Introduction
Hyperparathyroidism is characterized by
calcium-insensitive hyper-secretion of parathyroid hormone and the
development of tumors from parathyroid cells. The majority of
tumors in primary hyperparathyroidism are sporadic, but ~5% are
associated with hereditary cancer syndromes (1). Cases of primary hyperparathyroidism
(80–85%) are due to parathyroid adenomas, and 10–15% are attributed
to primary chief cell hyperplasia (2).
A molecular analysis of parathyroid hyperplasia,
adenoma and carcinoma has been reported (3), and cyclin D1, calcium sensing
receptor and vitamin D receptor genes are known to play a role in
tumor development in parathyroid glands (4,5).
Overexpression of cyclin D1, a key regulator of the cell cycle, has
been implicated in the pathogenesis of 20–40% of sporadic
parathyroid adenomas (6). In
addition, loss of chromosome segment 1p is strongly associated with
parathyroid adenoma and carcinoma, but not with hyperplasia
(2,3,7,8).
Other findings relevant to parathyroid pathogenesis are mutations
of the HRPT2 gene (1q24-32) or MEN gene (11q13) (9,10).
Germline mutations of the HRPT2 gene have been described in
parathyroid carcinoma, especially in HPT-JT syndrome (11–14),
and have been implicated in the development of a high proportion of
parathyroid carcinomas (2).
Furthermore, microarray profiling has been used to examine
different types of parathyroid disease, and these have proved to be
excellent objects for understanding the molecular pathogenesis of
the parathyroid gland (3,4). However, the molecular events involved
in the formation of parathyroid tumors, especially in HPT-JT
syndrome, are poorly understood. We therefore generated gene
expression profiles of the main types of primary parathyroid
disease: sporadic parathyroid adenoma, and parathyroid carcinoma in
HPT-JT syndrome due to an HRPT2 splicing mutation (hereinafter
referred to as HPT-JT syndrome). As a control we also profiled
normal parathyroid tissue.
Materials and methods
Tumor samples
Fresh tumor tissues were obtained from the patient
with parathyroid tumor in HPT-JT syndrome and one with a sporadic
parathyroid tumor (15). As a
control, normal parathyroid gland was obtained from excess tissues
after routine parathyroid auto-transplantation during
thyroidectomy. Sporadic parathyroid adenoma and the parathyroid
tumors in HPT-JT syndrome were snap-frozen in liquid nitrogen
immediately after surgery and stored at −80°C until use. The
parathyroid tumor in HPT-JT syndrome was classified as a carcinoma
(15), and confirmed to harbour a
germ-line HRPT2 splicing mutation (15,16).
The histology of the parathyroid carcinoma was classified according
to the WHO guidelines (4). Patient
data are summarized in Table I.
Approval for this study was obtained from the Human Research Ethics
Committees of the participating institutions.
 | Table IClinical and genetic data for the
three patients in this study. |
Table I
Clinical and genetic data for the
three patients in this study.
| Pt | Age (years) | Gender | Ca (mg/dl) | P (mg/dl) | iPTH (pg/ml) | Size (cm) | Wt (gm) | HDx | HRPT2 mutation |
|---|
| KSN | 54 | Female | 9.1 | 4.3 | - |
0.3*0.2 | - | Normal | None |
| CJN | 57 | Female | 11.9 | 2.0 | 624.28 |
3.5*2.0 | 6.0 | Sporadic | None |
| KKT | 22 | Male | 14.1 | 1.4 | 1110.51 |
4.0*2.5 | 7.5 | HPT-JT
syndrome | IVS2-1G>A |
Preparation of RNA
Total RNA was extracted from frozen tissues using
TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA) and
purified using Qiagen RNeasy spin columns (Qiagen, Inc., Valencia,
CA) according to the protocols recommended by Illumina (San Diego,
CA) for DASL applications. After DNase digestion and clean-up
procedures, RNA samples were quantified, aliquoted and stored at
−80°C until use. For quality control, RNA purity and integrity were
evaluated by denaturing gel electrophoresis, OD 260/280 ratio and
analysed on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo
Alto, CA).
DASL (cDNA mediated annealing, selection,
extension and ligation) assay
A 0.25–1.0 μg samples of total RNA were converted
into cDNA using biotinylated random primers, oligodeoxythymidine
primers and Illumina reagents. The resulting biotinylated cDNA was
annealed to assay oligonucleotides and bound to
streptavidin-conjugated paramagnetic particles to be able to select
cDNA/oligo complexes. After oligo hybridization, mis-hybridized and
non-hybridized oligos were washed away, and bound oligos were
extended and ligated to generate templates for amplification with
shared PCR primers. The fluorescence-labelled complementary strand
was hybridized at 45°C for 18 h to Illumina HumanRef-8 DASL
Expression BeadChips. After hybridization, the arrays were scanned
by laser confocal microscopy using an Illumina BeadArray Reader.
Array data export, processing and analysis were performed with
Illumina BeadStudio v3.1.3 (Gene Expression Module V3.3.8).
Raw data preparation and statistic
analysis
The exported raw array data were filtered by
detection P-value <0.05 (similar to noise signals) in at least
50% of samples. We applied a filtering criterion for data analysis
in which a higher signal value was required to obtain a detection
P-value <0.05. The selected gene signal value was transformed by
logarithm and normalized by a quantile method. Comparisons were
carried out using t-tests and Benjamini-Hochberg FDR (false
discovery rate) adjusted P-values (<0.05) and fold changes.
Subsequently Illumina HumanRef-8 DASL Expression BeadChip
expression data were re-analyzed using GenPlex software 3.0
(Istech, Inc., Korea). For primary data filtering, spots with a
P-call (detection call P-value <0.1) were selected, and the
remaining filtered data were used for further analysis. Quantile
normalization was used to normalize data.
Unsupervised and supervised analysis
Unsupervised hierarchical clustering of log ratios
was performed with Cluster 3.0, and the results were visualized
with Treeview software (Stanford University, Palo Alto, CA).
Pearson’s correlation, mean centering and average linkage were
applied in all clustering applications. For clustering, we used
average linkage clustering with standard correlation as the
similarity metric. Genes within 0.5 standard deviations of the
log-transformed ratios were discarded. To select specific and
robust gene sets associated with the normal parathyroid gland,
sporadic parathyroid adenoma and HPT-JT syndrome, we used the
combination analysis with Welch’s t-test and fold-change. On
Welch’s t-test and fold-change analysis, genes having P-values
<0.05 and showing fold-change >2.0 were selected.
KEGG pathway analysis
Molecular pathways associated with differentially
expressed genes were identified using the Kyoto Encyclopedia of
Genes and Genomes (KEGG) pathway database (http://www.genome.jp/kegg). This tool maps genes to
known pathways and provides a summary of the biological processes
affected. The above tool directs the specific genes to predicted
pathways and provides the summary of how the biological processes
have been affected. Then, the results of the biological processes
were shown by a bar graph, giving the P-value <0.05 that was
considered significant in KEGG.
Real-time PCR and RT-PCR
First strand cDNA was synthesized using 1 μg of
total RNA, oligo(dT) and SuperScript® II Reverse
Transcriptase (Invitrogen, Grand Island, NY). RT-PCR was carried
out using an iCycler (Bio-Rad, Hercules, CA). The RT-PCR conditions
were 95°C for 2 min, followed by 30 cycles at 95°C for 30 sec, 60°C
for 30 sec, 72°C for 30 sec and 72°C for 10 min for final
extension. Real-time RT-PCR for relevant genes was carried out
using a SYBR-Green PCR kit (Bio-Rad) with an Mx3000P™ Real-Time PCR
System (Stratagene, La Jolla, CA). The PCR primer sequences used
are shown in Table II. The PCR
conditions were 95°C for 10 min, followed by 45 cycles of 95°C for
15 sec, 60°C for 1 min and 72°C for 30 sec with a single
fluorescence measurement. For dissociation curves, reactions were
incubated at 95°C for 1 min, and lamped up from 55 to 95°C at a
heating rate of 0.1°C/sec, and fluorescence was measured
continuously. Gene expression was calculated according to the
2−ΔΔCt method using β-actin as an internal standard
(17).
| Table IIList of gene-specific primers. |
Table II
List of gene-specific primers.
| Gene | Primer |
|---|
| FGFR1 | F:
5′-CACCCGAGGCATTATTTGAC-3′
R: 5′-AAGTTCCTCCACAGGCACAC-3′ |
| FGF19 | F:
5′-AGATCAAGGCAGTCGCTCTG-3′
R: 5′-CGGATCTCCTCCTCGAAAGC-3′ |
| FGFR2 | F:
5′-GACAAAGACAAGCCCAAGGA-3′
R: 5′-TGACATAGAGAGGCCCATCC-3′ |
| RELA | F:
5′-TCTGCTTCCAGGTGACAGTG-3′
R: 5′-GCCAGAGTTTCGGTTCACTC-3′ |
| CHUK | F:
5′-GAAGGTGCAGTAACCCCTCA-3′
R: 5′-ATTGCCCTGTTCCTCATTTG-3′ |
| NTRK1 | F:
5′-GGACAACCCTTTCGAGTTCA-3′
R: 5′-CAAGGAGCAGCGTAGAAAGG-3′ |
| TCF7 | F:
5′-CCTTGATGCTAGGTTCTGGTG-3′
R: 5′-GCTTCTTGATGGTTGGCTTC-3′ |
| FGF3 | F:
5′-AGATAACGGCAGTGGAGGTG-3′
R: 5′-ATTATAGCCCAGCTCGTGGA-3′ |
| CACNA1A | F:
5′-AGTGAACAAAAACGCCAACC-3′
R: 5′-AAAGTAGCGCAGGTTCAGGA-3′ |
| FGF22 | F:
5′-TTCTACGTGGCCATGAACCG-3′
R: 5′-GTGTTGTGGCCGTTCTCTTC-3′ |
| β-actin | F:
5′-GAGCTACGAGCTGCCTGAC-3′
R: 5′-GGATGCCACAGGACTCCA-3′ |
| PLCB4 | F:
5′-ATCTGGAAGGGCGGATAGTT-3′
R: 5′-CATTGGACTGACGTTGTTGG-3′ |
| CAMK2D | F:
5′-AAGGGTGCCATCTTGACAAC-3′
R: 5′-TGCTTTCGTGCTTTCACATC-3′ |
| FGF1 | F:
5′-TCAGAGGACATGGCAAGGTA-3′
R: 5′-GGGAATGTCCCCAGGTTAAT-3′ |
| IL1B | F:
5′-TCCAGGGACAGGATATGGAG-3′
R: 5′-TCTTTCAACACGCAGGACAG-3′ |
| FGF7 | F:
5′-CAGTGGCAGTTGGAATTGTG-3′
R: 5′-CCTCCGTTGTGTGTCCATTT-3′ |
| TRAF2 | F:
5′-ACCAAGCTGGAAGCCAAGTA-3′
R: 5′-GTGAACACAGGCAGCACAGT-3′ |
| MAPK10 | F:
5′-CTTCCCAGATTCCCTCTTCC-3′
R: 5′-GCTGGGTCATACCAGACGTT-3′ |
| FGF17 | F:
5′-CAGATCCGCGAGTACCAACT-3′
R: 5′-TCACTCTCAGCCCCTTTGAT-3′ |
| FGFR4 | F:
5′-TTTCCCCTATGTGCAAGTCC-3′
R: 5′-GTAGGAGAGGCCGATGGAAT-3′ |
| CACNA1H | F:
5′-TACTCGTTGGACGGACACAA-3′
R: 5′-AAGCACAGCAGAAGGACGTT-3′ |
Immunohistochemistry (IHC)
Tissues were processed for paraffin embedding, and
3-μm sections were prepared and mounted on glass slides. The
mounted sections were pretreated with 10 mM sodium citric acid at
95°C for 10 min for antigen retrieval, and then with 0.3%
H2O2 in methanol for 30 min to permeabilize
them. The sections were blocked using 2.5% horse serum, and then
incubated with rabbit anti-FGFR1 antibody (Cell Signaling, Danvers,
MA). After rinsing, they were incubated sequentially in
biotinylated anti-rabbit antibody and then with ABC complex
(Vector, Burlingame, CA) for 1 h at room temperature.
Immunoreactivity was visualized by incubation in
3′,3-diaminobenzidine (DAB) solution (Vector) for 50 sec at room
temperature. The sections were counterstained with Harris
haematoxylin, dehydrated, cleared, mounted and viewed under a light
microscope (AX70; Olympus, Tokyo, Japan). For immunofluorescence
assays, the mounted sections were blocked with 2.5% horse serum,
and incubated with rabbit anti-FGFR1 antibody (Cell Signaling).
After rinsing, they were incubated with anti-rabbit antibody.
Nuclei were stained with 4′,6-diamino-2-phenylindole (DAPI)
(ImmunoBioscience, Santa Clara, CA) and viewed under a fluorescence
microscopy (Axiovert 200, Carl Zeiss, Oberkochen, Germany).
Results
Transcriptome scans identified
large-scale gene expression changes between HPT-JT syndrome,
sporadic parathyroid adenoma and normal parathyroid gland
Initially we performed a molecular classification
analysis to determine whether our spotted-oligonucleotide
microarray system was able to differentiate HPT-JT syndrome,
sporadic parathyroid adenoma, and normal parathyroid gland by
molecular profiling. We conducted an average linkage unsupervised
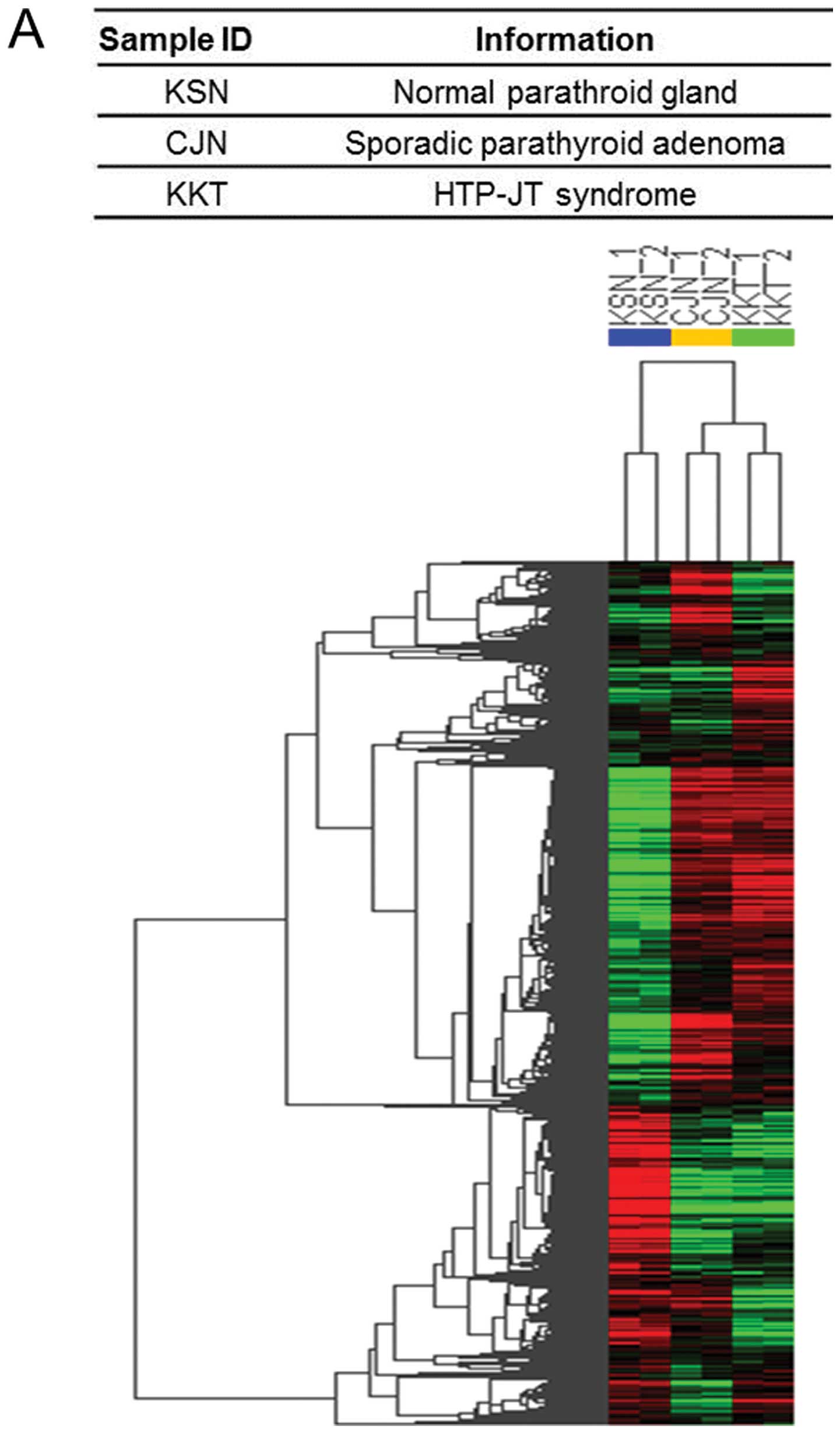
hierarchical clustering analysis. In Fig. 1A red and green indicate transcript
expression levels above and below the median of the sample. We
selected 5697 genes (>+2-fold, 3375 and <−2-fold, 2322 genes,
P<0.05) in HPT-JT syndrome and 5328 genes (>+2-fold, 3345 and
<−2-fold, 1983 genes, P<0.05) in sporadic parathyroid adenoma
relative to normal parathyroid gland according to the minimal
filtering criteria (Fig. 1B). Venn
diagram analysis of gene signatures common to HPT-JT syndrome and
sporadic parathyroid adenoma revealed that 2065 of the genes were
de-regulated relative to the normal parathyroid gland only in
HPT-JT syndrome, and 1696 genes only in sporadic parathyroid
adenoma (Fig. 1C). These data
suggest that thousands of genes either contributed to or were
affected by the development of tumor in parathyroid gland.
Identification of molecular signature in
HPT-JT syndrome and sporadic parathyroid adenoma
By applying the combination of Welch’s t-test and
fold-change analysis to the microarray data, we identified an
upregulated specific molecular signature of 1101 genes in HPT-JT
syndrome, and a downregulated specific molecular signature of 964
genes. Similarly we obtained an upregulated specific molecular
signature of 1071 genes in sporadic parathyroid adenoma and a
downregulated specific molecular signature of 625 genes. A total of
2274 genes were upregulated in both HPT-JT syndrome and sporadic
parathyroid adenoma, and 1358 genes were downregulated in both
(Fig. 1C).
Identification of the molecular pathways
in HPT-JT syndrome and sporadic parathyroid adenoma
Using the pathway mining tool in the KEGG pathway
database we characterized the biological processes in which the
2065 genes in the HPT-JT syndrome signature and the 1696 genes in
the sporadic parathyroid adenoma signature participated (Fig. 2). MAPK, regulation of actin, and
calcium signaling pathways were the most significant signaling
pathways associated specifically with HPT-JT syndrome (Fig. 2A), while calcium signaling, MAPK
and, insulin signaling were the most significant signaling pathways
associated with sporadic parathyroid adenoma (Fig. 2B). We sub-classified, according to
KEGG, 16 of the genes associated with pathways that were
specifically de-regulated in HPT-JT syndrome (Fig. 2C), and 15 of the genes in sporadic
parathyroid adenoma (Fig. 2D). In
Fig. 2C and D solid and open bars
represent genes up- or downregulated, respectively. Genes
overexpressed only in HPT-JT syndrome were FGFR1, FGF19, FGFR2,
RELA, CHUK, MKNK2 and NFATC4, and genes underexpressed only in
HPT-JT syndrome were FGF22, CACNA1A, FGF3, TCF7, NTRK1, EGFR,
CACNA1G, ATF4 and FGF12. The PLCB4, CAMK2D, FGF1, TNF, IL1B, FGF7,
PHKB, PRKAR2A and IRAK1 were overexpressed only in sporadic
parathyroid adenoma, while CACNA1H, FGFR4, FGF17, MAPK10, TRAF2 and
CAMK2B were underexpressed. It is evident that the genes
de-regulated in HPT-JT syndrome are quite different from those
de-regulated in sporadic parathyroid adenoma (Tables III and IV).
| Table IIISignificant de-regulated pathways and
genes associated with HPT-JT syndrome. |
Table III
Significant de-regulated pathways and
genes associated with HPT-JT syndrome.
| KEGG pathway
(HPT-JT syndrome) | Genesa | P-value |
|---|
| MAPK signaling
pathway | FGFR1,
FGF19, FGFR2, CHUK, MKNK2,
NFATC4, FGF12, ATF4, CACNA1G, EGFR, NTRK1, FGF3, CACNA1A,
FGF22 | 0 |
| Regulation of actin
cytoskeleton | FGFR1,
FGF19, FGFR2, FGF12, EGFR, FGF3, FGF22 | 9.48E-10 |
| Prostate
cancer | FGFR1,
FGFR2, RELA, CHUK, ATF4, EGFR, TCF7 | 2.99E-08 |
| Apoptosis | RELA,
CHUK, NTRK1 | 1.85E-08 |
| Significant
de-regulated pathways and genes associated with sporadic
parathyroid adenoma. |
|---|
|
|---|
| KEGG pathway
(sporadic parathyroid adenoma) | Genesa | P-value |
|---|
| Calcium signaling
pathway | PLCB4,
CAMK2D, PHKB, CAMK2B, CACNA1H | 0 |
| MAPK signaling
pathway | FGF1,
TNF, IL1B, FGF7, TRAF2, MAPK10, FGF17, FGFR4,
CACNA1H | 3.51E-14 |
| Insulin signaling
pathway | PHKB,
PRKAR2A, MAPK10 | 6.41E-09 |
| Apoptosis | TNF,
IL1B, PRKAR2A, IRAK1, TRAF2 | 2.09E-10 |
| Wnt signaling
pathway | PLCB4,
CAMK2D, CAMK2B, MAPK10 | 1.07E-06 |
| Regulation of actin
cytoskeleton | FGF1,
FGF7, FGF17, FGFR4 | 3.51E-05 |
| Toll-like receptor
signaling pathway | TNF,
IL1B, IRAK1, MAPK10 | 2.45E-07 |
| Table IVGenes frequently de-regulated in
HPT-JT syndrome. |
Table IV
Genes frequently de-regulated in
HPT-JT syndrome.
| Unigene | Symbol | Gene name | Fold changea | Molecular
function |
|---|
| Hs.264887 | FGFR1 | Fibroblast growth
factor receptor 1 | 1.81 | Mitogenesis and
differentiation |
| Hs.249200 | FGF19 | Fibroblast growth
factor 19 | 1.74 | Tumor growth and
invasion |
| Hs.533683 | FGFR2 | Fibroblast growth
factor receptor 2 | 1.36 | Mitogenesis and
differentiation |
| Hs.502875 | RELA | v-rel
reticuloendotheliosis viral oncogene homolog A (avian) | 1.35 | Ubiquitous
transcription factor |
| Hs.198998 | CHUK | Conserved
helix-loop-helix ubiquitous kinase | 1.33 | Ubiquination
pathway, thereby activating the transcription factor |
| Hs.515032 | MKNK2 | MAP kinase
interacting serine/threonine kinase 2 | 1.28 | Oncogenic
transformation and malignant cell proliferation |
| Hs.77810 | NFATC4 | Nuclear factor of
activated T-cells, cytoplasmic, calcineurin-dependent 4 | 1.28 | A member of the
nuclear factors of activated T cells DNA-binding transcription
complex |
| Hs.584758 | FGF12 | Fibroblast growth
factor 12 | −1.50 | Regulate cardiac
Na(+) and Ca(2+) channel currents |
| Hs.496487 | ATF4 | Activating
transcription factor 4 | −1.50 | Encodes a
transcription factor |
| Hs.591169 | CACNA1G | Calcium channel,
voltage-dependent, T type, α 1G subunit | −1.63 | Cell motility, cell
division and cell death |
| Hs.488293 | EGFR | Epidermal growth
factor receptor | −1.81 | Cell
proliferation |
| Hs.406293 | NTRK1 | Neurotrophic
tyrosine kinase, receptor, type 1 | −2.00 | Cell
differentiation |
| Hs.573153 | TCF7 | Transcription
factor 7 (T-cell specific, HMG-box) | −2.30 | Lymphocyte
differentiation |
| Hs.37092 | FGF3 | Fibroblast growth
factor 3 | −3.05 | Cell growth,
morphogenesis, tissue repair, tumor growth and invasion |
| Hs.501632 | CACNA1A | Calcium channel,
voltage-dependent, P/Q type, α 1A subunit | −3.21 | Hormone or
eurotransmitter release and gene expression |
| Hs.248087 | FGF22 | Fibroblast growth
factor 22 | −4.19 | Cell growth,
morphogenesis, tissue repair, tumor growth and invasion |
| Genes frequently
de-regulated in sporadic parathyroid adenoma. |
|---|
|
|---|
| Unigene | Symbol | Gene name | Fold changea | Molecular
function |
|---|
| Hs.472101 | PLCB4 | Phospholipase C, β
4 | 2.29 | Intracellular
transduction of many extracellular signals in the retina |
| Hs.144114 | CAMK2D |
Calcium/calmodulin-dependent protein
kinase (CaM kinase) II δ | 2.22 | Alternative
splicing results in multiple transcript variants |
| Hs.483635 | FGF1 | Fibroblast growth
factor 1 | 1.88 | Embryonic
development, cell growth, morphogenesis, tissue repair, tumor
growth and invasion |
| Hs.241570 | TNF | Tumor necrosis
factor | 1.86 | Cell proliferation,
differentiation, apoptosis, lipid metabolism and coagulation |
| Hs.126256 | IL1B | Interleukin 1,
β | 1.56 | Cell proliferation,
differentiation and apoptosis |
| Hs.567268 | FGF7 | Fibroblast growth
factor 7 | 1.41 | Embryonic
development, cell growth, morphogenesis, tissue repair, tumor
growth and invasion |
| Hs.78060 | PHKB | Phosphorylase
kinase, β | 1.29 | The δ subunit
mediates the depen- dence of the enzyme on calcium
concentration |
| Hs.631923 | PRKAR2A | Protein kinase,
cAMP-dependent, regulatory, type II, α | 1.28 | Regulate protein
transport from endosomes to the Golgi apparatus and further to the
endoplasmic reticulum (ER) |
| Hs.522819 | IRAK1 | Interleukin-1
receptor-associated kinase 1 | 1.23 | IL1-induced
upregulation of the transcription factor NF-κB |
| Hs.351887 | CAMK2B |
Calcium/calmodulin-dependent protein
kinase (CaM kinase) II β | −1.34 | Different cellular
localizations and interact differently with calmodulin |
| Hs.522506 | TRAF2 | TNF
receptor-associated factor 2 | −1.45 | Mediator of the
anti-apoptotic signals from TNF receptors |
| Hs.125503 | MAPK10 | Mitogen-activated
protein kinase 10 | −1.56 | Proliferation,
differentiation, trans-cription regulation and development |
| Hs.248192 | FGF17 | Fibroblast growth
factor 17 | −1.78 | Embryonic
development cell growth, morphogenesis, tissue repair, tumor growth
and invasion |
| Hs.165950 | FGFR4 | Fibroblast growth
factor receptor 4 | −2.28 | Mitogenesis and
differentiation |
| Hs.459642 | CACNA1H | Calcium channel,
voltage-dependent, T type, α 1H subunit | −2.39 | Influx of calcium
ions into the cell upon membrane polarization |
Experimental validation of the molecular
signature in HPT-JT syndrome and sporadic parathyroid adenoma using
comparative real-time PCR and RT-PCR
In order to validate the gene expression data
obtained by the DASL-assay, we selected the genes highly
de-regulated in both HPT-JT syndrome and sporadic parathyroid
adenoma shown in Fig. 2C and D,
and performed comparative real-time reverse transcriptase-PCR
analysis and RT-PCR. Levels of transcription of 10 selected genes
up- and downregulated in HPT-JT syndrome are shown in Fig. 3A and B, respectively, and those of
10 genes up- and downregulated in sporadic parathyroid adenoma in
Fig. 3C and D. Comparative
real-time RT-PCR data showed that FGFR1, FGFR2, FGF19, RELA and
CHUK were upregulated in HPT-JT syndrome relative to sporadic
parathyroid adenoma, in good agreement with the microarray data
(Fig. 2C). Similarly NTRK1 and
FGF22 were downregulated in agreement with the microarray data, but
the findings for TCF7, FGF3, CACNA1A were ambiguous, and their
levels of expression were low (Figs.
3B and 4A). In sporadic
parathyroid adenoma, the genes for PLCB4, FGF1, IL1B, FGF7, MAPK10,
FGFR4 did not differ significantly between the two samples, whereas
for CAMK2D, TRAF2, FGF17, CACNA1H the data of real-time PCR and
RT-PCR conflicted with the microarray findings (Figs. 3C and D, 4A). The most highly expressed gene in
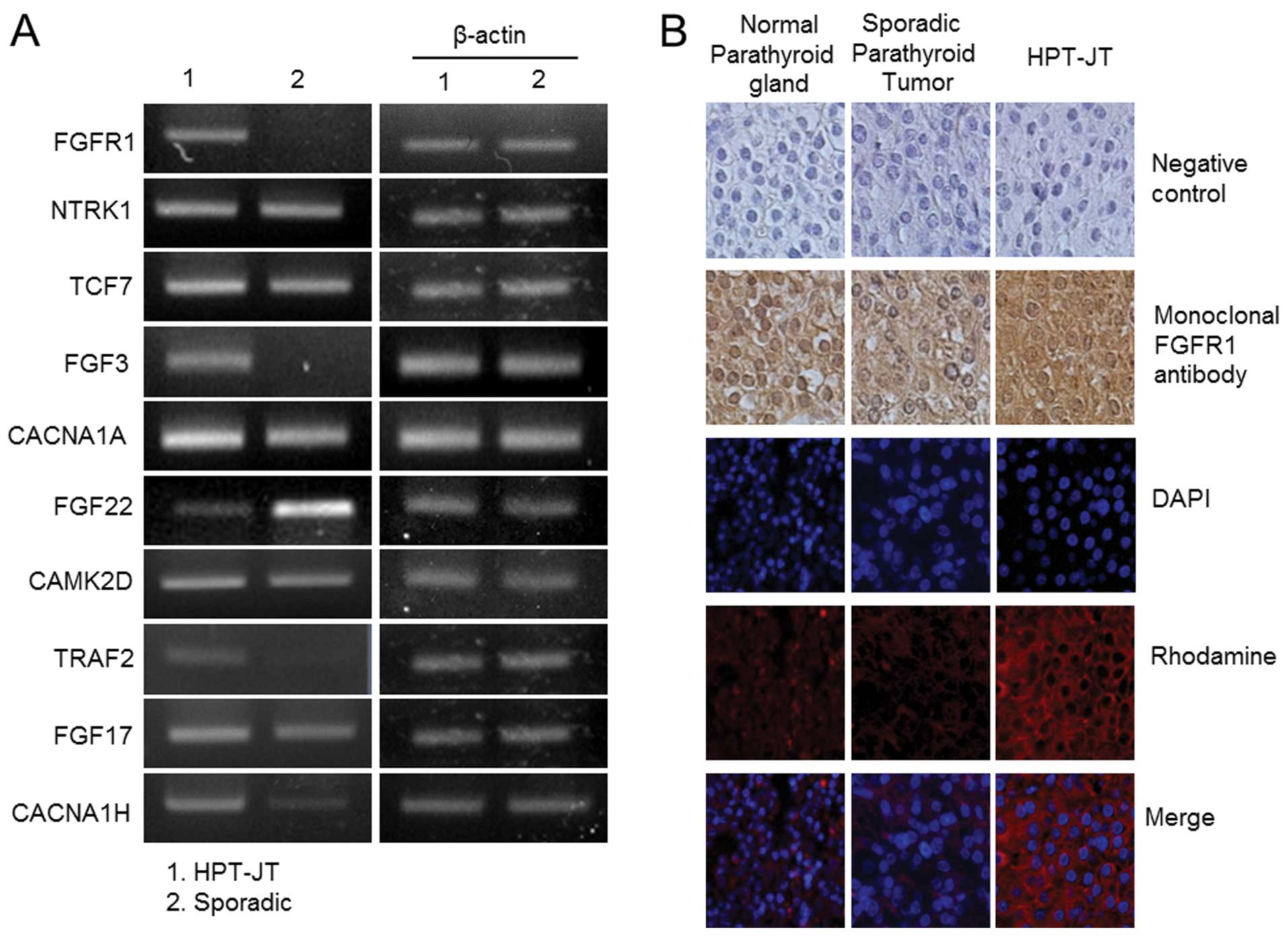
HPT-JT syndrome, FGFR1, was further validated by
immunohistochemistry. Immunohistochemical staining with FGFR1
antibody revealed strong expression in HPT-JT syndrome, but no
detectable expression in sporadic parathyroid adenoma or normal
parathyroid gland (Fig. 4B). The
finding that FGFR1 protein expression was significantly upregulated
in HPT-JT syndrome suggests that FGFR1 does indeed play a role in
parathyroid carcinogenesis.
Discussion
Parathyroid carcinoma is a rare malignant tumor
responsible for less than 1% of cases of hyperparathyroidism. An
increased incidence of this carcinoma has been reported in patients
with HPT-JT syndrome (2). The
incidence of parathyroid carcinoma in HPT-JT syndrome is reported
to be as high as 15% (18,19). Germline mutations of the HRPT2 gene
have been described in parathyroid carcinoma and in HPT-JT
parathyroid carcinoma syndrome (11–14),
but the function of the HRPT2 gene is unknown. Previously,
we were able to amplify the mutated allele generated by a splice
acceptor site mutation of HRPT2 in HPT-JT syndrome (15), and we quantified the aberrantly
spliced HRPT2 mRNA resulting from the splicing abnormality
by real-time RT-PCR (16). Loss of
HRPT2 expression was found to alter the expression of several
target genes that are associated with cell growth and cell death
(20).
In this study, to clarify the molecular mechanisms
involved in the development of parathyroid carcinomas in HPT-JT
syndrome harbouring a splicing HRPT2 mutation, we undertook gene
expression profiling using Illumina DASL matrices. We identified
sixteen genes in the HPT-JT syndrome involved in regulation of MAPK
signaling, regulation of actin cytoskeleton, prostate cancer and
apoptosis pathways, and 15 genes in sporadic parathyroid adenoma
involved in calcium signaling, MAPK signaling, insulin signaling,
apoptosis and Wnt signaling (Fig.
2, Table III). Our data
suggest that increased expression of fibroblast growth factor
receptor type 1 (FGFR1) is highly relevant to parathyroid
carcinogenesis in HPT-JT syndrome harbouring an HRPT2 splicing
mutation. FGF signaling mediated by FGFR involves a classic
receptor tyrosine kinase signaling pathway and its de-regulation at
various points can result in malignancy (21). FGF signaling is involved in
multiple developmental processes including embryonic mesodermal
patterning (22). In the adult, it
contributes to tissue homeostasis, as well as tissue repair,
angiogenesis and inflammation (23,24).
Elevated levels of FGFR1 have been found in a number of cancers,
including prostate, breast and sarcoma (25–28).
One study detected frequent focal amplification of FGFR1 in
non-small cell lung carcinoma cell lines, and these cell lines were
dependent on FGFR1 activity for growth (29,30).
FGFR1 is frequently upregulated in prostate cancer (29,31).
Klotho, which is expressed in the kidney, pituitary and parathyroid
glands, converts FGFR1 (a canonical receptor for various FGFs) into
a specific receptor for FGF-23 (32,33).
The parathyroid cells expressing Klotho and FGFR1 are responsive to
FGF-23, both in vivo and in vitro (33,34).
However, studies on FGFR-Klotho signaling in parathyroid glands
show conflicting results (33).
One current hypothesis suggests that FGFR1 upregulation destroys
the subtle interplay between epithelial and mesenchymal cells
(30), and FGFR1 has also been
suggested to have tumor suppressor properties, since downregulated
expression of FGFR2 in particular has been found in many cancers
(21,35–37).
However, it is not clear whether FGFR2 is a tumor suppressor, since
it is also found to be upregulated in gastric, pancreatic and lung
cancers (21). Further studies are
necessary to clarify the role of FGFR1 in parathyroid glands. Our
results may provide insight into the pathogenesis of parathyroid
neoplasia in the HPT-JT syndrome.
In summary, our gene expression profiling
experiments suggest that upregulation of FGFR1 expression is
associated with parathyroid carcinogenesis in HPT-JT syndrome due
to an HRPT2 splicing mutation. Hence FGF signaling molecules may
provide useful targets for treatment.
Acknowledgements
This study was supported by Basic Science Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Education, Science and Technology
(2011-0008886).
References
|
1
|
Starker LF, Akerstrom T, Long WD, et al:
Frequent germ-line mutations of the MEN1, CASR, and HRPT2/CDC73
genes in young patients with clinically non-familial primary
hyperparathyroidism. Horm Cancer. 3:44–51. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
DeLellis RA: Parathyroid carcinoma: an
overview. Adv Anat Pathol. 12:53–61. 2005. View Article : Google Scholar
|
|
3
|
Hunt JL, Carty SE, Yim JH, Murphy J and
Barnes L: Allelic loss in parathyroid neoplasia can help
characterize malignancy. Am J Surg Pathol. 29:1049–1055.
2005.PubMed/NCBI
|
|
4
|
Haven CJ, Howell VM, Eilers PH, et al:
Gene expression of parathyroid tumors: molecular subclassification
and identification of the potential malignant phenotype. Cancer
Res. 64:7405–7411. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yano S, Sugimoto T, Tsukamoto T, et al:
Decrease in vitamin D receptor and calcium-sensing receptor in
highly proliferative parathyroid adenomas. Eur J Endocrinol.
148:403–411. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arnold A, Shattuck TM, Mallya SM, et al:
Molecular pathogenesis of primary hyperparathyroidism. J Bone Miner
Res. 17(Suppl 2): N30–N36. 2002.PubMed/NCBI
|
|
7
|
Szabo E, Carling T, Hessman O and Rastad
J: Loss of heterozygosity in parathyroid glands of familial
hypercalcemia with hypercalciuria and point mutation in calcium
receptor. J Clin Endocrinol Metab. 87:3961–3965. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Valimaki S, Forsberg L, Farnebo LO and
Larsson C: Distinct target regions for chromosome 1p deletions in
parathyroid adenomas and carcinomas. Int J Oncol. 21:727–735.
2002.PubMed/NCBI
|
|
9
|
Cetani F, Pardi E, Giovannetti A, et al:
Genetic analysis of the MEN1 gene and HPRT2 locus in two Italian
kindreds with familial isolated hyperparathyroidism. Clin
Endocrinol (Oxf). 56:457–464. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tanaka C, Uchino S, Noguchi S, et al:
Biallelic inactivation by somatic mutations of the MEN1 gene in
sporadic parathyroid tumors. Cancer Lett. 175:175–179. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cetani F, Pardi E, Borsari S, et al:
Genetic analyses of the HRPT2 gene in primary hyperparathyroidism:
germline and somatic mutations in familial and sporadic parathyroid
tumors. J Clin Endocrinol Metab. 89:5583–5591. 2004. View Article : Google Scholar
|
|
12
|
Hobbs MR, Pole AR, Pidwirny GN, et al:
Hyperpara-thyroidism- jaw tumor syndrome: the HRPT2 locus is within
a 0.7-cM region on chromosome 1q. Am J Hum Genet. 64:518–525. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Howell VM, Haven CJ, Kahnoski K, et al:
HRPT2 mutations are associated with malignancy in sporadic
parathyroid tumours. J Med Genet. 40:657–663. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shattuck TM, Valimaki S, Obara T, et al:
Somatic and germ-line mutations of the HRPT2 gene in sporadic
parathyroid carcinoma. N Engl J Med. 349:1722–1729. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moon SD, Park JH, Kim EM, et al: A Novel
IVS2-1G>A mutation causes aberrant splicing of the HRPT2 gene in
a family with hyperparathyroidism-jaw tumor syndrome. J Clin
Endocrinol Metab. 90:878–883. 2005.
|
|
16
|
Moon S, Kim JH, Shim JY, et al: Analysis
of aberrantly spliced HRPT2 transcripts and the resulting proteins
in HPT-JT syndrome. Mol Genet Metab. 100:365–371. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
18
|
Carpten JD, Robbins CM, Villablanca A, et
al: HRPT2, encoding parafibromin, is mutated in
hyperparathyroidism-jaw tumor syndrome. Nat Genet. 32:676–680.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hewitt KM, Sharma PK, Samowitz W and Hobbs
M: Aberrant methylation of the HRPT2 gene in parathyroid carcinoma.
Ann Otol Rhinol Laryngol. 116:928–933. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang P, Bowl MR, Bender S, et al:
Parafibromin, a component of the human PAF complex, regulates
growth factors and is required for embryonic development and
survival in adult mice. Mol Cell Biol. 28:2930–2940. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ahmad I, Iwata T and Leung HY: Mechanisms
of FGFR-mediated carcinogenesis. Biochim Biophys Acta.
1823:850–860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
De Moerlooze L, Spencer-Dene B, Revest JM,
Hajihosseini M, Rosewell I and Dickson C: An important role for the
IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in
mesenchymal-epithelial signalling during mouse organogenesis.
Development. 127:483–492. 2000.PubMed/NCBI
|
|
23
|
Turner N and Grose R: Fibroblast growth
factor signalling: from development to cancer. Nat Rev Cancer.
10:116–129. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Haugsten EM, Wiedlocha A, Olsnes S and
Wesche J: Roles of fibroblast growth factor receptors in
carcinogenesis. Mol Cancer Res. 8:1439–1452. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kwabi-Addo B, Ozen M and Ittmann M: The
role of fibroblast growth factors and their receptors in prostate
cancer. Endocr Relat Cancer. 11:709–724. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Freier K, Schwaenen C, Sticht C, et al:
Recurrent FGFR1 amplification and high FGFR1 protein expression in
oral squamous cell carcinoma (OSCC). Oral Oncol. 43:60–66. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Meyer KB, Maia AT, O’Reilly M, et al:
Allele-specific up-regulation of FGFR2 increases susceptibility to
breast cancer. PLoS Biol. 6:e1082008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jacquemier J, Adelaide J, Parc P, et al:
Expression of the FGFR1 gene in human breast-carcinoma cells. Int J
Cancer. 59:373–378. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Murphy T, Darby S, Mathers ME and
Gnanapragasam VJ: Evidence for distinct alterations in the FGF axis
in prostate cancer progression to an aggressive clinical phenotype.
J Pathol. 220:452–460. 2010.PubMed/NCBI
|
|
30
|
Acevedo VD, Gangula RD, Freeman KW, et al:
Inducible FGFR-1 activation leads to irreversible prostate
adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer
Cell. 12:559–571. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Armstrong K, Ahmad I, Kalna G, et al:
Upregulated FGFR1 expression is associated with the transition of
hormone-naive to castrate-resistant prostate cancer. Br J Cancer.
105:1362–1369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Urakawa I, Yamazaki Y, Shimada T, et al:
Klotho converts canonical FGF receptor into a specific receptor for
FGF23. Nature. 444:770–774. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Imanishi Y, Nagata Y and Inaba M:
Parathyroid diseases and animal models. Front Endocrinol
(Lausanne). 3:782012.
|
|
34
|
Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V,
et al: The parathyroid is a target organ for FGF23 in rats. J Clin
Invest. 117:4003–4008. 2007.PubMed/NCBI
|
|
35
|
Amann T, Bataille F, Spruss T, et al:
Reduced expression of fibroblast growth factor receptor 2IIIb in
hepatocellular carcinoma induces a more aggressive growth. Am J
Pathol. 176:1433–1442. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Diez de Medina SG, Chopin D, El Marjou A,
et al: Decreased expression of keratinocyte growth factor receptor
in a subset of human transitional cell bladder carcinomas.
Oncogene. 14:323–330. 1997.PubMed/NCBI
|
|
37
|
Naimi B, Latil A, Fournier G, Mangin P,
Cussenot O and Berthon P: Down-regulation of (IIIb) and (IIIc)
isoforms of fibroblast growth factor receptor 2 (FGFR2) is
associated with malignant progression in human prostate. Prostate.
52:245–252. 2002. View Article : Google Scholar : PubMed/NCBI
|