Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignancies and the third major cause of cancer death
worldwide (1,2). Despite the recent advances in
diagnosis and treatment, the general prognosis of HCC still remains
extremely dismal because of high postoperative metastatic
recurrence (3). Increasing
evidence suggests that epithelial-mesenchymal transition (EMT)
represents one important source of HCC cells with highly invasive
capability (4).
EMT is a biological conversion process that
transforms polarized epithelial cells to the mesenchymal phenotype
(5,6). The completion of EMT is signaled by
the loss of cell-cell adhesion, the degradation of the underlying
basement membrane, and the acquisition of migratory or invasive
properties (5,6). The molecular hallmark of EMT is the
loss of epithelial marker E-cadherin and upregulation of the
mesenchymal markers Vimentin, Snail, Slug, and others. In many
cases, the involved factors are also used as biomarkers to
demonstrate the passage of a cell through EMT (7). Multiple signaling pathways regulate
the EMT process, which includes extracellular signal-regulated
protein kinases (ERKs), mitogen-activated protein kinase (MAPK),
phosphatidylinositol 3-kinase (PI3K)/Akt, Smads, RhoB, and
β-catenin signaling (8).
Identification of the key molecules and the signaling pathways that
lead to activation of EMT programs during this disease process
provides new insights into the plasticity of cellular phenotypes
and possible therapeutic interventions.
Mammalian sterile-20-like kinase 4 (MST4) is an
important member of the GCK (also known as MAP4K2) subfamily
(9) and belongs to the mammalian
sterile-20 (Ste20)-like (MST) kinases (10), which play multiple roles in the
regulation of signaling pathways governing cell mitosis,
homeostasis, polarity, migration, apoptosis, proliferation, and
differentiation (9–15). MST4 has low expression in the
normal liver (16–18) and in some hepatoma cell lines such
as PLC5 (16), whereas it is
highly expressed in other hepatoma cells, such as SK-Hep1 (16). Studies imply that the expression of
MST4 in different types of hepatoma cell lines is in disagreement.
Overexpression of MST4 enhances ERK activity, leading to increased
cell proliferation and cellular transformation (19,20).
Active MST4 can also stabilize to bind with GM130 (a cis-Golgi
matrix protein) and regulate cell migration and polarization
(21). In prostate cancer, MST4
accelerates the proliferation of prostate cancer cells and enhances
anchorage-independent growth (20). In a previous study, we analyzed the
expression profiles of HCCs without or with intrahepatic metastasis
and found MST4 to be an important candidate gene for metastatic HCC
(22). Based on these studies, we
speculate that MST4 is a promoter of tumor progression and
metastasis. However, little is known about the expression of MST4
in HCC samples and its role in the prognosis of HCC.
The present study shows that the high expression of
MST4 is detected in highly invasive HCC cells and human HCC
specimens with vascular invasion. Furthermore, Kaplan-Meier
analysis suggests that high MST4 levels are associated with
significantly worse clinical outcomes after surgical excision for
HCC. Upregulation of MST4 in vitro significantly promotes
cell proliferation, colony formation, and invasion of HCC by
promoting EMT through the activation of ERK signaling pathways. The
combination of MST4 and phosphorylated ERK (p-ERK) has better power
to predict the outcomes of HCC. We report for the first time, that
MST4 is a pivotal factor that facilitates HCC migration and
metastasis and may become a potential adjuvant treatment target for
aggressive HCC.
Materials and methods
Patients and clinical samples
A total of 178 primary HCC samples were randomly
selected from patients who received curative resection for HCC at
the Liver Cancer Institute and Zhongshan Hospital, Fudan University
(Shanghai, China) from February 2006 to November 2006. This study
was approved by the Zhongshan Hospital Research Ethics Committee.
Follow-up procedures have been described previously (23). Each patient was followed-up until
March 2012, with a median follow-up of 56 months (range, 2–69
months). The clinical characteristics of HCC patients are presented
in Table I.
 | Table IComparison of clinicopathologic
profiles between HCC patients with high MST4 and low MST4
levels. |
Table I
Comparison of clinicopathologic
profiles between HCC patients with high MST4 and low MST4
levels.
| MST4 level | |
|---|
|
| |
|---|
| High (n=61) | Low (n=117) | |
|---|
|
|
| |
|---|
| Variables | Patients, n | % | Patients, n | % | P-value |
|---|
| Age, years |
| ≤50 | 20 | 32.8 | 46 | 39.3 | 0.418 |
| >50 | 41 | 67.2 | 71 | 60.7 | |
| Sex |
| Female | 8 | 13.1 | 26 | 22.2 | 0.164 |
| Male | 53 | 86.9 | 91 | 77.8 | |
| HBeAg |
| Negative | 7 | 11.5 | 8 | 6.8 | 0.394 |
| Positive | 54 | 88.5 | 109 | 93.2 | |
| GGT |
| Negative | 35 | 57.4 | 63 | 53.8 | 0.751 |
| Positive | 26 | 42.6 | 54 | 46.2 | |
| Cirrhosis |
| Absent | 48 | 78.7 | 91 | 77.8 | 1.000 |
| Present | 13 | 21.3 | 26 | 22.2 | |
| AFP |
| ≤400 ng/ml | 40 | 65.6 | 79 | 67.5 | 0.867 |
| >400 ng/ml | 21 | 34.4 | 38 | 32.5 | |
| Tumor size |
| ≤5 cm | 42 | 68.9 | 67 | 57.3 | 0.147 |
| >5 cm | 19 | 31.1 | 50 | 42.7 | |
| No. of tumor
nodules |
| Single | 48 | 78.7 | 100 | 85.5 | 0.293 |
| Multiple | 13 | 21.3 | 17 | 14.5 | |
| Tumor capsule |
|
Well-differentiated | 33 | 54.1 | 62 | 53.0 | 1.000 |
| Poorly
differentiated | 28 | 45.9 | 55 | 47.0 | |
| Microvascular
invasion |
| Negative | 36 | 59.0 | 81 | 69.2 | 0.186 |
| Positive | 25 | 41.0 | 36 | 30.8 | |
| Tumor
differentiation stage |
| I–II | 42 | 68.9 | 86 | 73.5 | 0.598 |
| III–IV | 19 | 31.1 | 31 | 26.5 | |
| Clinical stage |
| I | 45 | 73.8 | 80 | 68.4 | 0.494 |
| II | 16 | 26.2 | 37 | 31.7 | |
| UICC TNM stage |
| Low (stage
I/II) | 53 | 86.9 | 105 | 89.7 | 0.620 |
| High (stage
III) | 8 | 13.1 | 12 | 10.3 | |
Tissue microarray and
immunohistochemistry
Preparation of tissue microarray and
immunohistochemistry procedures were performed as described
(23). Antibodies used were
anti-human MST4 (rabbit, polyclonal, 1:100; Proteintech) and
anti-human p-ERK (rabbit, polyclonal, 1:200; Cell Signaling
Technology). Two independent pathologists who did not have
information regarding the patient characteristics assessed the
immunohistochemistry staining. The immunohistochemistry staining
intensity was graded on a scale from 0 to 3 (0, no staining; 1,
weak staining; 2, mild staining; 3, strong staining). The staining
extent was graded on a scale from 0 to 3 based on the percentage of
immunoreactive tumor cells (0%; 1–25%; 26–50%; 51–100%). The final
score was obtained by multiplying the staining extent score by the
staining intensity score. If the final score was <3, then it was
defined as low expression. If the final score was >3, then it
was defined as high expression.
Plasmid constructs
Flag-tagged and HA-tagged full-length MST4 cDNA were
cloned in pcDNA3 (Invitrogen) to create pFlag-MST4 and pHA-MST4,
respectively. Lentiviral vectors pLKO.1 TRC (Addgene plasmid 10879)
and pWPI.1 (Addgene plasmid 12254) were used for producing
recombinant lentiviruses. For RNA interference of MST4, DNA
fragments encoding hairpin precursors for shMST4 group 1
(5′-CCTCTTGGTGTAT AGTATTTA-3′) and shMST4 group 2 (5′-CCAGATTGCTA
CCATGCTAAA-3′) were inserted into pLKO.1 TRC, respectively. A
scrambled siRNA precursor (Scr) of similar GC content as shMST4
group 1 and shMST4 group 2, but with no sequence identity with
MST4, was used as a negative control. For overexpression of MST4,
flag-tagged MST4 cDNA was cloned in pWPI.1.
Cell lines, transfection, and
lentiviruses
The human hepatic non-tumor cell line LO2, the
embryonic kidney cell line HEK293T, and HCC cell lines SMMC7721,
PLC, and Hep3B were obtained from Cell Bank of Shanghai Institutes
of Biological Sciences, Chinese Academy of Sciences (Shanghai,
China). MHCC-97H and MHCC-LM3 cell lines were established at the
Liver Cancer Institute and Zhongshan Hospital, Fudan University.
All cells were cultured in DMEM high-glucose supplemented with 10%
fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C
with 5% CO2.
The expression plasmids were transfected into cells
using Lipofectamine 2000 (Invitrogen) according to the
manufacturer’s instructions. Helper plasmids pSPAX2 (Addgene
plasmid 12260) and pMD2.G (Addgene plasmid 12259) were
cotransfected with pLKO.1-based or pWPI.1-based plasmids in HEK293T
cells to package recombinant lentiviruses. Supernatants from
cotransfections were used for infection of cultured cells.
Quantitative reverse-transcription
polymerase chain reaction
The detected cells were harvested in 6-well plates.
The total RNA of each group was isolated using TRIzol (Gibco
Laboratories). Reverse-transcription was performed with a
PrimeScript reverse-transcription reagent kit (Takara
Biotechnology, Dalian, China). Gene-specific primers for human MST4
and GAPDH were generated by Sangon Biotech (Shanghai, China). The
following primer sequences were used: MST4:
F-5′-TTCGAGCTGGTCCATTTGATG-3′, R-5′-TGA ATGCAGATAGTCCAGACCT-3′; and
GAPDH: F-5′-TGTG GGCATCAATGGATTTGG-3′, R-5′-ACACCATGTATTCCG
GGTCAAT-3′. The iCycler and iQ real-time polymerase chain reaction
(Bio-Rad, Hercules, CA, USA) and SYBR-Green PCR Master Mix (Toyobo,
Osaka, Japan) were used for amplification and detection. The steps
of amplification were 95°C for 30 sec, 60°C for 30 se and 72°C for
30 sec for 40 cycles. The data were analyzed by using the real-time
polymerase chain reaction analysis software Bio-Rad iQ5. All
experiments were performed in triplicate.
Cell proliferation assay
Cell proliferation was measured by Cell Counting
Kit-8 (Dojindo, Kumamoto, Japan). According to the instructions,
cultured cells were seeded onto 96-well plates at 5×103
per well (n=5 for each time-point) in a final volume of 100 μl. At
indicated time-points, CCK-8 solution (10 μl) was added to each
well and the absorbance at 450 nm was monitored with Tecan Infinite
M200 NanoQuant Daily (Mannedorf, Switzerland).
Colony formation assay
To detect clonogenesis ability, the cells
(1×103 cells) were suspended with DMEM containing 10%
FBS and cultured in a 6-well plate at 37°C with 5% CO2.
After 14 days, colonies were photographed and counted. All
experiments were performed in triplicate.
Invasion assay
The invasive ability of HCC cells was detected by
24-well transwell chambers with 8-μm pores of polycarbonate
membranes (Costar, Cambridge, MA, USA) coated with Matrigel (BD
Pharmingen, San Jose, CA, USA). The upper chamber was filled with
FBS-free DMEM medium and the bottom chamber was filled with DMEM
containing 10% FBS as a chemoattractant. Upper chambers containing
cells (5×104) were filled and incubated at 37°C. After
48 h, cells migrating to the bottom of the membrane were stained
with Giemsa (Sigma Chemical, St. Louis, MO, USA). Then, the cells
were imaged and counted with a microscope (Leica, UK). All
experiments were performed in triplicate.
Immunofluorescence assay
Cells on the cover slips were fixed with 4%
paraformaldehyde for 30 min and permeabilized in 1% Triton X-100
for 15 min at room temperature. After blocking with 1% bovine serum
albumin for 30 min, the cells were incubated with primary
antibodies for 4 h. The following antibodies were used: human
anti-E-cadherin (mouse monoclonal antibody, 1:50; Santa Cruz),
Vimentin (mouse monoclonal antibody, 1:50; Santa Cruz), and human
anti-MST4 (rabbit, polyclonal, 1:100; Proteintech). Then, samples
were incubated with Alexa 488-conjugated and 594-conjugated
secondary antibodies (Invitrogen) for 1 h at room temperature.
Slides were mounted in medium containing DAPI (Vector Laboratories,
Burlingame, CA, USA) and analyzed under a fluorescence
microscope.
Western blotting
The proteins were extracted with RIPA buffer
containing protease inhibitors, separated on a denaturing 10%
SDS-polyacrylamide gel, and transferred onto polyvinylidene
difluoride membrane. After blocking with 5% non-fat milk in
phosphate-buffered saline containing 0.05% Tween-20, the membrane
was incubated with primary antibodies overnight at 4°C, followed by
incubating with peroxidase secondary antibodies for 1 h at 37°C.
The following antibodies were used: anti-human MST4 (rabbit,
polyclonal, 1:500; Proteintech); anti-human GAPDH (rabbit,
polyclonal, 1:1000; Cell Signaling Technology); anti-human p-ERK,
ERK, E-cadherin, N-cadherin, Vimentin, Snail, and Slug, and the
secondary antibody belonging to the EMT Antibody Sampler kit
(rabbit, polyclonal, 1:1,000; Cell Signaling Technology). Western
blotting was detected and analyzed with Image Acquisition using
Image Quant LAS 4000 (GE Healthcare Life Sciences, MI, USA) and
Quantity One software (Bio-Rad Laboratories), respectively.
Statistical analysis
Statistical analysis was performed with SPSS 13.0
(SPSS, Chicago, IL, USA). All data are presented as the mean ±
standard deviation. The Student’s t-test and one-way analysis of
variance were applied to analyze the difference of two groups or
more than two groups, respectively. Kaplan-Meier analysis was used
for survival analysis and log-rank test was used to compare patient
survival between subgroups. A Cox proportional hazards model was
adopted for multivariate analysis. Receiver-operating
characteristic (ROC) curve analysis was applied to assess the
predictive values of variables. P<0.05 was considered
statistically significant for all tests.
Results
High expression of MST4 in HCC cell lines
and specimens with invasive and metastatic potential
Our previous cDNA array study showed that MST4 had
higher expression level in HCC with metastasis than those without
metastasis (22), indicating the
correlation between MST4 level and metastatic potential of HCC. In
this study, we first examined the protein expression of MST4 in the
normal liver cell line and HCC cell lines with different metastatic
abilities. Western blot analysis showed that MST4 was detected in
higher levels in HCC cell lines with high metastatic potential
(MHCC97H and MHCCLM3) than those cell lines with low-metastatic
potential (Hep3B, PLC, and SMMC7721) and normal liver cell line L02
(Fig. 1A)
We then tested the mRNA and protein levels of MST4
in HCC samples with vascular invasion (VIHCC) and with non-vascular
invasion (non-VIHCC), and found that they were much higher in VIHCC
than that in non-VIHCC (P<0.05) (Fig. 1B and C), which is in agreement with
our previous cDNA array results. Immunohistochemistry staining
showed that MST4 expression was negative in normal and peritumoral
liver tissue, relatively positive in non-VIHCC, and strongly
positive in VIHCC (Fig. 1D). These
results indicate that high expression of MST4 is closely associated
with aggressive metastasis in HCC.
High MST4 protein expression in HCC is
associated with worse clinical outcomes
We then evaluated the association of MST4 expression
and the outcomes of 178 HCC patients with 5-year follow-up after
operation using immunostaining and tissue microarrays. The
clinicopathologic characteristics of the patients are shown in
Table I. MST4 staining was mainly
found in the cytoplasm of tumor cells and around the nucleus but
was absent in most stromal cells (Fig.
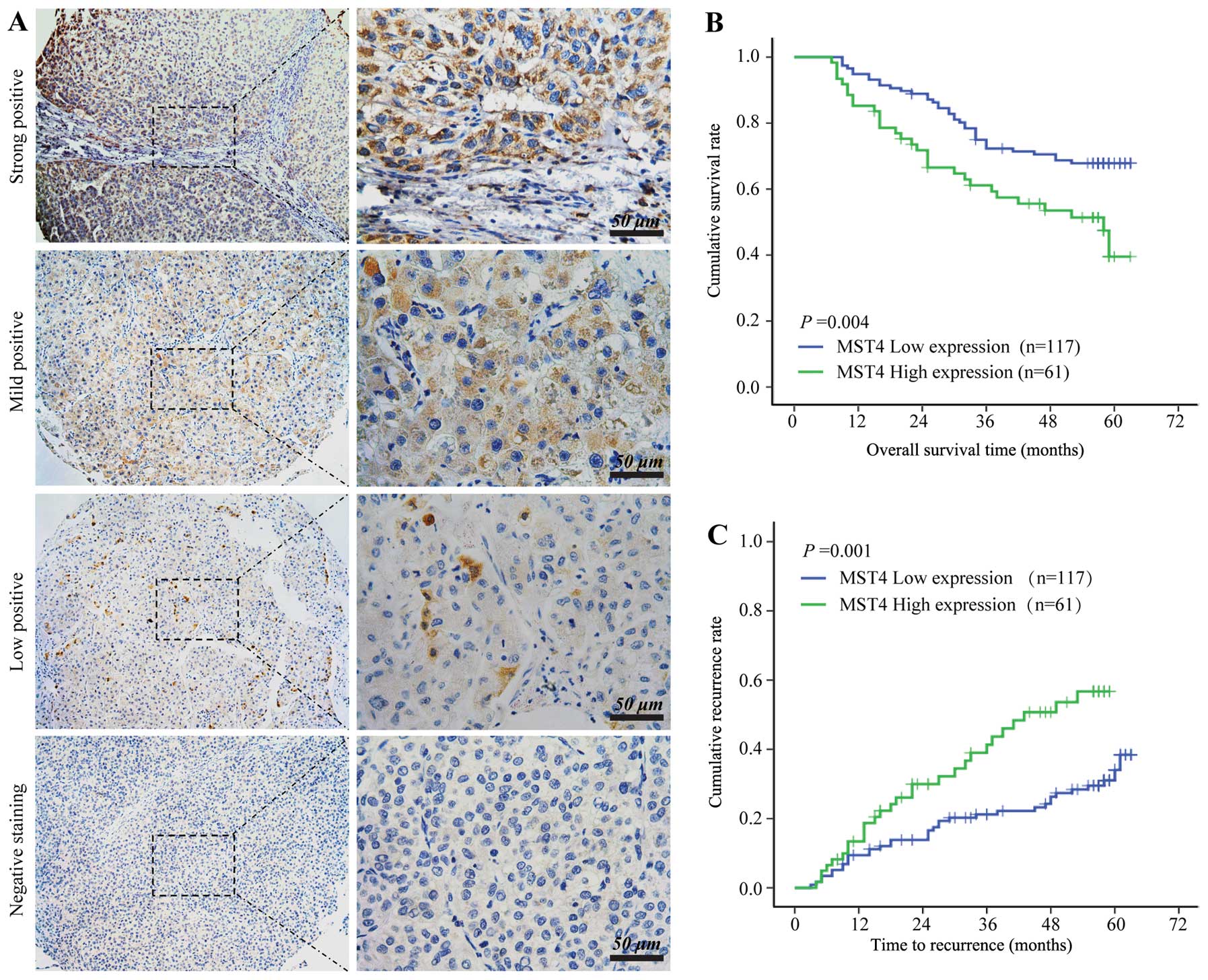
2A). According to the HCC staining scores, all the samples were
stratified into two grades: strong and mild MST4-positive staining
were defined as high MST4 expression (n=61), and low MST4-positive
staining and MST4-negative staining were defined as low MST4
expression (n=117).
Statistical analysis showed that the high MST4
expression group displayed significantly worse overall survival
(OS) (median OS, 58 months versus >63 months; log-rank=8.12;
P=0.004) (Fig. 2B) and shortened
time to tumor recurrence (TTR) (median TTR, 43.0 months versus
>63 months; log-rank 10.31; P=0.001) (Fig. 2C) compared to the low MST4
expression group. Large tumor size, microvascular invasion,
multiple tumors, and advanced TNM classification of malignant
tumors stage were found to be associated with worse OS and
shortened TTR in univariate analysis. Multivariate analysis results
revealed that MST4 intensity in tumors is an independent
prognosticator for both OS (relative risk, 1.863; P=0.012) and TTR
(relative risk, 2.348; P=0.001) (Table II), suggesting that MST4 is a
valuable predictor of clinical outcomes in HCC patients.
| Table IIUnivariate and multivariate analysis
of factors associated with survival and recurrence. |
Table II
Univariate and multivariate analysis
of factors associated with survival and recurrence.
| Overall
survival | Time to
recurrence |
|---|
|
|
|
|---|
| Univariate | Multivariate | Univariate | Multivariate |
|---|
|
|
|
|
|
|---|
| Features | P-value | HR | 95% CI | P-value | P-value | HR | 95% CI | P-value |
|---|
| Age |
| ≤50 vs. >50
years | 0.967 | | | NA | 0.545 | | | NA |
| Sex |
| Female vs.
male | 0.771 | | | NA | 0.983 | | | NA |
| Hepatitis B
antigen |
| Negative vs.
positive | 0.471 | | | NA | 0.869 | | | NA |
| Liver
cirrhosis |
| No vs. yes | 0.291 | | | NA | 0.033 | 3.074 | 1.223–7.725 | 0.017 |
| AFP |
| ≤400 vs. >400
ng/ml | 0.093 | | | NA | 0.791 | | | NA |
| Tumor size |
| ≤5 vs. >5
cm | 0.003 | 1.734 | 1.028–2.924 | 0.039 | 0.013 | | | NS |
| No. of tumor |
| Single vs.
multiple | 0.052 | | | NA | 0.010 | | | NS |
| Tumor
encapsulation |
| None vs.
complete | 0.336 | | | NA | 0.067 | | | NA |
| Microvascular
invasion |
| No vs. yes | 0.004 | | | NS | 0.027 | | | NS |
| Tumor
differentiation stage |
| I–II vs.
III–IV | 0.063 | | | NA | 0.815 | | | NA |
| TNM stage |
| I vs. II vs.
IIIa | 0.006 | 1.631 | 1.164–2.285 | 0.004 | 0.001 | 1.692 | 1.209–2.368 | 0.002 |
| MST4 |
| High vs. low
expression | 0.004 | 1.863 | 1.148–3.023 | 0.012 | 0.001 | 2.348 | 1.405–3.924 | 0.001 |
| p-ERK |
| High vs. low
expression | 0.038 | | | NS | 0.029 | 2.363 | 1.013–2.758 | 0.044 |
| Both MST4 and
p-Erk | 0.001 | | | NA | <0.001 | | | NA |
MST4 enhanced proliferation and invasive
potential of HCC cells in vitro
Considering the close association of high MST4
expression and high recurrence rate of HCC, we examined whether
high MST4 level plays a key role in HCC proliferation and invasion.
To verify this inference, lentivirus-mediated knockdown in highly
invasive MHCC-97H cells was performed to assess the functional
involvement of MST4 in HCC cell proliferation, colony formation,
and invasion in vitro. We generated three MST4-specific
shRNAs (shMST4) and infected MHCC-97H cells with a lentivirus
expressing shMST4 precursor, and found the second group of shMST4
induced >80% reduction which was used for further study
(Fig. 3A, left panel). Compared to
the scrambled shRNA group (SCR group) and wild-type group (WT
group), the proliferation and the clone formation in the MST4
knockdown group (KD group) were restrained significantly
(P<0.01) (Fig. 3B, left panel
and Fig. 3C, upper panel).
Furthermore, depletion of MST4 also led to dramatic decline in
invasiveness potential (transwell assay, P<0.01) (Fig. 3D, lower panel). We generated the
SMCC7721 clone with stable lentivirus-mediated overexpression of
MST4 (MST4 group) and the SMCC7721 empty vector as the control
group (MOCK group) and found that exogenous expression of MST4 in
the MST4 group significantly enhanced HCC cell proliferation,
colony formation, and invasion compared with wild-type and mock
groups(Fig. 3). Taken together,
these results suggest that MST4 plays an important role in HCC
proliferation and invasion.
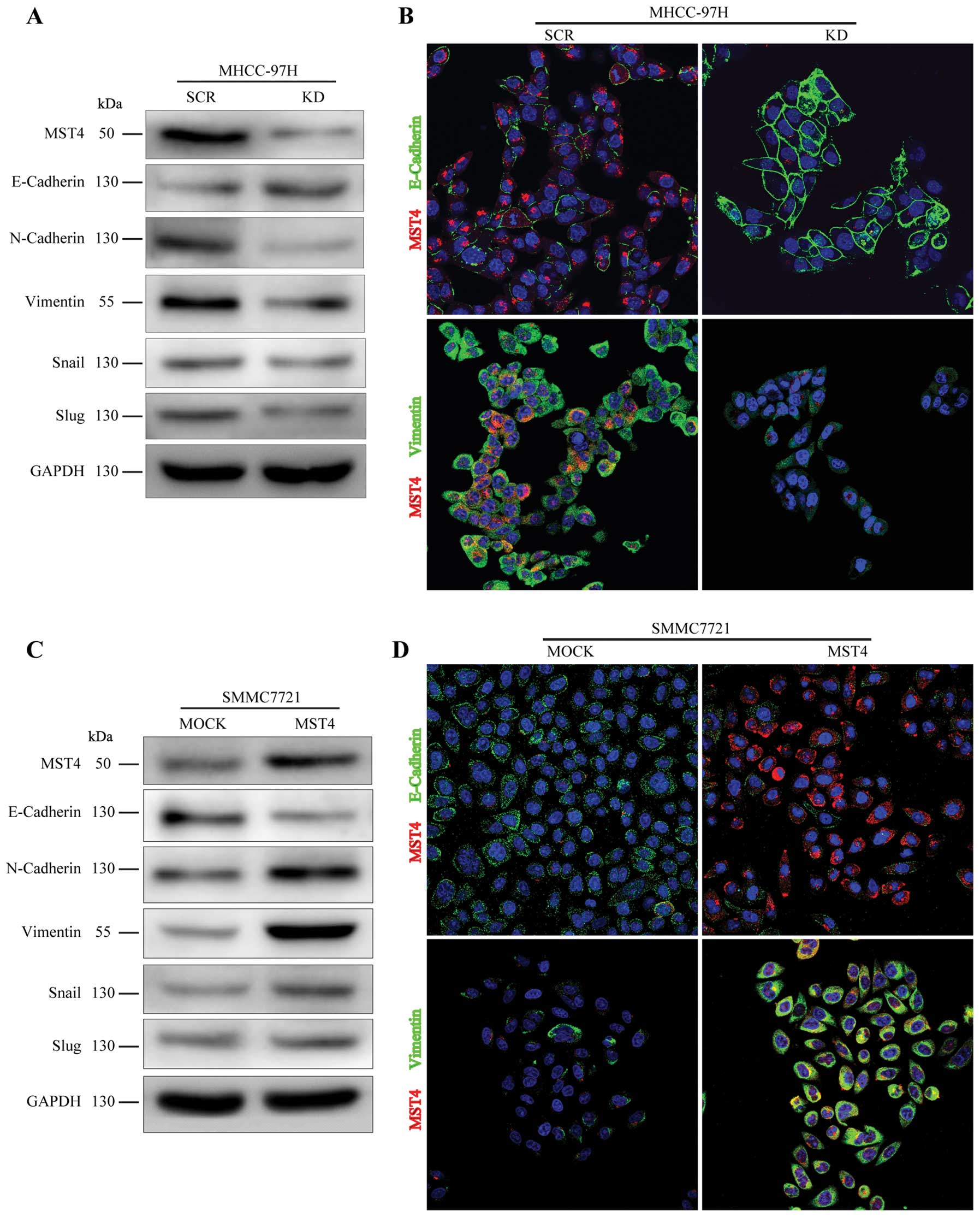
MST4 promotes mesenchymal transformation
in HCC cells
EMT is an important process in tumor proliferation
and invasion. To investigate whether MST4 altered the EMT process
in HCC metastatic progression, we examined the expression level of
E-cadherin, N-cadherin, and Vimentin, the cell markers of EMT, in
HCC cells. As shown in Fig. 4A and
C, relative to the scrambled shRNA group (SCR group), decreased
expression of MST4 (KD group) resulted in upregulation of
E-cadherin and downregulation of N-cadherin and Vimentin, whereas
overexpression of MST4 (MST4 group) led to downregulation of
E-cadherin and upregulation of N-cadherin and Vimentin, compared to
the control group (MOCK group). Immunofluorescence staining also
confirmed these results in Fig. 4B and
D. The expression of E-cadherin is negatively regulated by
transcription factors, such as Snail and Slug (24). As illustrated in Fig. 4A and C, the levels of Snail and
Slug were dramatically downregulated in the MHCC-97H knockdown
group and significantly upregulated after MST4 overexpression in
SMCC7721. These results indicate that MST4 may promote malignant
progression of HCC by inducing the EMT process.
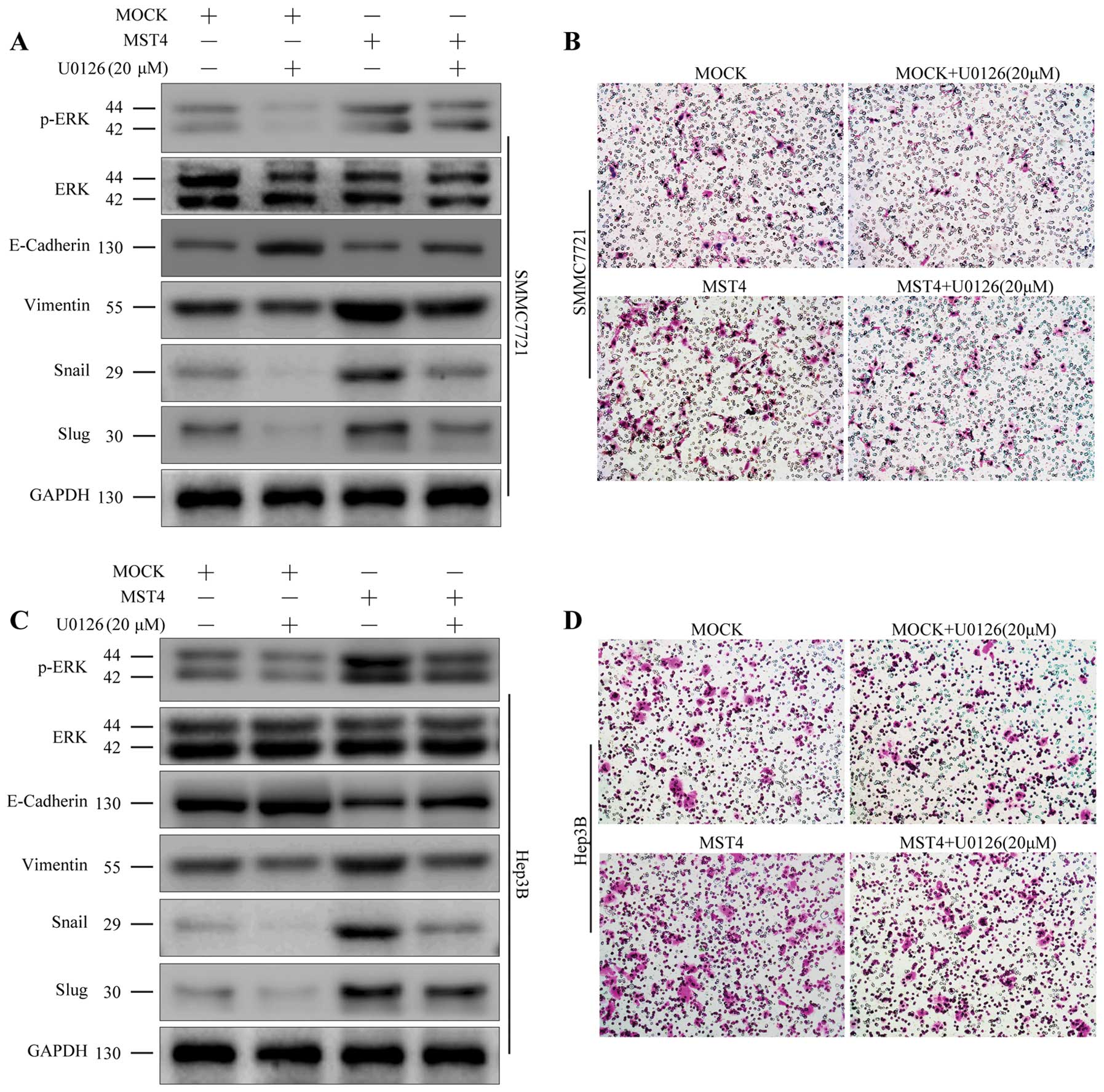
ERK pathway plays a critical role in
mediating MST4 function
Mounting evidence indicates that Snail and Slug play
crucial roles in initiating EMT process (25,26),
and they are both important downstream targets of the ERK signaling
pathway (27,28). To further investigate the
mechanisms of MST4 in promoting EMT process in HCC cells, we
examined the influence of MST4 overexpression on the ERK
pathway.
HCC cell lines SMMC7721 and Hep3B with MST4
overexpression were generated with stable lentivirus-mediated MST4
(MST4 group) and the empty vector (control MOCK group). As shown in
Fig. 5A and B, the ratio of p-ERK
to ERK was upregulated in the MST4 group compared to the mock
group. This trend was similar to that of the mesenchymal markers
such as Snail, Slug, and Vimentin, but was contrary to that of
E-cadherin.
To further confirm the key role of the p-ERK/ERK
pathway in the promotion of EMT with MST4, we used the p-ERK
inhibitor (U0126, 20 μM; Sigma-Aldrich) to decrease the activation
of ERK. As shown in Fig. 5, U0126
treatment partially reversed the MST4-induced increase in the ratio
of p-ERK to ERK and the mesenchymal markers but did not bring it
back down to control levels. In addition, U0126 treatment also
suppressed the migratory and invasive behavior of the SMMC7721 and
Hep3B cells with MST4 overexpression (Fig. 5B and D). These data revealed that
overexpression of MST4 promote the EMT process, at least in part,
through upregulation of the ERK signaling pathway.
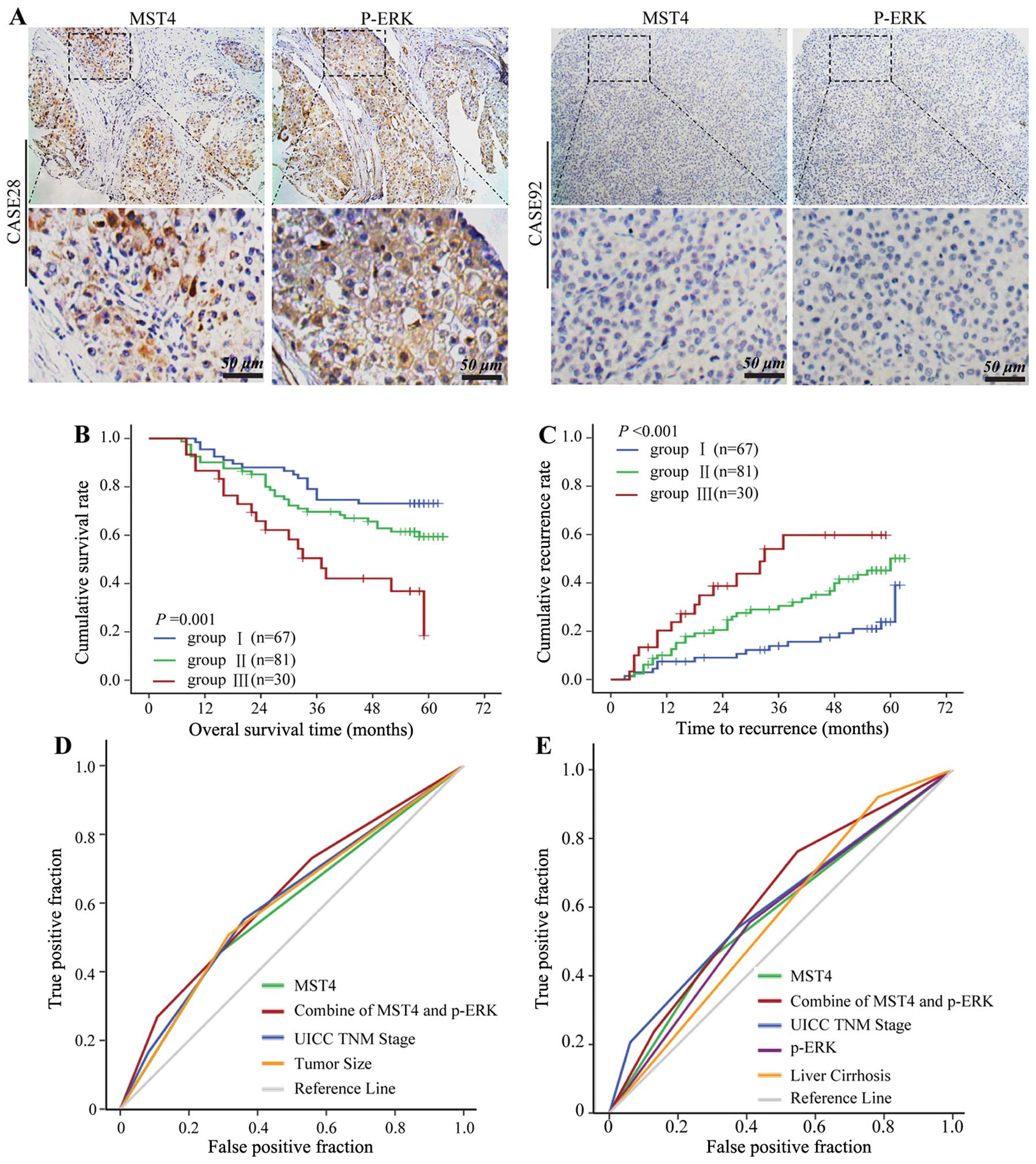
Combination of MST4 and p-ERK levels has
a better prognostic value for HCC
On the basis of the in vitro finding that
MST4 regulated phosphorylation of ERK in HCC, we further
investigated the relationship between MST4 and p-ERK in HCC tissue
microarray. Positive p-ERK staining was partly detected in
cytoplasmic and nuclear HCC cells (Fig. 6A), and p-ERK expression was
significantly correlated with MST4 expression (data not shown). To
further confirm the prognostic value of MST4 and p-ERK, HCC tissues
were classified into the following three groups according to the
positive staining value of MST4 and p-ERK: group 1 (n=67), low MST4
and low p-ERK expression; group 2 (n=81), either high MST4 or high
p-ERK expression; and group 3 (n=30), high MST4 and high p-ERK
expression. There were significant differences in OS (P=0.001)
(Fig. 6B) and TTR (P<0.001)
(Fig. 6C) among the three
groups.
Multivariate survival analysis results showed that
MST4 alone could predict death and recurrence precisely with the
areas under the receiver-operating characteristic curve of 0.584
[95% confidence interval (CI), 0.503–0.672] and 0.579 (95% CI,
0.502–0.668), respectively. The combination of MST4 and p-ERK
further elevated the area under the curve and was better for
predicting death (0.622; 95% CI, 0.532–0.707) (Fig. 6D) and recurrence (0.623; 95% CI,
0.537–0.708) (Fig. 6E) for
HCC.
Discussion
Currently, the molecular mechanism of HCC invasion
and metastasis is still not well-understood, and the high rate of
HCC metastatic recurrence is the main hindrance in further
improvement of overall survival after hepatectomy. It is urgent to
find new innovative therapeutic targets to improve HCC prognosis.
MST4, a member of the protein family that shares similarity with
Ste20, is an MST kinase (12,16–18),
which has been found to play important roles in regulating multiple
cell functions, such as cell polarity and proliferation (9,10),
but little is known about its oncogenic role in HCC progression. In
our previous gene expression profile study of HCC without or with
intrahepatic metastasis, MST4 was identified as an important
candidate gene for HCC metastasis (22). Here we elucidated that high MST4
expression was associated with malignance in HCC cell lines with
different metastatic potential, and in vitro experiments
found that MST4 played important roles in proliferation, colony
formation, and invasion in HCC cells. We also verified that high
expression of MST4 in HCC was associated with a high incidence of
intrahepatic metastasis, shorter time to progression, and poor
survival after hepatectomy, and univariate and multivariate
analysis results revealed that MST4 is an independent prognostic
indicator for both OS and TTR in HCC patients. These results
suggest that MST4 plays important roles in HCC proliferation,
invasion and metastasis.
Mounting evidence has indicated that EMT is an
important process for tumor invasion and metastasis. In this study,
downregulation of MST4 expression resulted in a sharp decline in
proliferation, colony formation, and invasion of the HCC cells with
high expression of E-cadherin and low expression of N-cadherin and
Vimentin. Conversely, overexpression of MST4 promoted the
metastatic potential of the HCC cells with accumulation of
N-cadherin and Vimentin and decrease of E-cadherin. Considering the
crucial role of EMT in the progression and metastasis of multiple
cancers and the influence of the expression of MST4 accompanied by
the change in mesenchymal marker Vimentin and epithelial marker
E-cadherin and N-cadherin, we proposed that MST4 is a key protein
in promoting the EMT process and is a promising molecule in shaping
the postoperative strategy for prevention of metastatic recurrence
for HCC.
Moreover, immunofluorescence staining confirmed that
the expression of E-cadherin was negatively regulated by
transcription factors Snail and Slug. The levels of Snail and Slug
were dramatically downregulated in the MST4 knockdown group and
significantly upregulated after MST4 overexpression. It is well
known that p-ERK/ERK signaling pathways upregulate Snail and Slug,
thereby leading to the downregulation of epithelial marker
E-cadherin (29,30). There is also evidence implying that
MST4 influences cell growth and transformation by modulating a
ras/raf-independent ERK pathway (16). We observed in our study that
exogenous expression of MST4 in SMMC7721 and Hep3B cells enhanced
cell proliferation and invasion with hyperactivation of ERK
signaling pathways. The inhibition of ERK signaling pathways,
through addition of specific inhibitor U0126, significantly
suppressed the expression of Snail and Slug without affecting MST4
expression levels and blocked MST4-mediated promotion of migration
and invasion. Moreover, the combination of MST4 and p-ERK in
receiver-operating characteristic curve analysis had better
prognostic value than MST4 alone for OS and accumulated recurrence
for HCC. These findings demonstrate that MST4 promotes EMT, at
least in part, through upregulation of the ERK signaling pathways,
and inhibitors of p-ERK might be a potent therapeutic approach
against HCC metastasis.
In conclusion, our results indicate that high
expression of MST4 is a major contributor to the invasive phenotype
of HCC by promoting the EMT process, and MST4 alone or combination
with p-ERK is a novel prognostic marker and potential therapeutic
target for HCC.
Acknowledgements
We thank Mr. Wei-De Zhang for assistance in
collecting patient data. This study was jointly supported by two
grants (nos. 81071993 and 81372654) from the National Natural
Science Foundation of China.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
3
|
Portolani N, Coniglio A, Ghidoni S, et al:
Early and late recurrence after liver resection for hepatocellular
carcinoma: prognostic and therapeutic implications. Ann Surg.
243:229–235. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu Y, Zhang JB, Qin Y, et al: PROX1
promotes hepatocellular carcinoma metastasis by way of
up-regulating hypoxia-inducible factor 1alpha expression and
protein stability. Hepatology. 58:692–705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Turley EA, Veiseh M, Radisky DC and
Bissell MJ: Mechanisms of disease: epithelial-mesenchymal
transition - does cellular plasticity fuel neoplastic progression?
Nat Clin Pract Oncol. 5:280–290. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tse JC and Kalluri R: Mechanisms of
metastasis: epithelial-to-mesenchymal transition and contribution
of tumor microenvironment. J Cell Biochem. 101:816–829. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shi Z, Jiao S, Zhang Z, et al: Structure
of the MST4 in complex with MO25 provides insights into its
activation mechanism. Structure. 21:449–461. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ling P, Lu TJ, Yuan CJ and Lai MD:
Biosignaling of mammalian Ste20-related kinases. Cell Signal.
20:1237–1247. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Delpire E: The mammalian family of sterile
20p-like protein kinases. Pflugers Arch. 458:953–967. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dan I, Watanabe NM and Kusumi A: The Ste20
group kinases as regulators of MAP kinase cascades. Trends Cell
Biol. 11:220–230. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hao W, Takano T, Guillemette J, Papillon
J, Ren G and Cybulsky AV: Induction of apoptosis by the Ste20-like
kinase SLK, a germinal center kinase that activates apoptosis
signal-regulating kinase and p38. J Biol Chem. 281:3075–3084. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nicke B, Bastien J, Khanna SJ, et al:
Involvement of MINK, a Ste20 family kinase, in Ras oncogene-induced
growth arrest in human ovarian surface epithelial cells. Mol Cell.
20:673–685. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Record CJ, Chaikuad A, Rellos P, et al:
Structural comparison of human mammalian ste20-like kinases. PLoS
One. 5:e119052010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin JL, Chen HC, Fang HI, Robinson D, Kung
HJ and Shih HM: MST4, a new Ste20-related kinase that mediates cell
growth and transformation via modulating ERK pathway. Oncogene.
20:6559–6569. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dan I, Ong SE, Watanabe NM, et al: Cloning
of MASK, a novel member of the mammalian germinal center kinase III
subfamily, with apoptosis-inducing properties. J Biol Chem.
277:5929–5939. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qian Z, Lin C, Espinosa R, LeBeau M and
Rosner MR: Cloning and characterization of MST4, a novel Ste20-like
kinase. J Biol Chem. 276:22439–22445. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma X, Zhao H, Shan J, et al: PDCD10
interacts with Ste20-related kinase MST4 to promote cell growth and
transformation via modulation of the ERK pathway. Mol Biol Cell.
18:1965–1978. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sung V, Luo W, Qian D, Lee I, Jallal B and
Gishizky M: The Ste20 kinase MST4 plays a role in prostate cancer
progression. Cancer Res. 63:3356–3363. 2003.PubMed/NCBI
|
|
21
|
Preisinger C, Short B, De Corte V, et al:
YSK1 is activated by the Golgi matrix protein GM130 and plays a
role in cell migration through its substrate 14-3-3zeta. J Cell
Biol. 164:1009–1020. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ye QH, Qin LX, Forgues M, et al:
Predicting Hepatitis B virus-positive metastatic hepatocellular
carcinomas using gene expression profiling and supervised machine
learning. Nat Med. 9:416–423. 2003. View
Article : Google Scholar
|
|
23
|
Zhu XD, Zhang JB, Zhuang PY, et al: High
expression of macrophage colony-stimulating factor in peritumoral
liver tissue is associated with poor survival after curative
resection of hepatocellular carcinoma. J Clin Oncol. 26:2707–2716.
2008. View Article : Google Scholar
|
|
24
|
Batlle E, Sancho E, Franci C, et al: The
transcription factor snail is a repressor of E-cadherin gene
expression in epithelial tumour cells. Nat Cell Biol. 2:84–89.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou BP, Deng J, Xia W, et al: Dual
regulation of Snail by GSK-3beta-mediated phosphorylation in
control of epithelial-mesenchymal transition. Nat Cell Biol.
6:931–940. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Doble BW and Woodgett JR: Role of glycogen
synthase kinase-3 in cell fate and epithelial-mesenchymal
transitions. Cells Tissues Organs. 185:73–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cross DA, Alessi DR, Cohen P, Andjelkovich
M and Hemmings BA: Inhibition of glycogen synthase kinase-3 by
insulin mediated by protein kinase B. Nature. 378:785–789. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cohen P and Frame S: The renaissance of
GSK3. Nat Rev Mol Cell Biol. 2:769–776. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hong KO, Kim JH, Hong JS, et al:
Inhibition of Akt activity induces the mesenchymal-to-epithelial
reverting transition with restoring E-cadherin expression in KB and
KOSCC-25B oral squamous cell carcinoma cells. J Exp Clin Cancer
Res. 28:28–39. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Conacci-Sorrell M, Simcha I, Ben-Yedidia
T, Blechman J, Savagner P and Ben-Ze’Ev A: Autoregulation of
E-cadherin expression by cadherin-cadherin interactions: the roles
of beta-catenin signaling, Slug, and MAPK. J Cell Biol.
163:847–857. 2003. View Article : Google Scholar : PubMed/NCBI
|