHz, MeOH) δ 8.39 (dd, 1H, J=2.0, 3.5 Hz), 8.07 (ddd, 1H,
J=1.0, 1.5, 8.0 Hz), 7.90 (ddd, 1H, J=1.0, 2.0, 8.0 Hz), 7.32 (t,
1H, J=7.8, 15.5 Hz), 6.44 (d, 1H, J=2.0 Hz), 6.23, (d, 1H, J=2.0
Hz), 3.81 (s, 3H); 13C NMR (125 MHz, MeOH) δ 180.06 (C),
166.38 (C), 163.27 (C), 158.59 (C), 155.46 (C), 140.97 (CH), 138.05
(CH), 133.78 (C), 131.50 (CH), 128.74 (CH), 106.21 (C), 100.03
(CH), 94.90 (CH), 94.68 (C), 60.93 (CH3).
3,5,7-Trihydroxy-2-(3-iodophenyl)-4H-chromen-4-one (
Fig. 2, 5c)
90% yield (18 mg, 0.045 mmol), dec. 143°C; IR (KBr,
cm−1) 3318.5 (b), 1690.8 (w), 1654.7 (s), 1601.9 (s),
1517.2 (m), 1375.6 (m), 1317.5 (m), 1168.3 (s); 1H NMR
(300 MHz, acetone) δ 12.10 (s, 1H), 8.62 (dd, 1H, J=1.7, 3.3 Hz),
8.27 (ddd, 1H, J=1.2, 1.7, 8.1 Hz), 7.88 (ddd, 1H, J=1.2, 1.7, 8.1
Hz), 7.40 (t, 1H, J=8.1, 15.9 Hz), 6.59 (d, 1H, J=2.1 Hz), 6.30 (d,
1H, J=2.1 Hz); 13C NMR (125 MHz, MeOH) δ 177.65 (C),
166.22 (C), 162.67 (C), 158.42 (C), 144.80 (C), 139.70 (CH), 139.09
(C), 137.31 (CH), 134.71 (C), 131.27 (CH), 127.79 (CH), 104.71 (C),
99.54 (CH), 94.66 (C), 94.59 (CH); HRMS (TOF) m/z calcd. for
C15H9IO5 (M+) 396.9573,
found 396.9591. Anal. Calcd. for
C15H9IO5 · H2O: C,
43.50%; H, 2.68%. Found: C, 44.39%; H 2.60%.
5,7-Dihydroxy-2-(4-iodophenyl)-3-methoxy-4H-chromen-4-one (
Fig. 2, 4d)
62% yield (31 mg, 0.076 mmol); 1H NMR
(500 MHz, MeOH) δ 7.92 (ddd, 2H, J=2.1, 4.3, 8.5 Hz), 7.83 (ddd,
2H, J=2.0, 4.3, 9.0 Hz), 6.42 (d, 1H, J=2.0 Hz), 6.23 (d, 1H, J=2.5
Hz), 2.14 (s, 3H); 13C NMR (125 MHz, MeOH) δ 180.05 (C),
166.38 (C), 163.25 (C), 158.56 (C), 156.30 (C), 140.86 (C), 139.15
(CH), 131.32 (C), 130.98 (CH), 106.17 (C), 100.00 (CH), 98.53 (C),
94.88 (CH), 60.86 (CH3).
3,5,7-Trihydroxy-2-(4-iodophenyl)-4H-chromen-4-one (
Fig. 2, 5d)
92% yield (18.4 mg, 0.046 mmol), dec. 154°C; IR
(KBr, cm−1) 3277.4 (b), 1686.5 (w), 1654.2 (s), 1623.6
(m), 1597.2 (s), 1499.5 (m), 1370.3 (m), 1316.8 (m), 1169.4 (s);
1H NMR (500 MHz, MeOH) δ 7.93 (ddd, 2H, J=2.0, 4.0, 9.0
Hz), 7.83 (ddd, 2H, J=1.5, 3.5, 8.5 Hz), 6.37 (d, 1H, J=2.0 Hz),
6.17 (d, 1H, J=2.0 Hz); 13C NMR (125 MHz, MeOH) δ 210.13
(C), 177.56 (C), 166.11 (C), 162.63 (C), 158.33 (C), 145.67 (C),
138.80 (CH), 132.22 (C), 130.21 (CH), 104.68 (C), 99.47 (CH), 96.85
(C), 94.55 (CH); HRMS (TOF) m/z calcd. for
C15H9IO5 (M+) 396.9573,
found 396.9592. Anal. Calcd. for
C15H9IO5 · H2O: C,
43.50%; H, 2.68%. Found: C, 44.12%; H, 2.62%.
5,7-Dihydroxy-3-methoxy-2-(p-tolyl)-4H-chromen-4-one (
Fig. 2, 4e)
20% yield (15 mg, 0.050 mmol); 1H NMR
(500 MHz, MeOH) δ 7.96 (d, 2H, J=8.5 Hz), 7.36 (d, 2H, J=8.0 Hz),
6.41 (d, 1H, J=2.0 Hz), 6.20 (d, 1H, J=2.0 Hz), 3.78 (s, 3H), 2.43
(s, 3H); 13C NMR (125 MHz, MeOH) δ 180.12 (C), 166.32
(C), 163.19 (C), 158.60 (C), 157.67 (C), 142.96 (C), 140.30 (C),
130.40 (CH), 129.40 (CH), 128.91 (C), 106.01 (C), 99.92 (CH), 94.84
(CH), 60.73 (CH3), 21.52 (CH3).
3,5,7-Trihydroxy-2-(p-tolyl)-4H-chromen-4-one
(
Fig. 2, 5e)
80% yield (4 mg, 0.014 mmol), dec. 139°C; IR (KBr,
cm−1) 3299.7 (b), 1700.9 (w), 1654.2 (s), 1598.1 (s),
1500.0 (m), 1371.6 (m), 1313.5 (m), 1166.6 (s); 1H NMR
(500 MHz, acetone) δ 12.20 (s, 1H), 8.17 (d, 2H, J=8.5 Hz), 7.40
(d, 2H, J=8.0 Hz), 6.58 (d, 1H, J=2.0 Hz), 6.30 (d, 1H, J=2.0 Hz),
2.43 (s, 3H); 13C NMR (125 MHz, acetone) δ 176.13 (C),
164.41 (C), 161.51 (C), 157.31 (C), 145.77 (C), 140.59 (C), 136.87
(C), 129.42 (CH), 128.64 (C), 127.82 (CH), 103.60 (C), 98.47 (CH),
93.91 (CH), 20.78 (CH3); HRMS (TOF) m/z calcd.
for C16H12O5 (M+)
285.0763, found 285.0783. Anal. Calcd. for
C16H12O5 · H2O:
C,63.57%; H, 4.67%. Found: C, 65.60%; H, 4.91%.
Galangin 3-methyl ether (
Fig. 2, 4f)
44% yield (32 mg, 0.113 mmol); 1H NMR
(300 MHz, acetone) δ 12.70 (s, 1H), 8.09 (m, 2H), 7.57 (m, 3H),
6.52 (d, 1H, J=2.0 Hz), 6.27 (d, 1H, J=2.0 Hz), 3.88 (s, 3H);
13C NMR (75 MHz, acetone) δ 178.91 (C), 164.34 (C),
162.48 (C), 157.27 (C), 155.72 (C), 139.50 (C), 131.02 (CH), 130.85
(C), 128.77 (CH), 128.52 (CH), 105.45 (C), 98.87 (CH), 93.95 (CH),
59.92 (CH3).
Galangin (
Fig.
2, 5f)
92% yield (12 mg, 0.044 mmol); 1H NMR
(500 MHz, MeOH) δ 8.19 (ddd, 2H, J=1.5, 3.0, 8.5 Hz), 7.51 (ddd,
2H, J=1.5, 7.0, 15.0 Hz), 7.45 ( ddd, 1H, J=1.5, 7.0, 16.0 Hz),
6.42 (m, 1H), 6.19 (m, 1H); 13C NMR (125 MHz, MeOH) δ
176.51 (C), 164.79 (C), 161.46 (C), 157.29 (C), 145.74 (C), 137.34
(C), 131.48 (C), 129.71 (CH), 128.27 (CH), 127.58 (CH), 103.50 (C),
98.21 (CH), 93.35 (CH).
General procedure for the synthesis of
aryl flavonol ethers through Suzuki-Miyaura cross-coupling
reactions
The procedure developed by Molander et al
(34) was used with minor
modifications. The organotrifluoroborate (0.073 mmol), the iodo
flavonol methyl ether (20 mg, 0.049 mmol), potassium carbonate (20
mg, 0.146 mmol) and Pd(OAc)2 (1 mg, 0.004 mmol) were
placed in a round bottom flask. These compounds were dissolved in 4
ml of methanol and heated to 65°C overnight (16 h) with stirring.
The solution was cooled to room temperature and 10% HCl was added
until the solution reached a pH of 5.0. The resulting precipitate
was isolated by filtration. The precipitate was extracted with
acetone until the extracts were no longer yellow and then the
acetone was removed under vacuum. The crude product was dissolved
in a minimal amount of acetone (0.5 ml) and excess hexanes (10 ml)
were added. The precipitate was isolated by filtration and this
process was repeated several times. The filtrate was dried under
vacuum to give a yellow precipitate. Deprotection of the flavonol
ethers was achieved as described earlier.
2-[(1,1′-Biphenyl)-3-yl]-5,7-dihydroxy-3-methoxy-4H-chromen-4-one
(Fig. 3, 7c)
68% yield (30 mg, 0.083 mmol); 1H NMR
(500 MHz, MeOH) δ 8.28 (dd, 1H, J=1.5, 3.0 Hz), 8.02 (ddd, 1H,
J=1.0, 1.5, 7.5 Hz), 7.79 (ddd, 1H, J=1.0, 2.3, 7.5 Hz), 7.67 (m,
2H), 7.63 (t, 1H, J=8.0, 16.0 Hz), 7.49 (m, 2H), 7.39 (ddd, 1H,
J=1.5, 3.5, 7.5 Hz), 6.43 (d, 1H, J=2.5 Hz), 6.21 (d, 1H, J=2.0
Hz), 3.83 (s, 3H); 13C NMR (125 MHz, MeOH) δ 180.10 (C),
163.20 (C), 158.78 (C), 157.35 (C), 143.07 (C), 141.67 (C), 140.73
(C), 137.50 (C), 132.39 (C), 130.69 (CH), 130.34 (CH), 130.10 (CH),
128.91 (CH), 128.28 (CH), 128.13 (CH), 127.99 (CH), 105.970 (C),
100.26 (CH), 95.12 (CH), 60.97 (CH3).
2-[(1,1′-Biphenyl)-3-yl]-3,5,7-trihydroxy-4H-chromen-4-one
(Fig. 3, 8c)
93% yield (18 mg, 0.051 mmol), dec. 138°C; IR (KBr,
cm−1) 3331.1 (b), 1686.1 (w), 1654.4 (s), 1603.6 (s),
1508.3 (m), 1375.4 (m), 1319.9 (m), 1169.0 (s); 1H NMR
(500 MHz, acetone) δ 8.54 (s, 1H), 8.25 (d, 1H, J=8.0 Hz), 7.80 (d,
1H, J=7.5 Hz), 7.75 (d, 2H, J=7.5 Hz), 7.68 (t, 1H, J=7.5, 15.0
Hz), 7.53 (m, 2H), 7.43 (t, 2H, J=7.5, 15.0 Hz), 6.61 (d, 1H, J=2.0
Hz), 6.30 (d, 1H, J=2.0 Hz); 13C NMR (125 MHz, acetone)
δ 176.32 (C), 164.75 (C), 161.78 (C), 157.41 (C), 145.41 (C),
141.61 (C), 140.74 (C), 132.09 (C), 129.39 (CH), 129.22 (CH),
128.77 (CH), 127.95 (CH), 127.28 (CH), 126.81 (CH), 126.38 (CH),
103.75 (C), 98.66 (CH), 94.08 (CH); HRMS (TOF) m/z calcd.
for C21H14O5 (M+)
347.0919, found 347.0967. Anal. Calcd. for
C21H14O5 · H2O: C,
69.23%; H, 4.43%. Found: C, 69.09%; H, 4.30%.
2-[(1,1′-Biphenyl)-4-yl]-5,7-dihydroxy-3-methoxy-4H-chromen-4-one
(Fig. 3, 7d)
67% yield (6 mg, 0.017 mmol); 1H NMR (500
MHz, acetone) δ 12.70 (s, 1H), 8.21 (ddd, 2H, J=2.0, 3.9, 4.0, 8.5
Hz), 7.87 (ddd, 2H, J=2.0, 3.0, 9.0 Hz), 7.77 (m, 2H), 7.52 (m,
2H), 7.43 (dddd, 1H, J=1.5, 2.5, 7.5, 15.0 Hz), 6.55 (d, 1H, J=2.0
Hz), 6.28 (d, 1H, J=2.0 Hz), 3.93 (s, 3H); 13C NMR (125
MHz, acetone) δ 179.62 (C), 165.04 (C), 163.25 (C), 158.01 (C),
156.04 (C), 144.07 (C), 140.60 (C), 130.44 (C), 129.90 (CH), 129.77
(CH), 128.97 (CH), 127.84 (CH), 127.83 (CH), 106.11 (C), 99.47
(CH), 94.56 (CH), 60.49 (CH3).
2-[(1,1′-Biphenyl)-4-yl]-3,5,7-trihydroxy-4H-chromen-4-one
(Fig. 3, 8d)
98% yield (6 mg, 0.016 mmol), dec. 148°C; IR (KBr,
cm−1) 3299.7 (b), 1702.1 (w), 1654.3 (s), 1625.1 (s),
1603.4 (s), 1508.0 (m), 1384.0 (m), 1314.4 (m), 1166.2 (s);
1H NMR (500 MHz, acetone) δ 12.10 (s, 1H), 8.36 (ddd,
2H, J=2.0, 4.3, 8.5 Hz), 7.87 (ddd, 2H, J=2.0, 3.5, 9.0 Hz), 7.77
(m, 2H), 7.51 (dddd, 2H, J=1.5, 4.0, 8.0, 15.0 Hz), 7.41 (dddd, 1H,
J=1.3, 2.6, 7.5, 15.0 Hz), 6.59 (d, 1H, J=2.0 Hz), 6.29 (d, 1H,
J=2.0 Hz); 13C NMR (125 MHz, acetone) δ 176.82 (C),
165.26 (C), 162.41 (C), 157.99 (C), 145.87 (C), 143.15 (C), 140.76
(C), 138.05 (C), 131.04 (C), 129.86 (CH), 129.01 (CH), 128.82 (CH),
127.79 (CH), 127.76 (CH), 104.33 (C), 99.24 (CH), 94.61 (CH); HRMS
(TOF) m/z calcd. for
C21H14O5 (M+) 347.0919,
found 347.0995. Anal. Calcd. for
C21H14O5 · H2O: C,
69.23%; H, 4.43%. Found: C, 68.21%; H, 4.27%.
Biological reagents
Quercetin, kaempferide, kaempferol and morin were
purchased from Indofine (Hillsborough, NJ, USA). Vi-cell reagent
Pak was purchased from Beckman Coulter (Fullerton, CA, USA). Alamar
Blue was purchased from BioSource International (Camarillo, CA,
USA).
Culture of prostate cancer and HIFF
cells
DU-145 and PC-3 were obtained from the American Type
Tissue Collection (Rockville, MD, USA). HIFF were a gift from Dr
Mary Sanchez-Lanier, Department of Microbiology, Washington State
University. HIFF are a fibroblast cell line that was isolated from
human infant foreskin under written consent from the minor’s parent
(approval #60277 from the Washington State University Institutional
Review Board). HIFF and the DU-145 cells were cultured in DMEM
(Hyclone, Logan, UT, USA) containing 1% penicillin/streptomycin
(Mediatech Incorporated, Herndon, VA, USA) and 10% FBS
(Equitech-Bio Inc., Kerrville, TX, USA). The PC-3 cells were
cultured in RPMI-1640 (Invitrogen Corp., Carlsbad, CA, USA)
containing 10% FBS and 1% penicillin/streptomycin. All of these
cells were seeded into 24-well plates and kept in a 37°C humidified
incubator with 5% CO2.
Treatment and measurement of cell
viability
Stock solutions of the natural product and synthetic
flavonol analogs were prepared by dissolving them in DMSO.
Preliminary dose-response studies were conducted in the prostate
cancer and HIFF cell lines with each flavonol to find the minimum
and maximum effective dose range and an approximate
EC50. The actual EC50 for each flavonol was
calculated from a minimum of 6 concentrations selected within the
linear portion of initial dose range curves and bracketing the
estimated EC50. Cells were seeded into 24-well plates 24
h prior to the experiment at the following cell densities: DU-145
(6,000 cells/well), PC-3 (6,500 cells/well) and HIFF (6,000
cells/well). The stock solutions of the flavonols were added to
each of the wells, except the control, to give the desired
concentration of the flavonols and a final concentration of 0.2%
DMSO and the plates were incubated for 72 h. DMSO did not alter the
growth or viability of DU-145, PC-3 or HIFF (data not shown).
Assays were run in quadruplicate. The reliability of three cell
viability assays was tested with the following reagents: i) Alamar
Blue: After removal of the culture medium, cells were washed with
PBS and incubated with Alamar Blue for 6 h. Fluorescence was
measured with an excitation at 530 nm and an emission at 590 nm.
ii) Crystal violet: After removal of the culture medium, the cells
were fixed with 200 μl of 4% formaldehyde at 4°C for 30 min, washed
with a 3:1 mixture of MeOH: AcOH, rinsed with 80% MeOH, and stained
with 0.1% crystal violet. After 1 h at room temperature, the wells
were gently rinsed with distilled water, dried, dissolved in 10%
AcOH and the absorbance was read at 570 nm. iii) Trypan blue
exclusion: The cells were trypsinized, washed with calcium free
phosphate buffered saline (PBS), centrifuged and resuspended in 500
μl of PBS. Cell viability was accurately determined using the
semi-automated Vi-Cell XR Cell Analyzer (Beckman Coulter).
Statistical analysis
The EC50 values of the flavonols in each
cell line were analyzed by one-way analysis of variance (ANOVA).
Comparisons of the EC50 values of the flavonols between
cell lines were evaluated by two-way ANOVA. Differences were
considered significant at P<0.05.
Results
Chemistry
A series of flavonol analogs were synthesized from
the starting material, ω-methoxyphloroacetophenone 1 (Fig. 2) (23,35,36).
Initially, these compounds were synthesized through a modified
multistep procedure by Tanaka et al (37), in which the
ω-methoxyphloroacetophenone 1 was esterified by the acid chloride 3
to produce a triester intermediate, which was subsequently cyclized
by refluxing in pyridine containing K2CO3 to
give the flavonol methyl ether 4. This sequence resulted in
moderate net yields of the flavonol methyl ethers. A major reason
for the modest yields was the poor recovery of the products from
the chromatographic purifications at each step. Therefore, we
explored an alternative approach, the ‘one pot’ procedure described
by Ichikawa et al (38)
followed by ester hydrolysis, to synthesizing the flavonol methyl
ether 4. Ichikawa et al used Et3N as both the
solvent and the base. This procedure resulted in slightly higher
yields (26–35%) of the flavonol methyl ether 4 than the previous
process. During these reactions we noted the low solubility of the
ω-methoxyphloroacetophenone in Et3N. Based on these
observations, the yields of compound 4 were increased through a
series of optimization reactions (solvent, temperature and reaction
time). These studies led to the use of Et3N as the base
and DMF as the solvent to give a homogenous solution, which
significantly increased the yields of the cyclized products. With
these optimal conditions, good yields were achieved in the
synthesis of the hydrophobic aromatic and halogenated flavonol
ethers 4a–d [3′-benzoyl (55%) and 4′-benzoyl (62%), 3′-iodo (64%)
and 4′-iodo (62%)]. Moderate to low yields were observed in the
synthesis of the smaller, less hydrophobic and non-substituted
flavonol ethers 4e–f [H-flavonol (44%) and 4′-methyl (20%)].
Demethylation of the methyl ethers of flavonols was known to occur
under strong, anhydrous Lewis Acid conditions (i.e.
AlBr3, CH3CN) (39), so we were pleasantly surprised to
find that deprotection of 4 could be achieved under the relatively
milder conditions of aqueous HBr in acetic acid (40), to form 5a–f in excellent yields
[3′-benzoyl (37) (84%) and
4′-benzoyl (84%), 3′-iodo (90%) and 4′-iodo (41) (92%), H-flavonol (42–45)
(92%) and 4′-methyl (80%) flavonols].
The remaining two analogs (3′- and 4′-phenyl) were
synthesized through a Suzuki-Miyaura palladium catalyzed
cross-coupling reaction using a procedure developed by Molander
et al (34) (Fig. 3). The 3′- and 4′-iodo flavonol
ethers 4c,d readily coupled with potassium phenyl trifluoroborate
using Pd(OAc)2 and potassium carbonate in methanol to
give high yields of both 3′-phenyl 7c (68%) and the 4′-phenyl 7d
(67%) flavonol ethers. These compounds were readily demethylated to
the 3′-phenyl 8c (93%) and 4′-phenyl 8d (98%) flavonols using the
previously determined reaction conditions.
Biological method validation of cell
viability assays
The reliability of various colorimetric and cell
counting assays in the presence of flavonols was determined by
comparing the metabolism-dependent Alamar Blue assay, the
DNA-binding crystal violet assay and the dye exclusion trypan blue
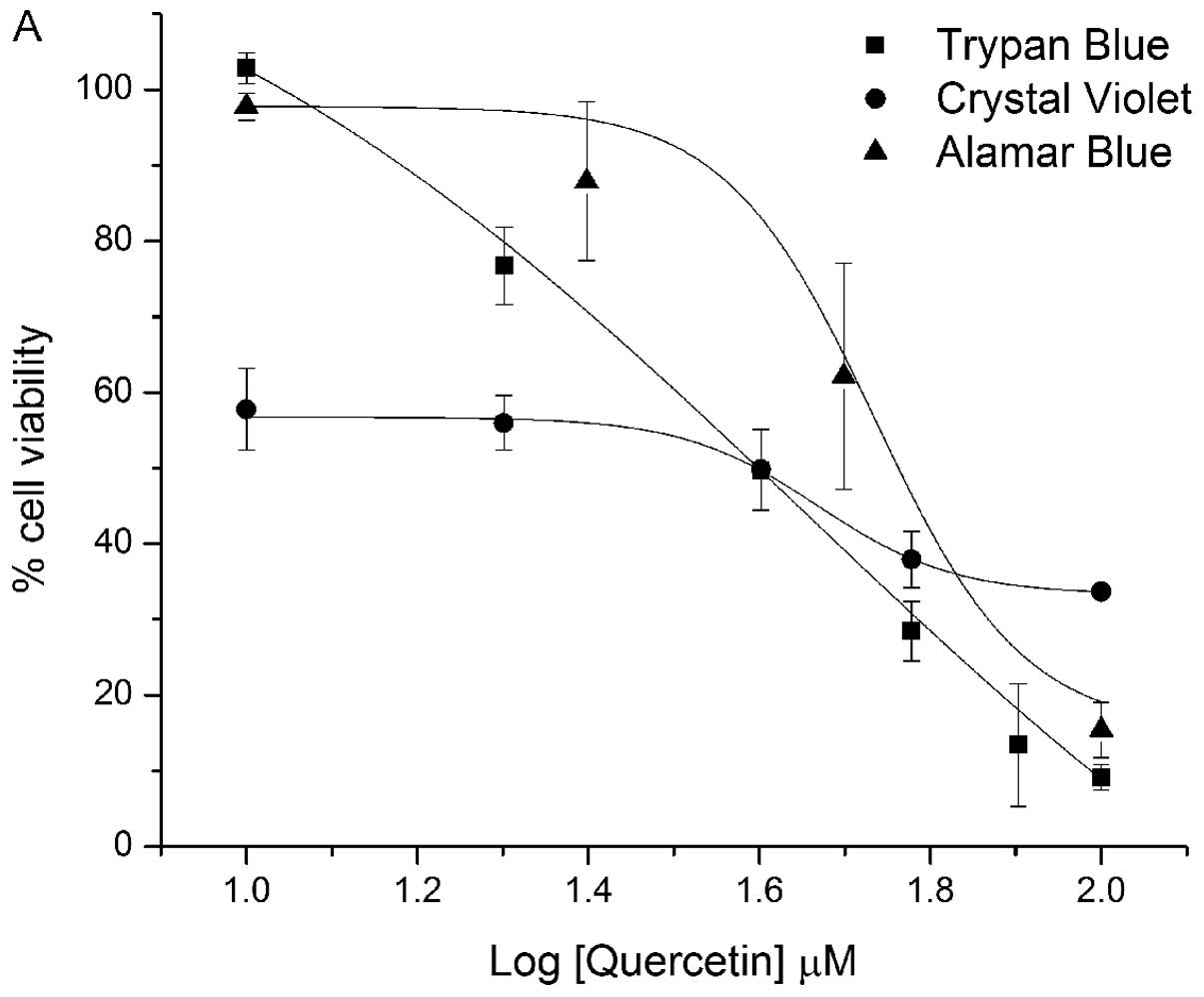
method. We found that flavonols reduced Alamar Blue in the absence
of cells and overestimated the number of viable prostate cancer
cells (Fig. 4). Flavonols also
interfered with the crystal violet assay by staining untreated
cells more intensely than flavonol treated cells. For many flavonol
treated cells, the maximum achievable cell viability was only 50%
compared to the control, even at non-cytotoxic concentrations. For
some flavonols, such as kaempferide, the maximum cell viability did
not ever reach 50%.
The trypan blue cell counting assay proved to be the
most reliable method for measuring cell viability in the presence
of flavonols with a minimal difference (≤10%) between the total
number of cells counted and the number of cells stained with trypan
blue. These results indicated that flavonols did not interfere with
the cell counting readings and provided a model by which to compare
the reliability of other assays such as Alamar Blue and crystal
violet. A comparison between trypan blue and Alamar Blue cell
viability curves showed a shift in the Alamar Blue curve to the
right, which is in agreement with previous studies that phenolic
compounds resulted in an overestimation of cell viability (Fig. 4). Quercetin had an EC50
value of 55.1 μM in Alamar Blue compared to 39.1 μM in trypan blue.
Kaempferide had an EC50 value of 30.1 μM in Alamar Blue
compared to 19.8 μM in trypan blue. We also found, for some
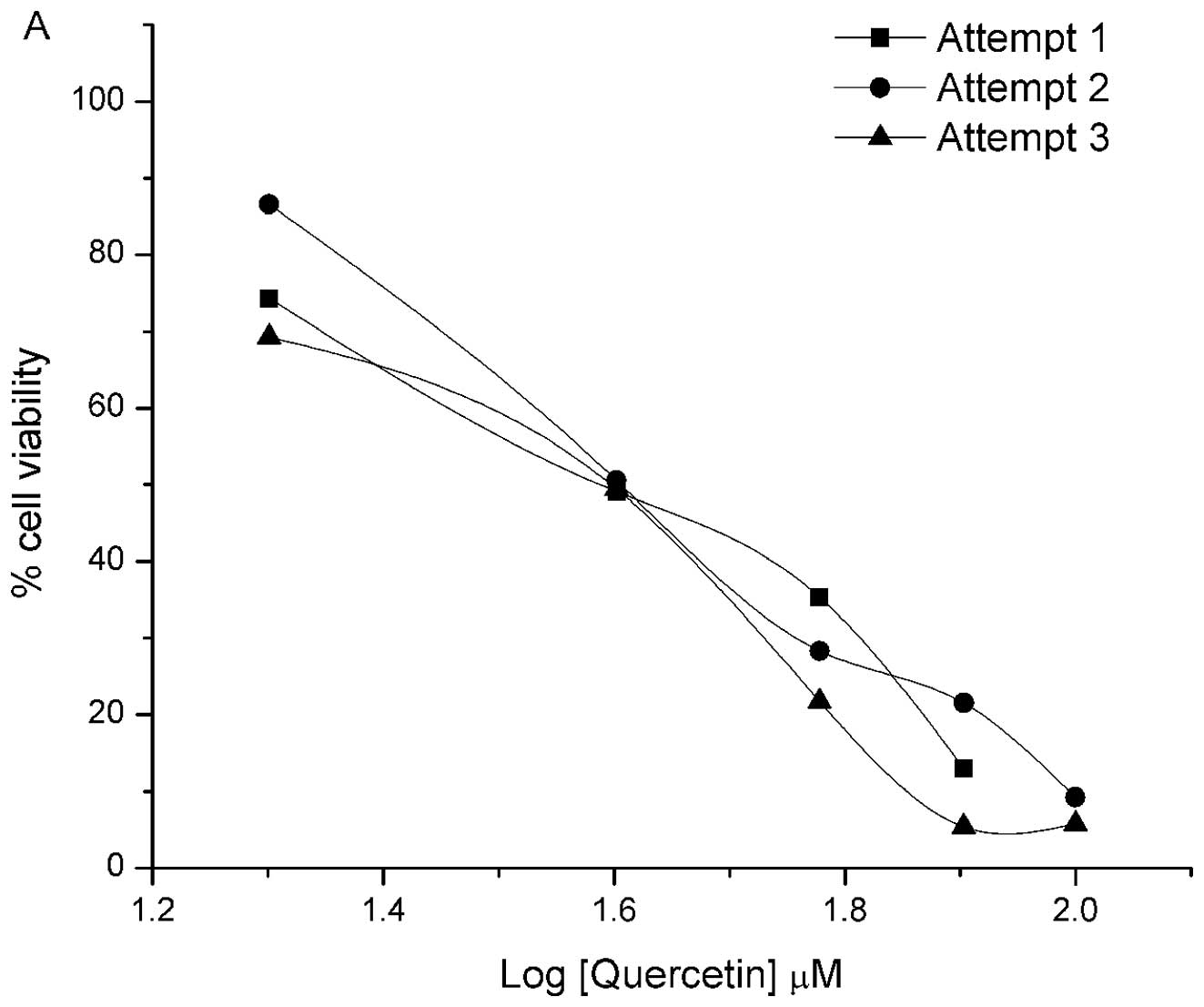
compounds, that the reproducibility of Alamar Blue was poor
(Fig. 5). This may have resulted
from inconsistent flavonol reduction of the metabolic compound
between analyses with fluctuating variables, such as the amount of
flavonols remaining in the well during incubation and cell numbers
between analyses. The major finding using the crystal violet assays
was that flavonols significantly masked the effects on cell
viability readings at lower concentrations (Fig. 4). However, at higher flavonol
concentrations this interference appeared to be minimal and
comparable to the trypan blue assay.
Effects of flavonols on the in vitro
viability of DU-145 and PC-3 prostate cancer cells
Flavonols with a carbonyl at C4, a double bond
between C2 and C3, and a hydroxyl group at position 3 of ring C,
were selected for this study to determine cytotoxicity towards
androgen-independent DU-145 and PC-3 human prostate cancer cells in
comparison to toxicity towards HIFF normal cells as determined
using the trypan blue exclusion assay. All of the flavonols tested
decreased the number of viable DU-145 and PC-3 cells. In DU-145
cells, the more hydrophobic and lipophilic flavonol analogs (3′-
and 4′-phenyl and 4′-iodo flavonols) were the most potent (Table I). The other hydrophobic and
lipophilic flavonols (3′-iodo, 3′- and 4′-benzoyl and 4′-methyl
flavonols) as well as the natural product, kaempferide, were
slightly less efficient at decreasing cell viability. The natural
product, galangin, moderately decreased viability, while quercetin
and kaempferol had the least effect.
 | Table IEC50 of DU-145 and
PC-3. |
Table I
EC50 of DU-145 and
PC-3.
| Flavonol | EC50
values in DU-145 cells | Flavonol | EC50
values in PC-3 cells |
|---|
| Quercetin |
39.11±2.87a | Quercetin |
32.87±4.10e |
| Kaempferol |
38.35±1.94a | Kaempferol |
33.29±2.96e |
| Galangin
(H-flavonol 5f) |
25.94±0.59b | Galangin
(H-flavonol 5f) |
17.11±1.29f |
| Kaempferide |
19.82±0.50c | Kaempferide |
14.88±0.78g |
| 4′-Benzoyl flavonol
(5b) |
16.61±0.70c | 3′-Iodo flavonol
(5c) |
10.54±0.72h |
| 3′-Benzoyl flavonol
(5a) |
14.81±0.71c | 4′-Benzoyl flavonol
(5b) |
9.97±0.36h |
| 3′-Iodo flavonol
(5c) |
14.53±1.02c | 4′-Methyl flavonol
(5e) |
9.89±1.24h |
| 4′-Methyl flavonol
(5e) |
14.30±0.53c | 3′-Benzoyl flavonol
(5a) |
9.31±0.58h |
| 4′-Iodo flavonol
(5d) |
7.51±0.27d | 3′-Phenyl flavonol
(8c) |
6.35±0.26i |
| 3′-Phenyl flavonol
(8c) |
6.33±0.58d | 4′-Phenyl flavonol
(8d) |
5.40±0.63j |
| 4′-Phenyl flavonol
(8d) |
5.57±0.60d | 4′-Iodo flavonol
(5d) |
4.13±0.16k |
In PC-3 cells, the more hydrophobic and lipophilic
analogs, 4′-iodo and 4′-phenyl flavonols, were the most effective
compounds at reducing cell viability (Table I). Other hydrophobic compounds
(3′-phenyl, 3′- and 4′-benzoyl, 4′-methyl flavonols, kaempferide
and galangin) were slightly less active, exhibiting EC50
values similar to each other. The least hydrophobic flavonols,
kaempferol and quercetin, were clearly identified as the least
efficacious compounds.
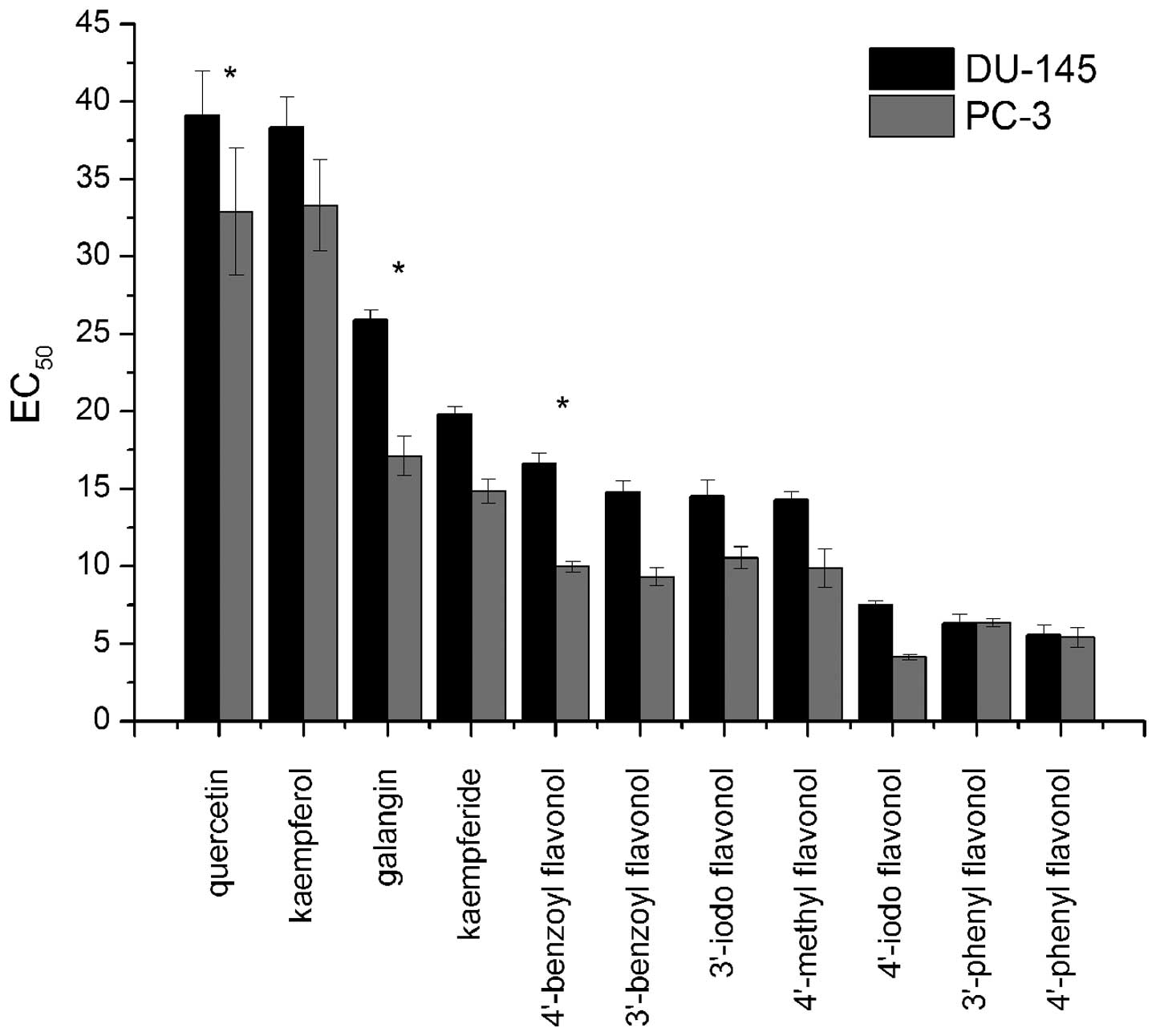
A comparison of the differences in the
EC50 values of the flavonols between the two prostate
cancer cell lines showed that only three compounds, quercetin,
galangin and 4′-benzoyl flavonol, exhibited differential
cytotoxicity (Fig. 6). All three
of these compounds were found to have a greater effect on viability
of PC-3 than on DU-145 cells (P<0.05). Of the flavonols tested,
4′-iodo, 3′-phenyl and 4′-phenyl flavonol proved to have the
greatest effect on cell viability, reducing the number of viable
prostate cells by 6.5-fold compared to the natural product,
quercetin.
The EC100 values for DU-145 and PC-3
showed a similar trend to their EC50 values (Table II). The more hydrophobic and
lipophilic analogs (3′- and 4′-phenyl, 3′- and 4′-iodo, 3′-benzoyl
and 4′-methyl flavonols) had the lowest EC100 values.
The natural products, kaempferide and galangin, were slightly less
efficient at achieving their maximum effect. Quercetin and
kaempferol were the least effective. A comparison of the
differences in EC100 values of flavonols between these
two cell lines showed that only two compounds, kaempferol and
4′-benzoyl, were statistically different (data not shown). Both
compounds were more effective against PC-3 than DU-145 cells
(P<0.05). From these results, we concluded that only a minimum
concentration (<25 μM) of the more hydrophobic and lipophilic
flavonol analogs (3′- and 4′-phenyl, 3′- and 4′-iodo, 3′-benzoyl
and 4′-methyl flavonols) was needed to achieve a maximum effect on
both the DU-145 and PC-3 cell lines.
| Table IIEC100 values of DU-145 and
PC-3. |
Table II
EC100 values of DU-145 and
PC-3.
| Flavonol | EC100
values in DU-145 cells | Flavonol | EC100
values in PC-3 cells |
|---|
| Quercetin |
85.42±6.11a | Quercetin |
77.13±3.40f |
| Kaempferol |
74.36±0.83a | Kaempferol |
57.62±6.40g |
| Galangin
(H-flavonol 5f) |
46.42±3.92b | Galangin
(H-flavonol 5f) |
43.92±4.14h |
| 4′-Benzoyl flavonol
(5b) |
41.04±2.18b | Kaempferide |
34.16±1.66h |
| Kaempferide |
39.19±0.87b | 4′-Benzoyl flavonol
(5b) |
22.31±0.91i |
| 3′-Iodo flavonol
(5c) |
24.61±2.06c | 4′-Methyl flavonol
(5e) |
20.38±1.77j |
| 4′-Methyl flavonol
(5e) |
23.75±2.53c | 3′-Iodo flavonol
(5c) |
19.54±1.08k |
| 3′-Benzoyl flavonol
(5a) |
20.43±0.41d | 3′-Benzoyl flavonol
(5a) |
17.86±1.56k |
| 3′-Phenyl flavonol
(8c) |
18.00±0.61d | 3′-Phenyl flavonol
(8c) |
11.33±1.38k |
| 4′-Phenyl flavonol
(8d) |
12.43±1.85d | 4′-Iodo flavonol
(5d) |
10.34±0.73l |
| 4′-Iodo flavonol
(5d) |
9.84±0.33e | 4′-Phenyl flavonol
(8d) |
8.75±0.45m |
A comparison of the difference between the
EC100 and EC50 value of the flavonols in
DU-145 showed that only a small increase in concentration (10 μM or
less) was needed to achieve maximal effect for the hydrophobic and
lipophilic analogs (4′-phenyl, 3′-benzoyl, 4′-methyl, 3′- and
4′-iodo flavonols) (Table III).
The natural products, galangin and kaempferide, as well as the
flavonol analog, 4′-methyl flavonol, had a slightly larger range
(<25 μM) with 3′-phenyl flavonol overlapping the two categories.
Quercetin and kaempferol were the least effective and the range
between the EC100 and EC50 values was at
least a 36 μM.
| Table IIIDifference between EC100
and EC50 values in DU-145 and PC-3. |
Table III
Difference between EC100
and EC50 values in DU-145 and PC-3.
| Flavonol | EC100 -
EC50 values in DU-145 cells | Flavonol | EC100 -
EC50 values in PC-3 cells |
|---|
| Quercetin |
46.31±8.55a | Quercetin |
44.26±4.29h |
| Kaempferol |
36.01±2.02b | Galangin
(H-flavonol 5f) |
26.81±5.31i |
| 4′-Benzoyl flavonol
(5b) |
24.43±1.10c | Kaempferol |
24.33±4.63i |
| Galangin
(H-flavonol 5f) |
20.48±0.46d | Kaempferide |
19.28±2.10j |
| Kaempferide |
19.37±2.02d | 4′-Benzoyl flavonol
(5b) |
12.34±0.99k |
| 3′-Phenyl flavonol
(8c) |
11.67±1.11e | 4′-Methyl flavonol
(5e) |
10.49±3.29k |
| 3′-Iodo flavonol
(5c) |
10.08±0.84f | 3′-Iodo flavonol
(5c) |
9.00±1.06l |
| 4′-Methyl flavonol
(5e) |
9.45±1.89f | 3′-Benzoyl flavonol
(5a) |
8.55±2.06l |
| 4′-Phenyl flavonol
(8d) |
6.86±0.18f | 4′-Iodo flavonol
(5d) |
6.21±0.59l |
| 3′-Benzoyl flavonol
(5a) |
5.62±5.50f | 3′-Phenyl flavonol
(8c) |
4.98±1.54l |
| 4′-Iodo flavonol
(5d) |
2.33±2.05g | 4′-Phenyl flavonol
(8d) |
3.35±0.93m |
In PC-3 cells, a 10 μM or less difference between
the EC100 and EC50 values was found in the
more hydrophobic and lipophilic flavonol analogs (4′-phenyl,
3′-benzoyl, 3′-phenyl, 3′- and 4′-iodo flavonols) (Table III). The less hydrophobic natural
products, kaempferol, galangin and kaempferide, required a slightly
larger range (<25 μM) to achieve a maximal effect with the
4′-methyl and 4′-benzoyl flavonol overlapping the two categories.
Quercetin was the least effective and the range between the
EC100 and EC50 values was 44 μM.
A comparison of the differences in the
EC100 and EC50 values of flavonols between
these two cell lines showed that only two compounds, kaempferol and
4′-benzoyl flavonol, were statistically different (data not shown).
These results indicate that hydrophobic and lipophilic flavonol
analogs tend to reach their maximal effect at lower concentrations
than natural flavonols and that this maximal effect is achieved
over a more narrow effective concentration range (with 4′-benzoyl
flavonol being the exception in DU-145). Narrow ranges between the
EC50 and EC100 values are beneficial because
they reduce the likelihood that the agents will be toxic to normal
cells.
Effects of flavonols on the in vitro
viability of HIFF normal cells
HIFF cells were used to determine the toxicity of
flavonols on normal cells. Flavonoid research has commonly been
conducted at concentrations of 50 μM or greater (5,46).
Based on this information, we first examined the effects of
flavonols on HIFF cells at 50 μM concentrations. At this
concentration, we found that all flavonols tested caused a decrease
in cell viability (Table IVA).
Quercetin was the least inhibitory and all of the synthesized
derivatives exhibited major inhibitory activity at the 50 μM
concentration.
| Table IVViability of HIFF cells treated with
flavonols. |
Table IV
Viability of HIFF cells treated with
flavonols.
| A, Viability of
cells treated with 50 μM flavonol |
|---|
|
|---|
| Flavonol | N | % Viability of HIFF
(50 μM)a |
|---|
| Quercetin | 3 | 84.47±2.21% |
| Kaempferol | 3 | 73.82±1.38% |
| Kaempferide | 3 | 52.17±1.17% |
| Galangin (5f) | 3 | 47.95±1.38% |
| 3′-Iodo flavonol
(5c) | 4 | 46.79±2.48% |
| 4′-Iodo flavonol
(5d) | 3 | 34.77±0.99% |
| 4′-Methyl flavonol
(5e) | 3 | 37.50±2.27% |
| 3′-Benzoyl flavonol
(5a) | 3 | 34.80±0.20% |
| 3′-Phenyl flavonol
(8c) | 3 | 34.63±2.74% |
| 4′-Phenyl flavonol
(8d) | 5 | 34.42±3.67% |
| 4′-Benzoyl flavonol
(5b) | 3 | 32.33±2.92% |
|
| B, Viability at the
average combined EC50 concentration of DU-145 and PC-3
for each flavonol |
|
| Flavonol | N | Flavonol (μM) | % Viability of
HIFFa |
|
| 4′-Benzoyl flavonol
(5b) | 3 | 13 | 110.92±3.20% |
| 3′-Iodo flavonol
(5c) | 4 | 13 | 105.98±5.79% |
| Kaempferol | 4 | 36 | 105.57±5.66% |
| Quercetin | 6 | 36 | 94.01±7.67% |
| Galangin (5f) | 4 | 20 | 92.63±5.58% |
| 3′-Benzoyl flavonol
(5a) | 6 | 12 | 90.36±4.85% |
| Kaempferide | 3 | 17 | 83.91±1.31% |
| 4′-Phenyl flavonol
(8d) | 4 | 6 | 82.09±5.48% |
| 4′-Iodo flavonol
(5d) | 3 | 6 | 70.21±2.70% |
| 4′-Methyl flavonol
(5e) | 3 | 12 | 65.00±4.12% |
| 3′-Phenyl flavonol
(8c) | 3 | 6 | 53.08±9.54% |
HIFF cell viability was also examined at the
average flavonol EC50 concentrations (for DU-145 and
PC-3) for individual compounds. At this concentration prostate
cancer cells would have a cell viability of 50%. Kaempferol,
4′-benzoyl and 3′-iodo flavonols had no affect on the cell
viability of HIFF cells (Table
IVB). The natural products, quercetin and galangin, and the
flavonol analog, 3′-benzoyl flavonol, caused a slight decrease in
the viability of HIFF cells. A moderate effect was observed for
kaempferide and 4′-phenyl flavonol. Three flavonols, 4′-methyl,
4′-iodo and 3′-phenyl flavonol exhibited the strongest decrease in
cell viability. The activity of flavonols towards HIFF cells did
not correlate with the activity of flavonols towards the prostate
tumor cells. A comparison of the flavonols, which minimally
decreased the viability of HIFF cells and efficiently decreased the
viability in prostate cancer cells at both the EC50 and
EC100 values, lead us to conclude that 3′-iodo and
3′-benzoyl flavonols are likely to be the most promising
therapeutic agents.
Discussion
For decades, flavonoids have been extensively
studied for their ability to act as anti-oxidants, enzyme
inhibitors and growth regulators. More recently, flavonols have
become available as supplements. In preparing to examine the
effects of new flavonol analogs on cell viability of human prostate
cancer cells, we found that many previous reports failed to use
reliable methods to examine this effect and in addition, a number
of studies examined the effects of non-biologically relevant
concentrations of flavonoids on cell viability. Cell viability and
proliferation (47–50) are commonly measured through
metabolic [MTT, MTS, Alamar Blue and CTG (Cell-Titer Glo)], DNA or
protein-binding (crystal violet, Hoechst 33342, propidium iodide
and rhodamine 123) and cell-based (trypan blue) assays.
Metabolic assays measure the metabolism of the
colorimetric, fluorescent and/or luminescent compound in live
cells. MTT (51), MTS (52) and Alamar Blue (53) are reduced by NAD(P)H-dependent
oxidoreductase enzymes, while CTG (54) is reduced by cellular ATP. Several
of these agents have previously proven unreliable in the presence
of flavonoids. For example, flavonoids reduce MTT (33,55,56)
and MTS (57), a second generation
tetrazolium dye, in the absence of cells and CTG (22) overestimates the number of viable
cells for some polyphenolic compounds. In this study, we found that
flavonoids also reduce Alamar Blue in the absence of cells and
overestimate the number of viable cells. For NAD(P)H-dependent
assays this interference is likely due to the ability of flavonoids
to mimic the reducing agent, NAD(P)H. Flavonoids are known
anti-oxidants, capable of preventing oxidative damage by being
oxidized themselves (58–60). Several anti-oxidants (flavonoids,
ascorbic acid, vitamin E, ubiquinone, hydroquinone and
N-acetylcysteine) also reduce MTT in the absence of cells
suggesting that MTT and other metabolic-dependent agents are not
reliable assays for analyzing agents that have anti-oxidant
activity (33,61–64).
For ATP-dependent assays, flavonoids likely mimic ATP by binding to
the ATP binding site (2,29,31).
Several flavonols, such as quercetin, kaempferol and galangin are
known to exhibit this property.
Flavonoids also interfere with the staining of DNA
(Hoechst 33342, propidium iodide and crystal violet) and proteins
(rhodamine 123). For example, the flavonol, quercetin, previously
was found to inhibit the binding of Hoechst 33342 to DNA and to
activate the binding of rhodamine 123 to proteins (31,65).
The anthocyanin, cyanidin-3-rutinoside, prevented the binding of
propidium iodide to DNA (66).
These dyes have commonly been used in flow cytometry studies. We
found that crystal violet stained the DNA of untreated cells more
intensely than flavonol treated cells. In several cases, the
maximum achievable cell viability was only 50% of the control at
nontoxic concentrations. The interference of the flavonoids with
the ability of crystal violet to bind the DNA might have resulted
from intercalation of the flavonols into DNA, since flavonols such
as quercetin and kaempferol are known to exhibit this property
(67). Rhodamine 123 measures
mitochondrial membrane potential, which is regulated by ATP
(68). Therefore, if quercetin
binds to the ATP binding site it would disrupt the membrane
potential and would interfere with the accumulation of rhodamine
123 inside the membrane. Additionally, quercetin enhances the rate
of rhodamine 123 transport into P-glycoproteins (69).
In order for these assays to be reliable, excess
flavonoids must be removed before the addition of the metabolic
agent. Previous investigators have suggested removing the culture
medium before the addition of the metabolic agent (33). However, flavonoids are minimally
soluble in biological buffers. Therefore, this method is
inefficient at removing all of the flavonoids in solution and in
addition, does not account for the flavonoids located within the
cell that can also interfere with the colorimetric agent. Liu et
al (70) previously found in
B12 cells, which are phenotypically similar to neural precursor
cells, that genistein reduced 60% more MTT than the control during
a 3 h incubation. Therefore, the impact that flavonoids have on
metabolic agents may vary depending on the amount and type of
flavonoid used.
Based on these results, we concluded that, though
popular, metabolic and DNA staining assays are not a reliable
method of measuring cell viability and proliferation in the
presence of flavonoids. The trypan blue cell counting assay
provides an alternative method to these assays, but has its own set
of limitations (71–73). Some color variations may be
undetectable to the human eye and debris can also be stained and
counted as a nonviable cell. For accurate results, early phase
cells (≤72 h) must be used to prevent an overestimation of the
number of viable cells and cells must be counted within 5 min of
the addition of the dye. An automatic cell viability analyzer such
as the Vi-Cell XR (Beckman Coulter Inc.) addresses most of these
limitations by more accurately detecting different sizes and shades
of stained cells, allowing rapid analysis (<3 min) and providing
more consistent results than the manual method (74). Additionally, the difference between
the total number of cells and the number of nonviable cells was
≤10%, implying that flavonols did not interfere with the cell
counting readings. Our results correlate well with previously
published trypan blue results. For example, Vijayababu et al
(75) found in PC-3 cells that
quercetin had an EC50 value of around 29 μM compared to
our 32.9 μM.
Polar natural flavonols (quercetin, kaempferol,
kaempferide and galangin) have commonly been the focus of flavonoid
cancer research. However, their in vitro anticancer effects
have not translated well to their effects in vivo, probably,
because absorption and bioavailability of dietary flavonoids (4–68
mg) are limited to the low micromolar range (76–78)
and previously, their effective and cytotoxic dosages have been
well above this concentration limit. For example, the flavonol
supplement quercetin has an EC50 value of 39.1 μM in
DU-145 and 32.9 μM in PC-3 cells. We found that increasing the
hydrophobicity and lipophilicity of a flavonol significantly lowers
the effective concentration of several analogs (4′-iodo, 3′-phenyl
and 4′-phenyl flavonol) into the desired low micromolar range thus
enhancing their therapeutic potential. However, information about
the absorption and bioavailability of these novel flavonols are
needed to provide a more complete picture of their ultimate
anticancer potential in vivo.
Acknowledgements
This study was supported in part by a grant from
the National Cancer Institute to G.G.M (CA101035) and Washington
State University’s College of Sciences.
References
|
1
|
Dorta DJ, Curti C and Rodrigues T: Effects
of flavonoids on mitochondria: An overview on pharmacological and
toxicological aspects. Mitochondrial Pharmacology and Toxicology.
Moreno AJM, Oliveira PJ and Palmeira CM: Transworld Research
Network; Kerala, India: pp. 147–161. 2006
|
|
2
|
Agullo G, Gamet-Payrastre L, Manenti S,
Viala C, Remesy C, Chap H and Payrastre B: Relationship between
flavonoid structure and inhibition of phosphatidylinositol
3-kinase: A comparison with tyrosine kinase and protein kinase C
inhibition. Biochem Pharmacol. 53:1649–1657. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Haddad AQ, Venkateswaran V, Viswanathan L,
Teahan SJ, Fleshner NE and Klotz LH: Novel antiproliferative
flavonoids induce cell cycle arrest in human prostate cancer cell
lines. Prostate Cancer Prostatic Dis. 9:68–76. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nair HK, Rao KV, Aalinkeel R, Mahajan S,
Chawda R and Schwartz SA: Inhibition of prostate cancer cell colony
formation by flavonoid quercetin correlates with modulation of
specific regulatory genes. Clin Diagn Lab Immunol. 11:63–69.
2004.PubMed/NCBI
|
|
5
|
Mokhtari MJ, Motamed N and Shokrgozar MA:
Evaluation of silibinin on the viability, migration, and adhesion
of the human prostate adenocarcinoma (PC-3) cell line. Cell Bio
Int. 32:888–892. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Daskiewicz JB, Depeint F, Viornery L, et
al: Effects of flavonoids on cell proliferation and caspase
activation in human colonic cell line HT29: an SAR study. J Med
Chem. 48:2790–2804. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Knowles LM, Zigrossi DA, Tauber RA,
Hightower C and Milner JA: Flavonoids suppress androgen-independent
human prostate tumor proliferation. Nutr Cancer. 38:116–122. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Skibola CF and Smith MT: Potential health
impacts of excessive flavonoid intake. Free Radic Biol Med.
29:375–383. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
MacGregor JT and Jurd L: Mutagenicity of
plant flavonoids: structural requirements for mutagenic activity in
Salmonella typhimurium. Mutat Res. 54:297–309. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Carver JH, Carrano AV and MacGregor JT:
Genetic effects of the flavonols quercetin, kaempferol, and
galangin on Chinese hamster ovary cells in vitro. Mutat Res.
113:45–60. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bjeldanes LF and Chang GW: Mutagenic
activity of quercetin and related compounds. Science. 197:577–578.
1977. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brown JP and Dietrich PS: Mutagenicity of
plant flavonols in Salmonella/mammalian microsome test:
activation of flavonol glycosides by mixed glycosidases from rat
cecal bacteria and other sources. Mutat Res. 66:223–240.
1979.PubMed/NCBI
|
|
13
|
Hillard JJ, Krause HM, Brenstein JI,
Fernandez JA, Nguyen V, Ohemeng KA and Barrett JF: A comparison of
active binding of 4-quinolones and novel flavone gyrase inhibitors
to DNA gyrase. Adv Exp Med Biol. 390:59–69. 1995.PubMed/NCBI
|
|
14
|
Guthrie N and Manthey JA:
Antiproliferative activities of citrus flavonoids against six human
cancer cell lines. J Agric Food Chem. 50:5837–5843. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
García-Mediavilla V, Crespo I, Collado PS,
Esteller A, Sánchez-Campos S, Tuñón MJ and González-Gallego J: The
anti-inflammatory flavones quercetin and kaempferol cause
inhibition of inducible nitric oxide synthase, cyclooxygenase-2,
and reactive C-protein, and down-regulation of the nuclear factor
kappaB pathway in Chang Liver cells. Eur J Pharmacol. 557:221–229.
2007.
|
|
16
|
Kaushik D, O’Fallon K, Clarkson PM, Dunne
CP, Conca KR and Michniak-Kohn B: Comparison of quercetin
pharmacokinetics following oral supplementation in humans. J Food
Sci. 77:H231–H238. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suomela JP, Ahotupa M, Yang B, Vasankari T
and Kallio H: Adsorption of flavonols derived from sea buckthorn
(Hippophae rhamnoides L.) and their effects on emerging risk
factors for cardiovascular disease in humans. J Agric Food Chem.
54:7364–7369. 2006.PubMed/NCBI
|
|
18
|
Manach C, Williamson G, Morand C, Scalbert
A and Rémésy C: Bioavailability and bioefficacy of polyphenols in
humans. I. Review of 97 bioavailability studies. Am J Clin Nutr.
81(Suppl 1): S230–S242. 2005.PubMed/NCBI
|
|
19
|
Del Rio DD, Borges G and Crozier A: Berry
flavonoids and phenolics: bioavailability and evidence of
protective effects. Br J Nutr. 104(Suppl 3): S67–S90.
2010.PubMed/NCBI
|
|
20
|
Gao S and Hu M: Bioavailability challenges
associated with development of anti-cancer phenolics. Mini Rev Med
Chem. 10:550–567. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Singh B, Mense SM, Bhat NK, Putty S,
Guthiel WA, Remotti F and Bhat HK: Dietary quercetin exacerbates
the development of estrogen-induced breast tumors in female ACI
rats. Toxicol Appl Pharmacol. 247:83–90. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yadegarynia S, Pham A, Ng A, et al:
Profiling flavonoid cytotoxicity in human breast cancer cell lines:
determination of structure-function relationships. Nat Prod Commun.
7:1295–1304. 2012.PubMed/NCBI
|
|
23
|
Chang H, Mi M, Ling W, et al: Structurally
related cytotoxic effects of flavonoids on human cancer cells in
vitro. Arch Pharm Res. 31:1137–1144. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li X, Wang X, Ye H, Peng A and Chen L:
Barbigerone, an isoflavone, inhibits tumor angiogenesis and human
non-small-cell lung cancer xenografts growth through VEGFR2
signaling pathways. Cancer Chemother Pharmacol. 70:425–437. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu TC, Yang YC, Huang PR, Wen YD and Yeh
SL: Genistein enhances the effect of trichostatin A on inhibition
of A549 cell growth by increasing expression of TNF receptor-1.
Toxicol Appl Pharmacol. 262:247–254. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wesolowska O, Wiśniewski J, Sroda-Pomianek
K, et al: Multidrug resistance reversal and apoptosis induction in
human colon cancer cells by some flavonoids present in citrus
plants. J Nat Prod. 75:1896–1902. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vaid M, Prasad R, Singh T, Jones V and
Katiyar SK: Grape seed proanthocyanidins reactivate silenced tumor
suppressor genes in human skin cancer cells by targeting epigenetic
regulators. Toxicol Appl Pharmacol. 263:122–130. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee DH, Szczepanski M and Lee YJ: Role of
Bax in quercetin-induced apoptosis in human prostate cancer cells.
Biochem Pharmacol. 75:2345–2355. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gamet-Payrastre L, Manenti S, Gratacap MP,
Tulliez J, Chap H and Payrastre B: Flavonoids and the inhibition of
PKC and PI 3-Kinase. Gen Pharmacol. 32:279–286. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee LT, Huang YT, Hwang JJ, et al:
Transinactivation of the epidermal growth factor receptor tyrosine
kinase and focal adhesion kinase phosphorylation by dietary
flavonoids; effect on invasive potential of human carcinoma cells.
Biochem Pharmacol. 67:2103–2114. 2004. View Article : Google Scholar
|
|
31
|
Di Pietro A, Conseil G, Perez-Victoria JM,
et al: Modulation by flavonoids of cell multidrug resistance
mediated by P-glycoprotein and related ABC transporters. Cell Mol
Life Sci. 59:307–322. 2002.PubMed/NCBI
|
|
32
|
Politzer P, Lane P, Concha MC, Ma Y and
Murray JS: An overview of halogen bonding. J Mol Model. 13:305–311.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bruggisser R, von Daeniken K, Jundt G,
Schaffner W and Tullberg-Reinert H: Interference of plant extracts,
phytoestrogens, and antioxidants with MTT tetrazolium assay. Planta
Med. 68:445–448. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Molander GA and Biolatto B:
Palladium-catalyzed Suzuki-Miyaura cross-coupling reactions of
potassium aryl- and heteroaryltrifluoroborates. J Org Chem.
68:4302–4314. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Khan N, Afaq F, Syed DN and Mukhtar H:
Fisetin, a novel dietary flavonoid, causes apoptosis and cell cycle
arrest in human prostate cancer LNCaP cells. Carcinogenesis.
29:1049–1056. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Harborne JB, Grayer RF, Porter LF, et al:
The Flavonoids Advances in Research Since 1980. Harborne, New York:
pp. 399–420. 1988
|
|
37
|
Tanaka H, Stohlmeyer MM, Wandless TJ and
Taylor LP: Synthesis of flavonol derivatives as probes of
biological processes. Tetrahedron Lett. 41:9735–9739. 2000.
View Article : Google Scholar
|
|
38
|
Ichikawa M, Pamukcu AM and Bryan GT: A
convenient method for the synthesis of kaempferol. Org Prep Proced
Int. 14:183–187. 1982. View Article : Google Scholar
|
|
39
|
Urgaonkar S and Shaw JT: Synthesis of
kaempferitrin. J Org Chem. 72:4582–4585. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Landini D, Montanari F and Rolla F:
Cleavage of dialkyl and aryl alkyl ethers with hydrobromic acid in
the presence of phase-transfer catalysts. Synthesis. 10:771–773.
1978. View Article : Google Scholar
|
|
41
|
Boumendjel A, Bois F, Beney C, Mariotte
AM, Conseil G and Di Pietro A: B-ring substituted
5,7-dihydroxyflavonols with high-affinity binding to P-glycoprotein
responsible for cell multidrug resistance. Bioorg Med Chem Let.
11:75–77. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Azimova SS and Vinogradova VIG: Galangin.
Natural Compounds: Flavonoids: Plant Sources, Structure and
Properties. Springer; New York: pp. 962013
|
|
43
|
Batirov EK, Kiyamitdinova F and Malikov
VM: Flavonoids of the epigeal part of Glycyrrhiza glabra.
Chem Nat Compd. 22:107–108. 1986. View Article : Google Scholar
|
|
44
|
Yuldashev MP: Flavonoids of the epigeal
part of Glycyrrhiza uralensis. Chem Nat Compd. 34:508–509.
1998. View Article : Google Scholar
|
|
45
|
Agrawal PK and Rastogi RP: 13C
NMR spectroscopy of flavonoids. Heterocycles. 16:21811981.
View Article : Google Scholar
|
|
46
|
Xu R, Zhang Y, Ye X, Xue S, Shi J, Pan J
and Chen Q: Inhibition effects and induction of apoptosis of
flavonoids on the prostate cancer cell line PC-3 in vitro. Food
Chem. 138:48–53. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Torkin R, Lavoie JF, Kaplan DR and Yeger
H: Induction of caspase-dependent, p53-mediated apoptosis by
apigenin in human neuroblastoma. Mol Cancer Ther. 4:1–11. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ferguson PJ, Kurowska E, Freeman DJ,
Chambers AF and Koropatnick DJ: A flavonoid fraction from cranberry
extract inhibits proliferation of human tumor cell lines. J Nutr.
134:1529–1535. 2004.PubMed/NCBI
|
|
49
|
Walle T, Ta N, Kawamori T, Wen X, Tsuji PA
and Walle UK: Cancer chemopreventive properties of orally
bioavailable flavonoids - methylated versus unmethylated flavones.
Biochem Pharmacol. 73:1288–1296. 2007. View Article : Google Scholar
|
|
50
|
Gosslau A, Chen M, Ho CT and Chen KY: A
methoxy derivative of resveratrol analogue selectively induced
activation of the mitochondrial apoptotic pathway in transformed
fibroblast. Br J Cancer. 92:513–521. 2005.
|
|
51
|
Berridge MV and Tan AS: Characterization
of the cellular reduction of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT):
subcellular localization, substrate dependence, and involvement of
mitochondrial electron transport in MTT reduction. Arch Biochem
Biophys. 303:474–482. 1993. View Article : Google Scholar
|
|
52
|
Dunigan DD, Waters SB and Owen TC: Aqueous
soluble tetrazolium/formazan MTS as an indicator of NADH- and
NADPH-dependent dehydrogenase activity. Biotechniques. 19:640–649.
1995.PubMed/NCBI
|
|
53
|
Maeda H, Matsu-Ura S, Yamauchi Y and
Ohmori H: Resaurin as an electron acceptor in glucose
oxidase-catalyzed oxidation of glucose. Chem Pharm Bull.
49:622–625. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Suszynski TM, Wildey GM, Falde EJ, et al:
The ATP/DNA ratio is a better indicator of islet cell viability
than the ADP/ATP ratio. Transplant Proc. 40:346–350. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wisman KN, Perkins AA, Jeffers MD and
Hagerman AE: Accurate assessment of the bioactivities of
redox-active polyphenolics in cell culture. J Agric Food Chem.
56:7831–7837. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Maioli E, Torricelli C, Fortino V,
Carlucci F, Tommassini V and Pacini A: Critical appraisal of the
MTT assay in the presence of rottlerin and uncouplers. Biol Proced
Online. 11:227–240. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang P, Henning SM and Heber D:
Limitations of MTT and MTS-based assays for measurement of
antiproliferative activity of green tea polyphenols. PLoS One.
5:e102022010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang D, Liu Y, Chu L, et al: Relationship
between the structures of flavonoids and oxygen radical absorbance
capacity values: a quantum chemical analysis. J Phys Chem A.
117:1784–1794. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Pourcel L, Routaboul JM, Cheynier V,
Lepiniec L and Debeaujon I: Flavonoid oxidation in plants: from
biochemical properties to physiological functions. Trends Plant
Sci. 12:29–36. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jacobs H, Moalin M, Bast A, van der Vijgh
WJ and Haenen GR: An essential difference between the flavonoids
monoHER and quercetin in their interplay with the endogenous
antioxidant network. PLoS One. 5:e138802010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang S, Yu H and Wickliffe JK: Limitations
of the MTT and XTT assays for measuring cell viability due to
superoxide formation induced by nano-scale TiO2. Toxicol
In Vitro. 25:2147–2151. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Stockert JC, Blázquez-Castro A, Cañete M,
Horobin RW and Villanueva A: MTT assay for cell viability:
Intracellular localization of the formazan product is in lipid
droplets. Acta Histochem. 114:785–796. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tranzer JP and Pearse AG: Cytochemical
demonstration of ubiquinones in animal tissues. Nature.
199:1063–1066. 1963. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Carmichael J, DeGraff WG, Gazdar AF, Minna
JD and Mitchell JB: Evaluation of a tetrazolium-based semiautomated
colorimetric assay: assessment of chemosensitivity testing. Cancer
Res. 47:936–942. 1987.PubMed/NCBI
|
|
65
|
Shapiro AB and Ling V: Positively
cooperative sites for drug transport by P-glycoprotein with
distinct drug specificities. Eur J Biochem. 250:130–137. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Bennett MD, Price HJ and Johnston JS:
Anthocyanin inhibits propidium iodide DNA fluorescence in
Euphorbia pulcherrima: implications for genome size
variation and flow cytometry. Ann Bot. 101:777–790. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tajmir-Riahi HA, Diamantoglou S, Kanakis
CD, Tarantilis PA and Polissiou MG: Flavonoids interactions with
DNA and RNA: binding modes and antioxidative effects. Acta Hort.
744:195–204. 2007.
|
|
68
|
Baracca A, Sqarbi G, Solaini G and Lenaz
G: Rhodamine 123 as a probe of mitochondrial membrane potential:
evaluation of proton flux through F(0) during ATP synthesis.
Biochim Biophys Acta. 1606:137–146. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Shapiro AB and Ling V: Effect of quercetin
on Hoechst 33342 transport by purified and reconstituted
P-glycoprotein. Biochem Pharmacol. 53:587–596. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Liu Y, Peterson DA, Kimura H and Schubert
D: Mechanism of cellular
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
reduction. J Neurochem. 69:581–593. 1997. View Article : Google Scholar
|
|
71
|
Altman SA, Randers L and Rao G: Comparison
of trypan blue dye exclusion and fluorometric assays for mammalian
cell viability determinations. Biotechnol Prog. 9:671–674. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Smith AL and Smith HV: A comparison of
fluorescein diacetate and propidium iodide staining and in vitro
excystation for determining Giardia intestinalis cyst
viability. Parasitology. 99:329–331. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Mascotti K, McCullough J and Burger SR:
HPC viability measurement: trypan blue versus acridine orange and
propidium iodide. Transfusion. 40:693–696. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Szabo SE, Monroe SL, Fiorino S, Bitzan J
and Loper K: Evaluation of an automated instrument for viability
and concentration measurements of cryopreserved hemapoietic cells.
Lab Hematol. 10:109–111. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Vijayababu MR, Arunkumar A, Kanagaraj P,
Venkataraman P, Krishnamoorthy G and Arunakaran J: Quercetin
downregulates matrix metalloproteinases 2 and 9 proteins in
prostate cancer (PC-3). Mol Cell Biochem. 287:109–116. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hollman PC and Katan MB: Absorption,
metabolism and health effects of dietary flavonoids in man. Biomed
Pharmacother. 51:305–310. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Hollman PC, van Trijp JM, Mengelers MJ, de
Vries JH and Katan MB: Bioavailability of the dietary antioxidant
flavonol quercetin in man. Cancer Lett. 114:139–140. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Hollman PC, van Trijp JM, Buysman MN, van
der Gaag MS, Mengelers MJ, de Vries JH and Katan MB: Relative
bioavailability of the antioxidant flavonoid quercetin from various
foods in man. FEBS Lett. 418:152–156. 1997. View Article : Google Scholar : PubMed/NCBI
|