Introduction
Taxanes, diterpenes originally isolated from the
bark of Taxus brevifolia, represent a relatively new group
of anticancer drugs. Paclitaxel, the first taxane used in
chemotherapy, came into clinical use in the early nineties and has
become one of the main anticancer drugs, especially for the
treatment of ovarian, breast and lung cancer, and Kaposi’s sarcoma.
In rapidly dividing cancer cells, paclitaxel and other taxanes bind
to β-tubulin subunit and stabilize it. In doing so, they destroy
the dynamic instability of microtubules, which is necessary for
their proper function. As a result, cells are arrested in M phase
of the cell cycle and subsequently undergo apoptosis (1). Nevertheless, cancer cell resistance
(either intrinsic or acquired) to these compounds often reverses
the benefits of taxane treatment.
The last decade has seen enormous efforts to
elucidated the mechanisms of resistance to anticancer drugs
(2–7). Nonetheless, elucidation is far from
complete; although some mechanisms of resistance have been
described that could provide useful markers of taxane
resistance.
Drug efflux pumps often top the list of basic
mechanisms of resistance that are common for many anticancer
compounds. Upregulation of the ABC transporter P-glycoprotein (Pgp)
(encoded by the ABCB1 gene) results in enhanced efflux of
anticancer drugs from cells. The mechanism has been well
established in resistance to taxanes (8–11).
Other significant ABC transporters connected with taxane resistance
include multidrug resistance-associated proteins 1 (ABCC1/MRP1)
(12), 2 (ABCC2/MRP2) (13) and 3 (ABCC3/MRP3) (14). Oddly, upregulation of these
proteins has not always been detected in taxane resistant cells
(15–17), which conflicts with
expectations.
Enhanced drug metabolism within the cell is
considered to be another possible explanation for reduced taxane
effectiveness in cancer cells. Two isoforms of cytochrome p450 are
mainly responsible for paclitaxel utilization, i.e. cyp3A4 and
cyp2C8 (18). In some cases,
overexpression of these isoforms have been found in taxane
resistant cancer cells (19,20).
Nevertheless, individual patient genetic variability could account
for the differences (21). Also
phase II metabolic enzyme glutathione S-transferase P1 (GSTP1) has
been reported to be connected with paclitaxel resistance. This
enzyme conjugates glutathione with various drugs causing their
deactivation. Localized inside the nucleus, GSTP1 can also protect
DNA against damage caused by anticancer drugs (22). Some studies have suggested that
expression of GSTP1 could be associated with higher survival rates
of cancer cells after taxane treatment (23–25).
Interestingly, GSTP1 has been shown to co-localize with another
possible marker of taxane resistance, Bcl-2, within the nucleus
(26–28).
Another mechanism of taxane resistance involve
various β-tubulin mutations which can weaken the binding of taxane
to β-tubulin and change the dynamics of the microtubule system
(29–31). Mutations of α-tubulin are also
believed to be connected with taxane resistance, but to a lesser
extent; their importance lies more in influencing the binding of
microtubule-associated proteins (MAPs) (32,33).
Molecules directly connected with cell survival and
apoptosis can also be involved. Expression of proteins, such as
survivin, an inhibitor of apoptosis known to interact with tubulin
(34,35), cyclin-dependent kinase inhibitor
p21 (35,36) or p53, a factor inducing its
expression (37,38), have been reported to influence cell
sensitivity to taxanes.
Even though the mechanisms and compounds mentioned
here is a heterogeneous group, there have been attempts to find
connections between various mechanisms and markers of taxane
resistance. De Hoon et al (22) have suggested that some mechanisms
of resistance in breast cancer cells might be connected to
expression and signaling of human epidermal growth factor receptor
2 (HER2) and subsequent activation of STAT3 as well as other
downstream effectors.
Despite the current knowledge concerning resistance
to taxanes, more studies are required to elucidate this phenomenon
and provide reliable biomarkers to reveal tumors where taxane
treatment (with all its side-effects) would have no impact. Indeed,
investigation of resistance mechanisms (insights into treatment
failure) remains a key task in cancer treatment (7,39).
To study further the mechanisms of resistance to
taxanes, we employed a proteomic approach using the SK-BR-3 cell
line (human breast adenocarcinoma) with acquired resistance to
paclitaxel. We compared protein expression patterns from paclitaxel
resistant cells to patterns from non-resistant cells and searched
for proteins with different expression.
Materials and methods
Materials
Immobiline DryStrips pH 3–11NL 18 cm, Protein
Extraction Buffer-III, 2-D Clean-Up Kit, 2-D Quant Kit, DeStreak
Reagent and IPG Buffer 3–11NL were obtained from GE Healthcare
(Uppsala, Sweden). Rabbit polyclonal antibody against serpin B4
(anti-SERPINB4 antibody defined by manufacturer also as
‘SCCA2/SCCA1 fusion protein antibody’: ab104338), mouse monoclonal
antibody against cytokeratin 18 (anti-cytokeratin 18 antibody:
ab55395), mouse monoclonal antibody against heat shock protein 27
[anti-Hsp27 antibody (8A7): ab78436] and rabbit polyclonal antibody
against GAPDH (anti-GAPDH antibody: ab9485) were obtained from
Abcam (Cambridge, UK). Mouse monoclonal antibody anti-actin (clone
AC-40: A3853) against human actin was obtained from Sigma-Aldrich
(St. Louis, MO, USA).
Cells and cell culture conditions
Human breast carcinoma cell line SK-BR-3 was
obtained from American Type Culture Collection (ATCC, Rockville,
MD, USA). Resistant SK-BR-3 cells were derived in our lab from
original sensitive SK-BR-3 cells by gradual adaptation to
paclitaxel. The concentration of paclitaxel in the culture medium
was increased approximately every 20th passage. Resistant SK-BR-3
cells display long-term proliferation in culture medium containing
100 nM paclitaxel. At this taxane concentration original sensitive
SK-BR-3 cells die within 72 h. This concentration of paclitaxel
approximates concentrations used in clinical practice (26,40).
The culture medium consisted of basic medium
supplemented with 10% heat-inactivated fetal bovine serum. The
basic medium was RPMI-1640 based medium containing extra
L-glutamine (300 μg/ml), sodium pyruvate (110 μg/ml), HEPES (15
mM), penicillin (100 U/ml) and streptomycin (100 μg/ml), as
previously described (41–43). Cells were maintained at 37°C in a
humidified atmosphere of 5% CO2 in air. For maintaining
taxane-resistant cells, the medium was supplemented with paclitaxel
(in DMSO) to a final concentration of 100 nM (final concentration
of DMSO in medium was below 0.1%) just prior to use. For
experiments, cells were harvested and seeded (approximately
5×106 in 15 ml of medium per sample). After 24-h
preincubation the culture medium was replaced with a fresh medium
and after another 24 h the cells were harvested.
Sample preparation for
2D-electrophoresis
Cells were trypsinized, washed with ice-cold PBS and
resuspended in Protein Extraction Buffer-III (GE Healthcare) (urea,
thiourea, ASB-16, CHAPS) containing 1% Protease-Inhibitor Mix G
(SERVA Electrophoresis GmbH, Heidelberg, Germany). A 2-D Clean-Up
Kit (GE Healthcare) was used for sample purification, per
manufacturer’s instructions. Briefly, the sample was precipitated
using Precipitant and Co-precipitant, incubated for 15 min at 4°C
and centrifuged. The pellet was then washed with Wash Buffer with
Wash Additive, incubated at −20°C for 30 min and centrifuged. After
brief air drying, the pellet was dissolved again in Protein
Extraction Buffer-III as described above. Protein concentrations
were determined using a 2-D Quant Kit (GE Healthcare), which is
compatible with both the detergents and thiourea present in Protein
Extraction Buffer-III.
2D-electrophoresis: isoelectric
focusing
Isoelectric focusing was carried out in an IPGphor
focusing unit (GE Healthcare, former Amersham Biosciences) using
3–11NL Immobiline DryStrips 18 cm (GE Healthcare). Each strip was
rehydrated for at least 24 h with 340 μl of the diluted protein
sample containing 400 μg of protein, 6.8 μl of bromophenol blue
solution (0.1%), 4 μl of DeStreak Reagent (GE Healthcare) and 6.8
μl of IPG buffer (pH 3.0–11.0; GE Healthcare) in Protein Extraction
Buffer-III as described above. After rehydration, strips were
focused at 20°C with current limited to 50 μA/strip using the
following conditions: 150 V for 1 h, gradient 150–300 V for 10 min,
300 V for 2 h, gradient 300–1,000 V for 10 min, 1,000 V for 2 h,
gradient 1,000–8,000 V for 1 h, 8,000 V for 15 h.
2D-electrophoresis: equilibration
After focusing, strips were equilibrated for 20 min
in equilibration buffer containing 6 M urea, 30% glycerol, 2% SDS,
50 mM Tris (stock solution of 1.5 M Tris-HCl, pH 8.8), 2%
dithiothreitol and then equilibrated for another 20 min in the same
buffer with 2.5% iodoacetamide instead of dithiothreitol. Then the
strips were placed on the top of gels and sealed using 0.5% agarose
solution containing bromophenol blue.
2D-electrophoresis: SDS-PAGE
The second dimension was performed using an Ettan
DaltSix Electrophoresis System (GE Healthcare, former Amersham
Biosciences). A total of 10% polyacrylamide gel (pH 8.8) with a 4%
stacking gel (pH 8.8) was used for protein separation. Gels were
run at a constant current starting with 35 mA/gel for 1 h and then
65 mA/gel till the blue line reached the bottom of the gels
(approximately 4 h). After running the second dimension, each gel
was washed 3×10 min in distilled water and stained overnight in 500
ml of colloidal Coomassie Blue solution per gel according to
(44).
Gel image and analysis
Stained gels were scanned using a calibrated UMAX
PowerLook 1120 scanner employing LabScan software (both from GE
Healthcare). Gel couples were analyzed using Image
MasterTM 2D Platinum 6.0 software (GE Healthcare, former
Amersham Biosciences). We analyzed differences in intensity between
corresponding spots on each set of gels (each set contained gel
with proteins from sensitive SK-BR-3 cells and gel with proteins
from resistant SK-BR-3). Spots with at least 2-fold average
difference in expression between sensitive and resistant cell
lysates were selected. Statistical significance of difference in
intensity of these spots was determined using the Student’s
t-test.
Enzymatic digestions
CBB-stained protein spots were excised from the gel,
cut into small pieces and destained using 50 mM 4-ethylmorpholine
acetate (pH 8.1) in 50% acetonitrile (MeCN). After complete
destaining, gels were washed with water, reduced in size by
dehydration in MeCN and reconstituted again in water. The
supernatant was removed and the gel was partly dried in a SpeedVac
concentrator. Gel pieces were then incubated overnight at 37°C in a
cleavage buffer containing 25 mM 4-ethylmorpholine acetate, 5% MeCN
and trypsin (100 ng; Promega). The resulting peptides were
extracted with 40% MeCN/0.1% trifluoroacetic acid (TFA).
MALDI mass spectrometry and protein
identification
An aqueous 50% MeCN/0.1% TFA solution of
α-cyano-4-hydroxycinnamic acid (5 mg/ml; Sigma-Aldrich, St. Louis,
MO, USA) was used as a MALDI matrix. Peptide mixture (1 μl) was
deposited on a MALDI plate, allowed to air-dry at room temperature
and overlaid with 0.4 μl of matrix. Mass spectra were measured
using an Ultraflex III MALDI-TOF (Bruker Daltonics, Bremen,
Germany); mass range of 700–4,000 Da, calibrated internally using
the monoisotopic [M+H]+ ions of trypsin auto-proteolytic
fragments (842.5 and 2,211.1 Da). The peak lists created using the
flexAnalysis 3.3 program were searched using an in-house MASCOT
search engine against SwissProt 2013_12 database subset of human
proteins with the following search settings: peptide tolerance of
30 ppm, missed cleavage site value set to one, variable
carbamidomethylation of cysteine, oxidation of methionine and
protein N-term acetylation. Proteins with MOWSE scores over the
threshold, 56 (calculated for the used settings) were considered as
identified. The identity of protein candidate was confirmed using
MS/MS analysis.
Western blot analysis
Western blot was carried as described previously
(41,43,45)
with minor modification. Briefly, whole cell lysates were separated
by SDS-PAGE using 9% acrylamide gels and blotted into 0.2 μm
nitrocellulose membrane for 2 h at 0.25 A using a Mini-Protean II
blotting apparatus (Bio-Rad). The membrane was blocked with 5% low
fat milk in TBS for 30 min. Tween-20 (0.1%) in TBS was used for
washing. The washed membrane was then incubated with the relevant
primary antibody. After incubation (overnight, room temperature),
the washed membrane was incubated for 1 h with the corresponding
horseradish peroxidase-conjugated secondary antibody (Santa Cruz
Biotechnology, Santa Cruz, CA, USA). The horseradish
peroxidase-conjugated secondary antibody was detected by enhanced
chemiluminescence using the SuperSignal reagent from Pierce
(Rockford, IL, USA) and a Gel Logic 4000 PRO device. Optical
density of bands was analyzed using Carestream Molecular Imaging
Software v. 5.2.0 (Carestream Health, Berlin, Germany).
Statistical analysis
Statistics was performed using Sigma Plot, version
11.00. Significant differences in intensity of spots (2-D gels) and
bends (western blot) were analyzed by Student’s t-test.
Results
Detection of proteins with changed
expression
In order to compare protein expression in the human
breast adenocarcinoma cell line (SK-BR-3) sensitive to paclitaxel
and in SK-BR-3 cells with acquired resistance to paclitaxel, we
prepared whole cell lysates of these cells. Lysates were prepared
using Protein Extraction Buffer. Samples were purified using
TCA-based precipitation and 400 μg of proteins of each sample were
used for two-dimensional electrophoresis (2-DE) (see Materials and
methods).
We prepared three pairs of 2-DE gels from three
independent sets of samples containing SK-BR-3 cells resistant to
paclitaxel and SK-BR-3 cells sensitive to paclitaxel. Gels were
stained using colloidal coomassie blue. The stained gels were
scanned and differences in intensity of corresponding spots in each
pair of gels were analyzed using ImageMaster 2D Platinum 6.0
software (see Materials and methods).
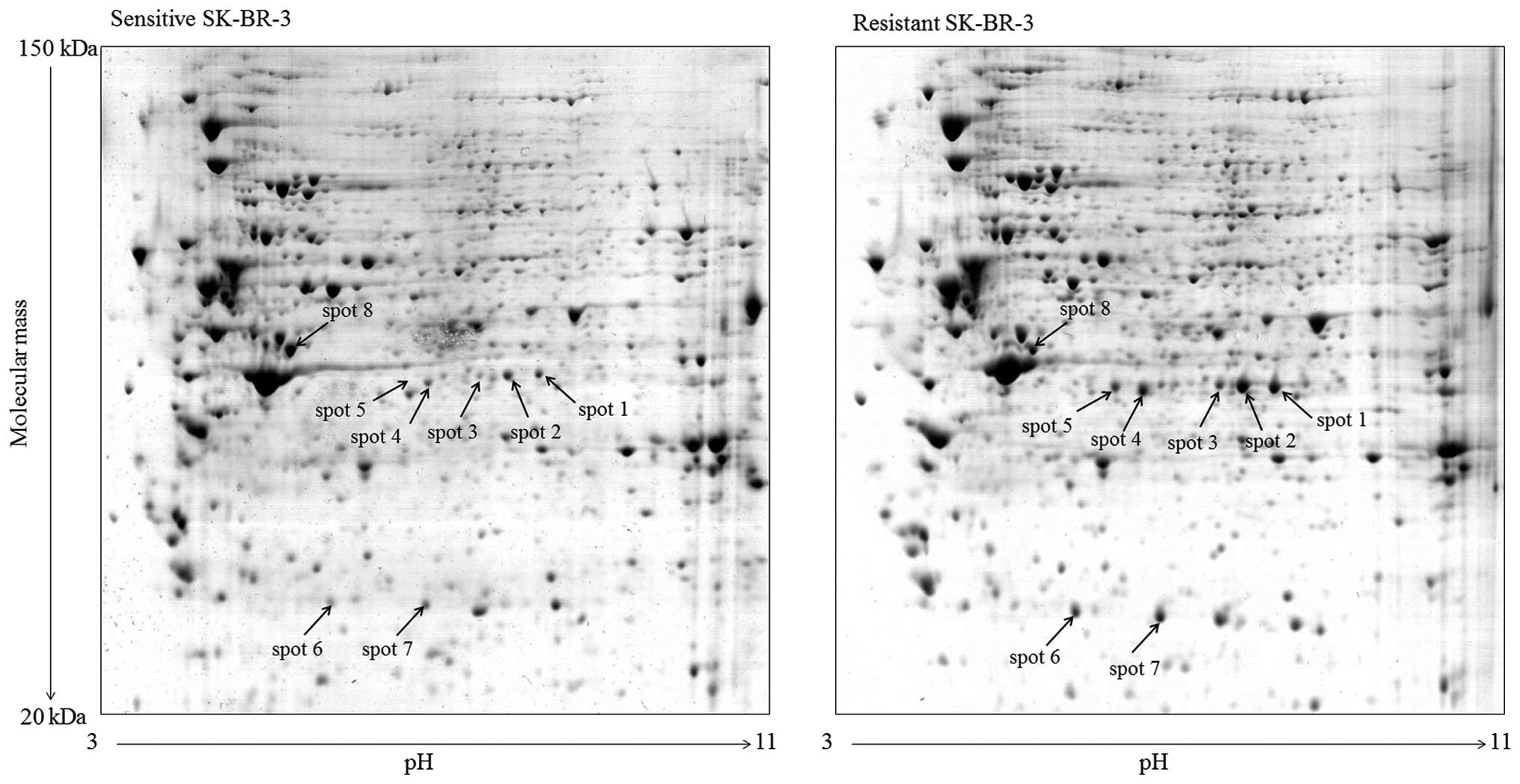
Approximately 700 stable spots were detected on each
2-DE gel. The expression profiles of proteins with isoelectric
point within pH range 3.0–11.0 and molecular mass of 20–150 kD were
highly reproducible for both sensitive and resistant cells as well
as for individual sets of samples. Differences in intensity of
corresponding spots were analyzed for each pair of gels and spots
with 2-fold higher or lower intensity, when comparing sensitive and
resistant cells, and considered as proteins with changed
expression. Eight spots with significantly changed expression were
detected (Figs. 1 and 2). These spots were cut from the gels and
subjected to MS analysis (see Materials and methods).
Identification of proteins with changed
expression
The identified proteins (Table I) with changed expression in
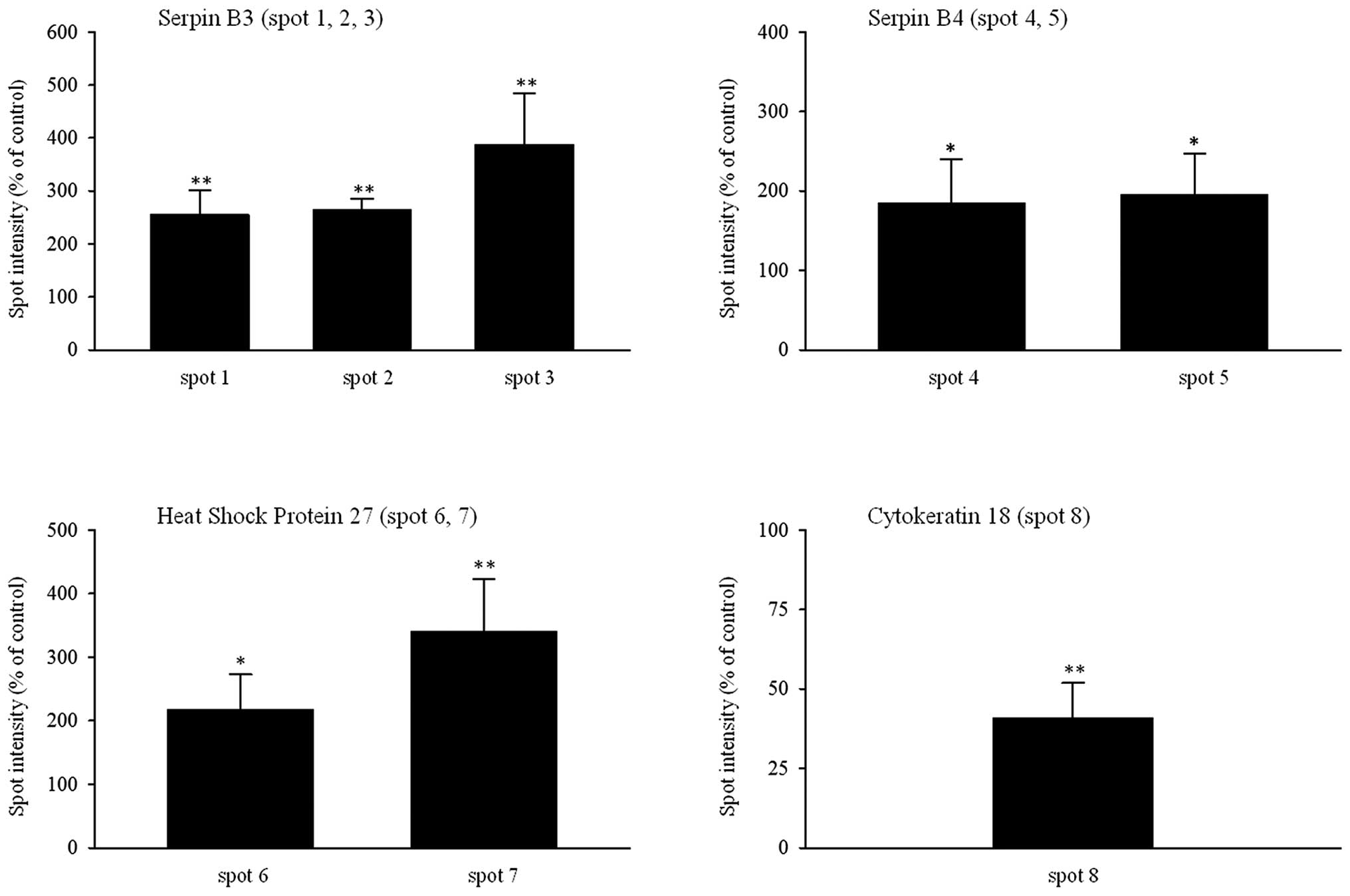
resistant SK-BR-3 cells were: serpin B3 (spots 1, 2, 3 with
intensity increased to 235, 264 and 387%, respectively), serpin B4
(spots 4 and 5 with intensity increased to 190 and 195%,
respectively), heat shock protein 27 (HSP27) (spots 6 and 7 with
intensity increased to 218 and 340%, respectively) and cytokeratin
18 (spot 8 with intensity decreased to 41%) (Fig. 2).
 | Table IResults of MS analysis. |
Table I
Results of MS analysis.
| No. | Protein name | SwissProt no. | M | UM | SC (%) | MS (score) | MW (kD) | pI-value |
|---|
| 1 | Serpin B3 | SPB3_HUMAN | 19 | 7 | 47 | FMFDLFQQFR
(55) | 45 | 6.4 |
| 2 | Serpin B3 | SPB3_HUMAN | 16 | 6 | 47 | FHCNHPFLFFIR
(41) | 45 | 6.4 |
| 3 | Serpin B3 | SPB3_HUMAN | 9 | 5 | 25 | SVDFANAPEESR
(63) | 45 | 6.4 |
| 4 | Serpin B4 | SPB4_HUMAN | 11 | 4 | 31 | STDFANAPEESR
(66)
TYQFLQEYLDAIKK (59) | 45 | 5.9 |
| 5 | Serpin B4 | SPB4_HUMAN | 8 | 3 | 16 | STDFANAPEESR
(55)
TYQFLQEYLDAIKK (57) | 45 | 5.9 |
| 6 | Heat shock protein
beta-1 | HSPB1_HUMAN | 8 | 4 | 41 | LFDQAFGLPR
(56) | 23 | 6.0 |
| 7 | Heat shock protein
beta-1 | HSPB1_HUMAN | 8 | 3 | 41 | LFDQAFGLPR
(48)
SNEITIPVTFESR (49) | 23 | 6.0 |
| 8 | Keratin, type I
cytoskeletal 18 | K1C18_HUMAN | 19 | 4 | 44 | TVQSLEIDLDSMR
(51)
SLGSVQAPSYGAR (43) | 48 | 5.3 |
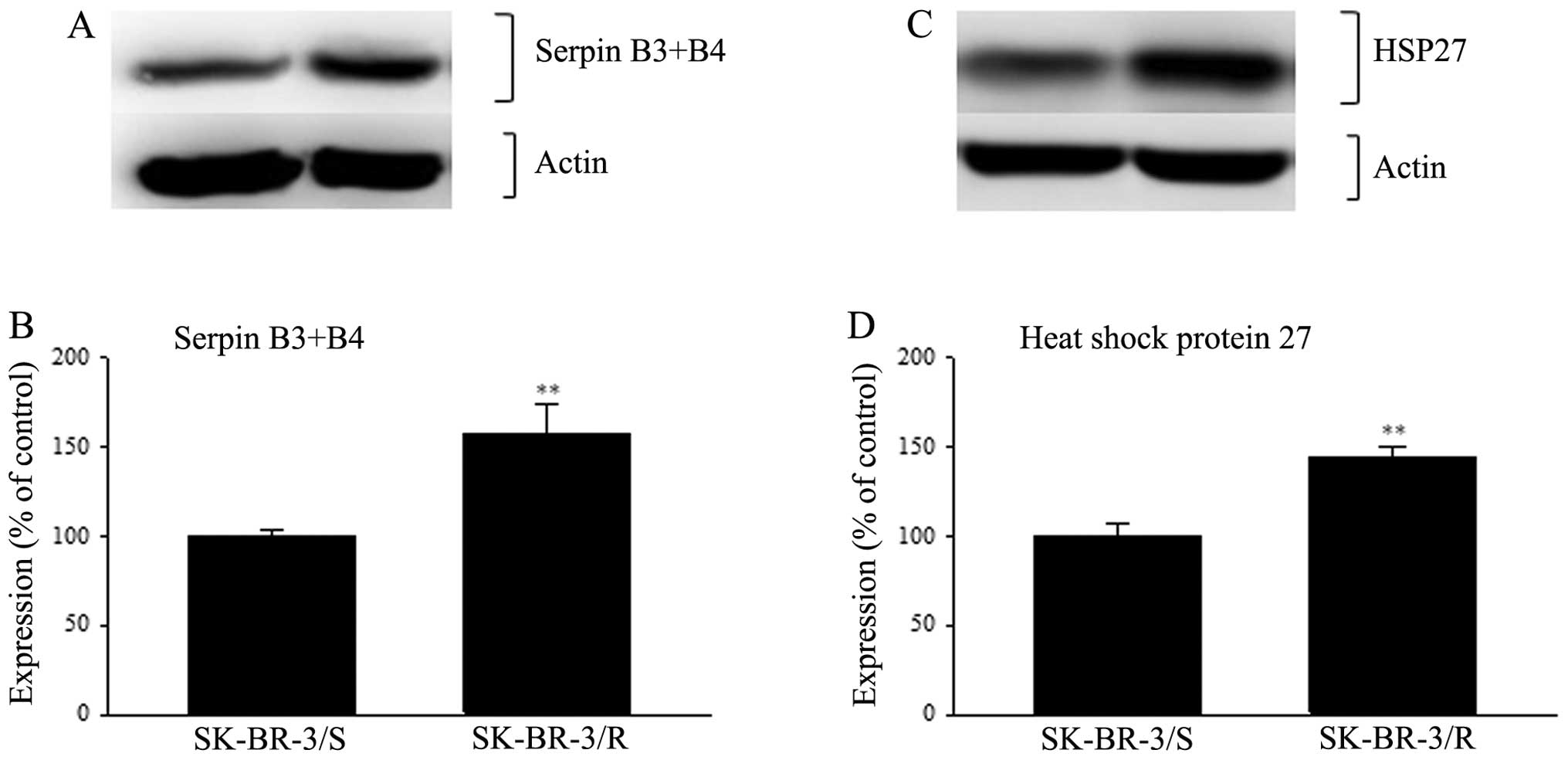
Western blot analysis of the new sets of samples
confirmed the detected changes in expression of all these proteins.
Serpin B3 and serpin B4 were detected as one protein. These two
proteins have a high homology (92%) and thus cross reactivity of
primary antibodies against them is highly probable if not
straightly admitted by manufacturer. The expression of serpin B3+B4
in SK-BR-3 cells resistant to paclitaxel detected using western
blotting was elevated up to 158% when compared to cells sensitive
to paclitaxel (p<0.01) (Fig.
3). The expression of HSP27 in resistant cells was increased to
144% in resistant cells compared to sensitive cells (p<0.01)
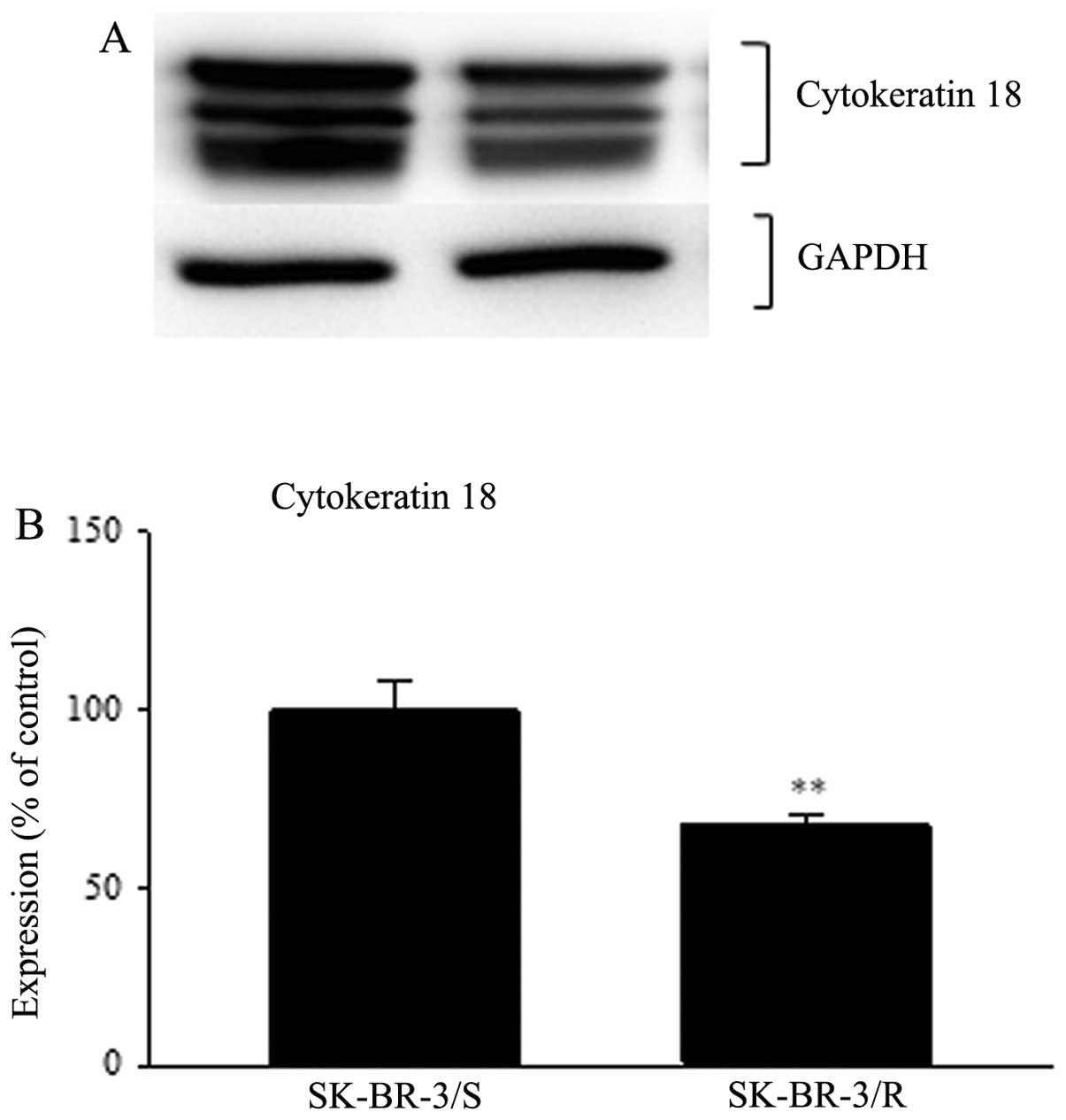
(Fig. 3). Western blot analysis of
cytokeratin 18 showed three bands very close together. We searched
for western blots of whole SK-BR-3 lysates using cytokeratin 18
antibody and found that more bands or one very wide band for
cytokeratin 18 has been previously published (http://www.scbt.com/datasheet-32722-cytokeratin-18-rck106-antibody.html;
accessed June 7, 2013). Thus, for densitometry analysis we took
these three bands as one. We detected decreased expression of
cytokeratin 18 down to 68% in resistant cells compared to sensitive
cells (p<0.01) (Fig. 4).
For serpin B3+B4 and HSP27, actin was used as a
loading control. For cytokeratin 18 GAPDH was used. The reason for
the different loading controls was that cytokeratin 18 has
approximately the same size as actin and both these proteins are
very strongly expressed. Therefore stripping off either cytokeratin
18 or actin failed to yield satisfactory results.
Discussion
Using western blot analysis, we found a significant
increase (158%) in the expression of serpin B3/B4 in human SK-BR-3
breast cancer cells resistant to paclitaxel compared to sensitive
SK-BR-3 cells (Fig. 3). The
increase is lower compared to 2-DE gels analysis, but still
significant (analyzed by Student’s t-test, p<0.01) (Fig. 2).
Serpin B3 and B4 share the same molecular weight
(92% identical sequence of amino acids) but have different pI
(serpin B3 represents a neutral form with pI>6.2, serpin B4
represents an acidic form with pI<6.2) (46). This corresponded with position of
their spots on our 2-DE gels (Fig.
1). Our gels revealed three spots for serpin B3 and two spots
for serpin B4. A higher number of serpin B3 protein species on 2-DE
gels was described previously by Ho et al (47). Spots of the same protein with
different pI, which we found in our study, could be the result of
various levels of phosphorylation of serpins. Because of their
almost identical sequence of amino acids, serpin B3 and serpin B4
are often analyzed together as serpin B3+B4.
Serpins (serine protease inhibitors) represent a
large group of proteins, most of them capable of inhibiting
proteases (serine proteases and in some cases also cysteine
proteases) (48). Serpin B3 and B4
are clade B serpins or ovoalbumin serpins (ov-serpins). Serpin B3
inhibits papain-like cysteine proteases (e.g., cathepsin-S, -K and
-L), while serpin B4 inhibits both serine proteases (e.g.,
cathepsin G) and cysteine proteases (e.g., Der p 1 and Der f 1)
(49). Both these serpins are
often overexpressed in cancer cells of epithelial origin (46,50).
Upregulation of serpin B3 and B4 has previously been
described as protective in cells exposed to radiation (51). Suppression of these proteins has
been shown to suppress tumor growth (52). Recently, serpin B3 was suggested as
a prognostic tool for docetaxel resistance in breast cancer
(53) and platinum resistance in
epithelial ovarian cancer (54)
and lung cancer (55).
The mechanism of pro-survival effect of serpin B3
and B4 on cancer cells is likely to be connected with their ability
to inhibit proteases. Cathepsin S, a known target for serpin B3, is
essential for proper presentation of antigens to immune cells.
Inhibition of its activity could lead to impaired recognition of
cancer cells by the immune system. Another cathepsin, cathepsin L,
promotes production of endostatin which has an anti-angiogenic
effect (56). Therefore,
inhibition of cathepsin L by serpin B3 could support angiogenesis.
Cathepsin G has been described as an agent opposing tumor cell-cell
adhesion (57). Thus, its
inhibition would benefit cancer cells. It is worth noting that some
of these cathepsins have been reported to have pro-cancer effects,
as well (58,59).
More complex explanations of upregulation of serpin
B3 and B4 in resistant cancer cells have also been suggested.
Serpin B3 is believed to be able to inhibit the lysosomal cell
death pathway (LCDP) induced by microtubule-stabilizing agents,
including paclitaxel (60),
through inhibition of LCDP mediators, e.g. cathepsins.
Interestingly, serpin B4 has been described to inhibit also
granzyme M-induced cell death, which is the main pathway used by
cytotoxic lymphocytes to kill tumor cells (61).
Upstream regulation of serpin B3 and B4 expression
has been suggested. Serpins B3+B4 were described as being activated
through STAT3 activation (62). De
Hoon et al have suggested signaling pathways connected with
resistance to paclitaxel (22).
They proceed from human epidermal growth factor receptor 2 (HER2)
through PI3K/Akt to i) activation of STAT3 and ii) upregulation of
P-glycoprotein and survivin, resulting in an overall increase in
cancer cell survival (22). Based
on these two studies, we suspect that increased expression of
serpins B3+B4 is connected to paclitaxel resistance via the
mechanism suggested by De Hoon et al (22).
In this study we confirmed that increased expression
of serpin B3 and B4 can potentially affect resistance to paclitaxel
and we hypothesize that the mechanism of serpin B3 and B4
upregulation involves activation of STAT3.
Another protein found to have significantly higher
expression (144%) in resistant SK-BR-3 cells compared to sensitive
cells was heat shock protein 27 (HSP27). Again, the increase in
HSP27 expression detected using western blot analysis was lower
than the increase found using 2D gels analysis (Figs. 2 and 3), however, it was nonetheless
statistically significant (Student’s t-test, p<0.01).
Heat shock protein 27 is an ATP-independent chaperon
and functions as a protective agent against protein aggregation
(63). It is highly expressed even
in normal cells under stress conditions like heat, oxidative stress
or exposure to chemotherapeutic agents (64). In some studies, HSP27 was thought
to protect cancer cells against cell death. Its overexpression was
reported to inhibit activation of procaspase-3 and procaspase-9
(65). It antagonizes Bax-mediated
mitochondrial changes (66) and it
is generally thought to block programmed cell death by directly
sequestering intermediates in the caspase-dependent apoptosis
pathway (67).
Thus, we hypothesize that HSP27 can positively
affect cell survival through inhibition of the inner
(mitochondrial) apoptotic pathway. Development of the ability to
prevent activation of this pathway could be crucial for survival of
SK-BR-3 cells when exposed to paclitaxel and thus could be part of
the resistance mechanism to taxanes in certain types of cancer
cells.
Cytokeratin 18 was the only protein with reduced
expression in resistant SK-BR-3 cells compared to sensitive cells.
The cytokeratin 18 molecule (together with cytokeratin 8) is a
component of the most common intermediate filaments in cells of
epithelial origin. Besides the many functions connected with its
role in intracellular scaffolding (e.g., structuring cytoplasm,
providing resistance against external stresses) and cellular
processes like mitosis or apoptosis, it also has a role in tumor
cell behavior (68). Additionally,
some studies have suggested the involvement of cytokeratin 18 in
certain signaling pathways (68,69).
Cytokeratin 18 was reported to be involved in
pro-survival PI3K/Akt pathway (69), which can counteract Fas-mediated
apoptosis (70). Fortier et
al reported overexpression of cytokeratin 18 (and cytokeratin
8) as a result of overexpression of Akt1 (71). This would suggest that higher
expression of cytokeratin 18 could be a marker of reduced
susceptibility to cell death (i.e., resistant cells); however, our
findings do not support this conclusion.
The tumor necrosis factor receptor 2 (TNFR2) is
another signaling pathway with which cytokeratin 18 can interact.
Binding of cytokeratin 18 to the cytoplasmic domain of tumor
necrosis factor receptor 2 (TNRF2) leads to changes in JNK
signaling and NF-κB activation. Cytokeratin 18 is also able to
negatively influence TNF-induced apoptosis (72). However, our results showed a
different pattern, which could mean that TNF-induced cell death
does not play as important role in paclitaxel-induced cell death in
SK-BR-3 cells.
Relative to our results, Meng et al (73) found a lower expression of
cytokeratin 18 promoting proliferation of breast cancer cells
MCF-7; however, they connected this effect to the modulation of
estrogen receptor-α pathway. Nevertheless, SK-BR-3 cells do not
express estrogen receptor-α. A trend similar to that in our study
was found in some primary breast carcinoma screenings where lower
expression of cytokeratin 18 as associated with a worse prognosis
(74,75).
The function and significance of cytokeratin 18 in
cancer cells needs to be elucidated more thoroughly; more precisely
we need to determine why upregulation of cytokeratin 18 indicates
reduced susceptibility to treatment in some cases, but not in
others.
Our study found four proteins (serpin B3, serpin B4,
heat shock protein 27, cytokeratin 18) with changed expression in
breast cancer cells (the type not expressing estrogen receptor but
overexpressing HER2) resistant to paclitaxel. These proteins
represent a heterogeneous group and do not seem to be directly
connected with each other (with serpin B3 and B4 as an exception).
This suggests that processes involved in cancer cells surviving
normally lethal doses of paclitaxel are both numerous and complex.
Nevertheless, even if revealed proteins interact with different
pathways of cell signaling, they still represent a group of
potential biomarkers that should be probably seen as a kind of
whole and tested together rather than considered and treated as
four individual biomarkers.
Acknowledgements
This study was supported by grant NT 13679-4 of the
Internal Grant Agency, Ministry of Health of the Czech Republic,
and by the Institutional Research Project of the Institute of
Microbiology (RVO61388971). We thank Thomas Secrest for the English
language revision of the manuscript.
References
|
1
|
Jordan MA: Mechanism of action of
antitumor drugs that interact with microtubules and tubulin. Curr
Med Chem Anticancer Agents. 2:1–17. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Villeneuve DJ, Hembruff SL, Veitch Z,
Cecchetto M, Dew WA and Parissenti AM: cDNA microarray analysis of
isogenic paclitaxel- and doxorubicin-resistant breast tumor cell
lines reveals distinct drug-specific genetic signatures of
resistance. Breast Cancer Res Treat. 96:17–39. 2006. View Article : Google Scholar
|
|
3
|
Galletti E, Magnani M, Renzulli ML and
Botta M: Paclitaxel and docetaxel resistance: Molecular mechanisms
and development of new generation taxanes. Chem Med Chem.
2:920–942. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stordal B and Davey R: A systematic review
of genes involved in the inverse resistance relationship between
cisplatin and paclitaxel chemotherapy: Role of BRCA1. Curr Cancer
Drug Targets. 9:354–365. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Smoter M, Bodnar L, Duchnowska R, Stec R,
Grala B and Szczylik C: The role of Tau protein in resistance to
paclitaxel. Cancer Chemother Pharmacol. 68:553–557. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vergara D, Tinelli A, Iannone A and Maffia
M: The impact of proteomics in the understanding of the molecular
basis of paclitaxel-resistance in ovarian tumors. Curr Cancer Drug
Targets. 12:987–997. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rebucci M and Michiels C: Molecular
aspects of cancer cell resistance to chemotherapy. Biochem
Pharmacol. 85:1219–1226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mechetner E, Kyshtoobayeva A, Zonis S, et
al: Levels of multidrug resistance (MDR1) P-glycoprotein expression
by human breast cancer correlate with in vitro resistance to taxol
and doxorubicin. Clin Cancer Res. 4:389–398. 1998.PubMed/NCBI
|
|
9
|
Pires MM, Emmert D, Hrycyna CA and
Chmielewski J: Inhibition of P-glycoprotein-mediated paclitaxel
resistance by reversibly linked quinine homodimers. Mol Pharmacol.
75:92–100. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thollet A, Vendrell JA, Payen L, et al:
ZNF217 confers resistance to the pro-apoptotic signals of
paclitaxel and aberrant expression of Aurora-A in breast cancer
cells. Mol Cancer. 9:2912010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y, Chen QF, Jin S, et al:
Up-regulation of P-glycoprotein is involved in the increased
paclitaxel resistance in human esophageal cancer radioresistant
cells. Scand J Gastroenterol. 47:802–808. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kwak JO, Lee SH, Lee GS, et al: Selective
inhibition of MDR1 (ABCB1) by HM30181 increases oral
bioavailability and therapeutic efficacy of paclitaxel. Eur J
Pharmacol. 627:92–98. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huisman MT, Chhatta AA, van Tellingen O,
Beijnen JH and Schinkel AH: MRP2 (ABCC2) transports taxanes and
confers paclitaxel resistance and both processes are stimulated by
probenecid. Int J Cancer. 116:824–829. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O’Brien C, Cavet G, Pandita A, et al:
Functional genomics identifies ABCC3 as a mediator of taxane
resistance in HER2-amplified breast cancer. Cancer Res.
68:5380–5389. 2008.PubMed/NCBI
|
|
15
|
Violini S, D’Ascenzo S, Bagnoli M, et al:
Induction of a multifactorial resistance phenotype by high
paclitaxel selective pressure in a human ovarian carcinoma cell
line. J Exp Clin Cancer Res. 23:83–91. 2004.PubMed/NCBI
|
|
16
|
Takano M, Otani Y, Tanda M, Kawami M,
Nagai J and Yumoto R: Paclitaxel-resistance conferred by altered
expression of efflux and influx transporters for paclitaxel in the
human hepatoma cell line, HepG2. Drug Metab Pharmacokinet.
24:418–427. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kars MD, Iseri OD and Gunduz U: A
microarray based expression profiling of paclitaxel and vincristine
resistant MCF-7 cells. Eur J Pharmacol. 657:4–9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Taniguchi R, Kumai T, Matsumoto N, et al:
Utilization of human liver microsomes to explain individual
differences in paclitaxel metabolism by CYP2C8 and CYP3A4. J
Pharmacol Sci. 97:83–90. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Garcia-Martin E, Pizarro RM, Martinez C,
et al: Acquired resistance to the anticancer drug paclitaxel is
associated with induction of cytochrome P4502C8. Pharmacogenomics.
7:575–585. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hendrikx J, Lagas JS, Rosing H, Schellens
JHM, Beijnen JH and Schinkel AH: P-glycoprotein and cytochrome P450
3A act together in restricting the oral bioavailability of
paclitaxel. Int J Cancer. 132:2439–2447. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Henningsson A, Sparreboom A, Sandstrom M,
et al: Population pharmacokinetic modelling of unbound and total
plasma concentrations of paclitaxel in cancer patients. Eur J
Cancer. 39:1105–1114. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
De Hoon JPJ, Veeck J, Vriens B, Calon TGA,
van Engeland M and Tjan-Heijnen VCG: Taxane resistance in breast
cancer: A closed HER2 circuit? Biochim Biophys Acta Rev Cancer.
1825:197–206. 2012.PubMed/NCBI
|
|
23
|
Arai T, Miyoshi Y, Kim SJ, et al:
Association of GSTP1 expression with resistance to docetaxel and
paclitaxel in human breast cancers. Eur J Surg Oncol. 34:734–738.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jardim BV, Moschetta MG, Gelaleti GB, et
al: Glutathione transferase pi (GSTpi) expression in breast cancer:
An immunohistochemical and molecular study. Acta Histochem.
114:510–517. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miyake T, Nakayama T, Naoi Y, et al: GSTP1
expression predicts poor pathological complete response to
neoadjuvant chemotherapy in ER-negative breast cancer. Cancer Sci.
103:913–920. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ferlini C, Raspaglio G, Mozzetti S, et al:
Bcl-2 down-regulation is a novel mechanism of paclitaxel
resistance. Mol Pharmacol. 64:51–58. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pan Z and Gollahon L: Paclitaxel
attenuates Bcl-2 resistance to apoptosis in breast cancer cells
through an endoplasmic reticulum-mediated calcium release in a
dosage dependent manner. Biochem Biophys Res Commun. 432:431–437.
2013. View Article : Google Scholar
|
|
28
|
Ali-Osman F, Brunner JM, Kutluk TM and
Hess K: Prognostic significance of glutathione S-transferase pi
expression and subcellular localization in human gliomas. Clin
Cancer Res. 3:2253–2261. 1997.PubMed/NCBI
|
|
29
|
Cheung CHA, Wu SY, Lee TR, et al: Cancer
cells acquire mitotic drug resistance properties through beta
I-tubulin mutations and alterations in the expression of
beta-tubulin isotypes. PLoS One. 5:e125642010. View Article : Google Scholar
|
|
30
|
Wang YH, Sparano JA, Fineberg S, et al:
High expression of class III beta-tubulin predicts good response to
neoadjuvant taxane and doxorubicin/cyclophosphamide-based
chemotherapy in estrogen receptor-negative breast cancer. Clin
Breast Cancer. 13:103–108. 2013. View Article : Google Scholar
|
|
31
|
Yin SH, Zeng CQ, Hari M and Cabral F:
Random mutagenesis of beta-tubulin defines a set of dispersed
mutations that confer paclitaxel resistance. Pharm Res.
29:2994–3006. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Edelman MJ and Shvartsbeyn M: Epothilones
in development for non-small-cell lung cancer: Novel anti-tubulin
agents with the potential to overcome taxane resistance. Clin Lung
Cancer. 13:171–180. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Diaz JF, Barasoain I and Andreu JM: Fast
kinetics of taxol binding to microtubules. Effects of solution
variables and microtubule-associated proteins. J Biol Chem.
278:8407–8419. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang M, Mukherjee N, Bermudez RS, et al:
Adenovirus-mediated inhibition of survivin expression sensitizes
human prostate cancer cells to paclitaxel in vitro and in vivo.
Prostate. 64:293–302. 2005. View Article : Google Scholar
|
|
35
|
Wang S, Huang X, Lee CK and Liu B:
Elevated expression of erbB3 confers paclitaxel resistance in
erbB2-overexpressing breast cancer cells via upregulation of
survivin. Oncogene. 29:4225–4236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lv KZ, Liu LQ, Wang LB, et al: Lin28
mediates paclitaxel resistance by modulating p21, Rb and Let-7a
miRNA in breast cancer cells. PLoS One. 7:e400082012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lu MS, Xiao L, Hu JL, Deng S and Xu Y:
Targeting of p38 mitogen-activated protein kinases to early growth
response gene 1 (EGR-1) in the human paclitaxel-resistance ovarian
carcinoma cells. J Huazhong Univ Sci Tech-Med. 28:451–455. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Arai K, Matsumoto Y, Nagashima Y and
Yagasaki K: Regulation of class II beta-tubulin expression by tumor
suppressor p53 protein in mouse melanoma cells in response to vinca
alkaloid. Mol Cancer Res. 4:247–255. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Farrell A: A close look at cancer. Nat
Med. 17:262–265. 2011. View Article : Google Scholar
|
|
40
|
Pan Z, Gao YL, Heng LS, et al: Amphiphilic
N-(2,3-dihydroxypropyl)-chitosan-cholic acid micelles for
paclitaxel delivery. Carbohydr Polym. 94:394–399. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ehrlichova M, Koc M, Truksa J, Naldova Z,
Vaclavikova R and Kovarr J: Cell death induced by taxanes in breast
cancer cells: Cytochrome c is released in resistant but not in
sensitive cells. Anticancer Res. 25:4215–4224. 2005.PubMed/NCBI
|
|
42
|
Ehrlichova M, Ojima I, Chen J, et al:
Transport, metabolism, cytotoxicity and effects of novel taxanes on
the cell cycle in MDA-MB-435 and NCI/ADR-RES cells.
Naunyn-Schmiedebergs Arch Pharmacol. 385:1035–1048. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nemcova-Furstova V, Balusikova K, Sramek
J, James RF and Kovar J: Caspase-2 and JNK activated by saturated
fatty acids are not involved in apoptosis induction but modulate ER
stress in human pancreatic beta-cells. Cell Physiol Biochem.
31:277–289. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dyballa N and Metzger S: Fast and
sensitive colloidal coomassie G-250 staining for proteins in
polyacrylamide gels. J Vis Exp. 30:1–5. 2009.PubMed/NCBI
|
|
45
|
Jelinek M, Balusikova K, Kopperova D, et
al: Caspase-2 is involved in cell death induction by taxanes in
breast cancer cells. Cancer Cell Int. 13:422013. View Article : Google Scholar
|
|
46
|
Vidalino L, Doria A, Quarta S, Zen M,
Gatta A and Pontisso P: Serpin B3, apoptosis and autoimmunity.
Autoimmun Rev. 9:108–112. 2009. View Article : Google Scholar
|
|
47
|
Ho KY, Huang HH, Hung KF, et al:
Cholesteatoma growth and proliferation: Relevance with serpin B3.
Laryngoscope. 122:2818–2823. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gettins PGW: Serpin structure, mechanism,
and function. Chem Rev. 102:4751–4803. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Izuhara K, Ohta S, Kanaji S, Shiraishi H
and Arima K: Recent progress in understanding the diversity of the
human ov-serpin/clade B serpin family. Cell Mol Life Sci.
65:2541–2553. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cataltepe S, Gornstein ER, Schick C, et
al: Co-expression of the squamous cell carcinoma antigens 1 and 2
in normal adult human tissues and squamous cell carcinomas. J
Histochem Cytochem. 48:113–122. 2000.PubMed/NCBI
|
|
51
|
Murakami A, Suminami Y, Hirakawa H, Nawata
S, Numa F and Kato H: Squamous cell carcinoma antigen suppresses
radiation-induced cell death. Br J Cancer. 84:851–858. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Suminami Y, Nagashima S, Murakami A, et
al: Suppression of a squamous cell carcinoma (SCC)-related serpin,
SCC antigen, inhibits tumor growth with increased intratumor
infiltration of natural killer cells. Cancer Res. 61:1776–1780.
2001.
|
|
53
|
Collie-Duguid ESR, Sweeney K, Stewart KN,
Miller ID, Smyth E and Heys SD: SerpinB3, a new prognostic tool in
breast cancer patients treated with neoadjuvant chemotherapy.
Breast Cancer Res Treat. 132:807–818. 2011. View Article : Google Scholar
|
|
54
|
Lim W, Kim HS, Jeong W, et al: Serpin B3
in the chicken model of ovarian cancer: A prognostic factor for
platinum resistance and survival in patients with epithelial
ovarian cancer. Plos One. 7:e498692012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kerr KM: Personalized medicine for lung
cancer: new challenges for pathology. Histopathology. 60:531–546.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Felbor U, Dreier L, Bryant RAR, Ploegh HL,
Olsen BR and Mothes W: Secreted cathepsin L generates endostatin
from collagen XVIII. EMBO J. 19:1187–1194. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kudo T, Kigoshi H, Hagiwara T, Takino T,
Yamazaki M and Yui S: Cathepsin G, a neutrophil protease, induces
compact cell-cell adhesion in MCF-7 human breast cancer cells.
Mediat Inflamm. 2009:8509402009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lankelma JM, Voorend DM, Barwari T, et al:
Cathepsin L, target in cancer treatment? Life Sci. 86:225–233.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Small DM, Burden RE, Jaworski J, et al:
Cathepsin S from both tumor and tumor-associated cells promote
cancer growth and neovascularization. Int J Cancer. 133:2102–2112.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Broker LE, Huisman C, Span SW, Rodriguez
JA, Kruyt FAE and Giaccone G: Cathepsin B mediates
caspase-independent cell death induced by microtubule stabilizing
agents in non-small cell lung cancer cells. Cancer Res. 64:27–30.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
De Koning PJA, Kummer JA, de Poot SAH, et
al: Intracellular serine protease inhibitor serpin B4 inhibits
granzyme M-induced cell death. PLoS One. 6:e226452011.PubMed/NCBI
|
|
62
|
Ahmed ST and Darnell JE: Serpin B3/B4,
activated by STAT3, promote survival of squamous carcinoma cells.
Biochem Biophys Res Commun. 378:821–825. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ehrnsperger M, Graber S, Gaestel M and
Buchner J: Binding of nonnative protein to Hsp25 during heat shock
creates a reservoir of folding intermediates for reactivation. EMBO
J. 16:221–229. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Schmitt E, Gehrmann M, Brunet M, Multhoff
G and Garrido C: Intracellular and extracellular functions of heat
shock proteins: repercussions in cancer therapy. J Leukoc Biol.
81:15–27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Garrido C, Bruey JM, Fromentin A, Hammann
A, Arrigo AP and Solary E: HSP27 inhibits cytochrome c-dependent
activation of procaspase-9. FASEB J. 13:2061–2070. 1999.PubMed/NCBI
|
|
66
|
Havasi A, Li ZJ, Wang ZY, et al: Hsp27
inhibits Bax activation and apoptosis via a phosphatidylinositol
3-kinase-dependent mechanism. J Biol Chem. 283:12305–12313. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ciocca DR, Arrigo AP and Calderwood SK:
Heat shock proteins and heat shock factor 1 in carcinogenesis and
tumor development: an update. Arch Toxicol. 87:19–48. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Weng YR, Cui Y and Fang JY: Biological
functions of cytokeratin 18 in cancer. Mol Cancer Res. 10:485–493.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Galarneau L, Loranger A, Gilbert S and
Marceau N: Keratins modulate hepatic cell adhesion, size and G1/S
transition. Exp Cell Res. 313:179–194. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Abreu MT, Arnold ET, Chow JYC and Barrett
KE: Phosphatidylinositol 3-kinase-dependent pathways oppose
Fas-induced apoptosis and limit chloride secretion in human
intestinal epithelial cells. Implications for inflammatory
diarrheal states. J Biol Chem. 276:47563–47574. 2001. View Article : Google Scholar
|
|
71
|
Fortier AM, Van Themsche C, Asselin E and
Cadrin M: Akt isoforms regulate intermediate filament protein
levels in epithelial carcinoma cells. FEBS Lett. 584:984–988. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Inada H, Izawa I, Nishizawa M, et al:
Keratin attenuates tumor necrosis factor-induced cytotoxicity
through association with TRADD. J Cell Biol. 155:415–425. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Meng YG, Wu ZQ, Yin XY, et al: Keratin 18
attenuates estrogen receptor alpha-mediated signaling by
sequestering LRP16 in cytoplasm. BMC Cell Biol. 10:962009.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Woelfle U, Sauter G, Santjer S, Brakenhoff
R and Pantel K: Down-regulated expression of cytokeratin 18
promotes progression of human breast cancer. Clin Cancer Res.
10:2670–2674. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Schaller G, Fuchs I, Pritze W, et al:
Elevated keratin 18 protein expression indicates a favorable
prognosis in patients with breast cancer. Clin Cancer Res.
2:1879–1885. 1996.PubMed/NCBI
|