Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignant diseases worldwide with the majority of cases
relating to hepatitis B virus (HBV) infection. Interactions between
the HBV-encoded X (HBx) protein and host factors contribute to the
development of HBV-associated HCC (1,2). HBx
is a multifunctional regulator that modulates transcription, signal
transduction pathways, cell cycle progression, DNA repair,
apoptosis, protein degradation pathways and genetic stability
through interaction with host factors (3,4).
During recent years evidence has accumulated that HBx protein has
both anti-apoptotic and pro-apoptotic actions (5,6). The
subcellular localization of HBx is primarily in the cytoplasm, with
a small fraction in the nucleus. In addition, a fraction of
cytosolic HBx localizes with mitochondria in different cell lines,
affecting mitochondrial physiology, metabolism and other relevant
functions (7,8). Some research groups have proved that
HBx is localized to the outer mitochondrial membrane, consistent
with the reported interaction of HBx with the voltage-dependent
anion channel (VDAC), a channel that spans the outer mitochondrial
membrane (9–11). In recent years, our studies have
revealed that HBx can also interact with the inner mitochondrial
membrane protein COXIII, by yeast two-hybrid system, mating
experiment and coimmunoprecipitation (12,13).
Cytochrome c oxidase (COX or complex IV) is
the terminal enzyme of the respiratory chain that catalyzes the
reduction of molecular oxygen and plays a pivotal role in the
generation of ATP and maintenance of mitochondrial membrane
potential (ΔΨm) (14,15). COXIII is one of the 3 mtDNA encoded
subunits of COX that functions in the process of growth,
differentiation, transcription and signal transduction by
supporting higher energy requirements for cells (16–18).
The effect of cytochrome oxidase decreases when the level of COXIII
is reduced (19). COX encoded gene
mutations or expression changes may make the cell abnormal that is
associated with the development of tumors mainly through an
increase in reactive oxygen species in mitochondria oxidative
phosphorylation (20–22).
Increasing evidence suggests that inflammation
contributes to HCC development due to the adverse effects of
inflammatory mediators such as proinflammatory cytokine and
reactive oxygen species (ROS). They play a key role on DNA repair,
DNA methylation, DNA oxidation and lipid peroxidation (23–25).
The main production site of ROS is identified to be mitochondria
(26–28). Cyclooxygenase-2 (COX-2) is a key
mediator of inflammatory responses that promotes the growth of HCC
cells by stimulating proliferation, angiogenesis, invasiveness and
inhibits apoptosis and immune response (29,30).
Notably, there is experimental evidence showing a relationship
between ROS and cyclooxygenase-derived products. Thus ROS can
promote cyclooxygenase expression and the COX/PG
(cyclooxygenase/prostaglandin) synthase pathways can induce the
generation of ROS (31).
Additionally, ROS from mitochondria plays a key role in COX-2
induction (28,32). However, it remains unknown how the
two inflammatory mediators ROS and COX-2 converge during
inflammation leading to HCC development.
In this study, we constructed a HepG2 cell line
stably expressing the HBx gene by lentivirus vectors to explore the
effect of HBx on the development of HBV associated HCC. We found
that HBx co-localized with the inner mitochondrial membrane
protein, COXIII, leading to changes of mitochondrial biogenesis and
morphology. The upregulation of COX-2 caused by HBx through
generation of mitochondria ROS promoted cell growth.
Materials and methods
Cells cultures
Human embryonic kidney 293T cell line was provided
by ATCC (UK). The human hepatoma cell line HepG2 cell was purchased
from Shanghai Cell Bank (Shanghai, China). Cells were cultured in
Dulbecco’s modified Eagle’s medium containing 10% fetal bovine
serum and 1% penicillin-streptomycin solution at 37°C in a 5%
CO2 incubator.
Antibodies and reagents
Anti-DYKDDDDK-Tag (anti-flag) antibody was purchased
from Abmart (Shanghai, China). Anti-COX-2 antibody was purchased
from Abcam (Co., USA). Anti-cytochrome c oxidase subunit III
(anti-COXIII) (N-20) antibody was purchased from Santa Cruz
Biotechnology (Santa Cruz, CA). Anti-β-actin antibodies were
purchased from ZSGB-BIO (Beijing, China). CFTM488-conjugated donkey
anti-mouse IgG (H+L), CFTM350-conjugated donkey anti-goat IgG
(H+L), N-acetylcysteine (NAC) and puromycin were obtained from
Sigma (St. Louis, MO). Mito Tracker Red was obtained from Molecular
Probes (Eugene, OR). NS-398 was purchased from Beyotime (Shanghai,
China). Cell counting kit-8 was from Dojindo Laboratories
(Kumamoto, Japan). Primers were synthesized by Biosune (Shanghai,
China).
Generation of the recombinant lentivirus
and establishment of the stably transfected HepG2 cell line
The HBx expression plasmid PcDNA3.1-x (HBV subtype
ayw) was a gift from Professor Michael J. Bouchard (Drexel
University College of Medicine, Philadelphia, PA). The lentivirus
packaging system: pLOV.CMV.eGFP.2A.EF1a.PuroR (pLOV), psPAX2 and
pMD2.G were provided by Neuron Biotech Co., Ltd. (Shanghai, China).
The lentivirus vector pLOV contains the gene coding for green
fluorescent protein (GFP), puromycin and the peptide DYKDDDDK
(flag). Full-length HBx (465 bp) was amplified from PcDNA3.1-x by
polymerase chain reaction (PCR) and then infused with flag epitope
at the N-terminus and next cloned into pLOV. The recombinant
lentiviral particles were generated by transient co-transfection
with 293T cells by the three-plasmid expression system, and
harvested by filtration through a 0.45-μm filter and
ultracentrifugation at 100,000 g for 2 h at 4°C. HepG2 cells at 60%
confluency were transfected with the recombinant lentivirus. Then
the cell clones were treated with 0.2 μg/ml puromycin for 10 days.
Positive clones expressing GFP and resistant to puromycin were
screened and named HepG2-HBx and HepG2-mock, respectively.
Indirect immunofluorescence assay
To eliminate the impact of green fluorescence caused
by GFP in HepG2-HBx cells, we used 100% methanol as a fixative. As
a result, green fluorescence disappeared (data not shown),
consistent with previous research (33). Cells were stained for 30 min with
150 nM Mito Tracker Red and fixed with a 100% ice methanol solution
and pre-incubated in blocking solution (5% donkey serum albumin in
PBS) and next incubated with primary antibodies (anti-flag,
anti-COXIII) at 4°C overnight. Then, the fluorescence-labeled
second antibodies (CFTM488-conjugated donkey anti-mouse,
CFTM350-conjugated donkey anti-goat) were added and incubated for
60 min at 37°C in the dark, and the sections were mounted using
glycerol. Fluorescence images were obtained by confocal laser
scanning microscopy (Leica SP5, Solms, Germany).
Isolation of mitochondria and measurement
of COX activity
Mitochondria were isolated using the mitochondrial
isolation kit for mammalian cells (Thermo Scientific, MA, USA)
according to the manufacturer’s instructions. Briefly, following
lysis of approximately 2×107 cells, cell debris and
nuclei were pelleted at 700 × g for 10 min at 4°C, followed by
centrifugation at 3,000 × g for 15 min at 4°C to pellet a
mitochondria-enriched fraction, and then 12,000 × g for 5 min at
4°C to pellet the isolated mitochondria. Protein concentrations
were measured using the bicinchoninic acid (BCA) assay and then
adjusted to 1 μg/μl. COX activity was determinated using the
Cytochrome c Oxidase Assay kit (GenMed, Shanghai, China)
according to the manufacturer’s instructions by an enzyme mark
instrument (Bio-Tek ELX-800, USA). Isolated mitochondria (10 μl)
were combined with 10 μl of lysis buffer, then mixed with 205 μl of
buffer solution in 96-well plates. The reaction was initiated by
the addition of 25 μl of reaction liquid, and the decrease in
absorbance at 550 nm was measured for 1 min. Activity was
calculated based on the following equation: Units/ml =
[(ΔAbs550/min for the sample - ΔAbs550/min for the blank) ×
dilution factor × total reaction volume]/[mitochondria isolate
volume x the difference in extinction coefficients between ferro-
and ferri-cytochrome c at 550 nm (21.84)]. One unit is the
amount that oxidizes 1 μmol reduced cytochrome c/min at pH
7.0 and 25°C.
Flow cytometric analysis
Cells in the three groups were harvested with
trypsin, and resuspended in PBS (1×106 cells/ml) for
analyzing ROS and mitochondrial membrane potential by flow
cytometry (Becton-Dickson Co., C6, San Jose, CA). i) The
mitochondrial membrane potential was evaluated using the cationic
fluorescent dye TMRM (Sigma). Cells incubated in 100 nM TMRM
solution, 20 min at 37°C in the dark, then analysed by a flow
cytometer. ii) Intracellular ROS levels were detected by staining
cells with 50 μM DihyDroeThidium (DHE) fluorescence probe (Vigorous
Biotechnology, Beijing, China) for 30 min in the dark, then
analysed by a flow cytometer.
Transmission electron microscopy
(TEM)
The morphological changes of cell and mitochondria
were observed by TEM. A total of 1×106 of cells were
collected and subjected to fixation with 3% fresh glutaraldehyde
and 1.5% paraformaldehyde solution at 4°C for 1 h, next to
post-fixation with 1% osmium tetroxide and 1.5% potassium
ferrocyanide solution at 4°C for 1.5 h, then dehydration was
carried out with a graded series of ethanol solution, and finally
the cells were to embedded in Epon-618. Ultrathin sections were cut
to stain with uranyl acetate and lead citrate, and observed under
TEM (Phillips EM208, WI, USA).
Reverse-transcriptase polymerase chain
reaction (RT-PCR) and western blotting
i) RT-PCR: total RNA was extracted using TRIzol
reagent. PCR amplification reactions were set up according to the
manufacturer’s protocol (Thermo Scientific). The primer sequences
of each gene and the PCR conditions are listed in Table I. PCR was carried out in a GeneAmp
PCR System (PE2400, USA). The results of electrophoresis were
scanned by a UVP scanner with Grab-IT software (Gel Doc 100,
Bio-Rad, USA) and gray scale of each band was calculated using
Image J software. ii) Western blotting: cells were harvested and
cell lysate was prepared. Protein concentration was measured using
BCA Protein Assay kit (Beyotime). Immunoblotting was performed
using specific primary antibodies then incubated with the
appropriate horseradish peroxidase-conjugated secondary antibodies
and detected using an ECL kit (ZSGB-BIO). Densitometry was
performed on the western blot images by using Image-J software and
expressed as arbitrary densitometric units normalised to β-actin
expression.
 | Table IPrimer sequence for the genes used in
RT-PCR analyses. |
Table I
Primer sequence for the genes used in
RT-PCR analyses.
| Gene | | Primer
sequences |
|---|
| HBV X (HBx) | Sense: | 5′-ATG CAA GCT TAT
GGC TGC TAG GCT GTA CTG-3′ |
| Antisense: | 5′-TGC GAA TTC TTA
GGC AGA GTG AAA AAG TTG-3′ |
| COXIII | Sense: | 5′-AGC CCA TGA CCC
CTA ACA-3′ |
| Antisense: | 5′-CCT CAT AGT TAG
TGG ACT CG-3′ |
| COX-2 | Sense: | 5′-TCA AGT CCC TGA
GCA TCT ACG-3′ |
| Antisense: | 5′-ACA TTC CTA CCA
CCA GCA ACC-3′ |
| β-actin | Sense: | 5′-GGC ATC GTG ATG
GAC TCC G-3′ |
| Antisense: | 5′-GCT GGA AGG TGG
ACA GCG A-3′ |
Cell proliferation assay
Cell proliferation was analyzed by the cell-counting
kit-8. Cells (3×103) were seeded in 96-well plates and
each well contained 100 μl of complete DMEM medium (6 replica wells
for each cell line). After 1, 2 and 3 days of incubation, the media
was removed and 100 μl of fresh complete DMDM medium containing
premixed CCK-8 (110 dilution in complete DMEM medium) was added to
each well. The plates were incubated at 37°C with 5% CO2
for 3 h, followed by shaking thoroughly for 1 min on a shaker. The
absorbance at 450 nm was measured using an ELISA reader, each day
of the 3-day experiment to calculate the growth curve.
Colony formation assay
Cell suspensions were transferred into 6-well plates
(500 cells/well), and each group contained 3 wells. After
incubation for 14 days, the cells were rinsed in PBS for twice and
stained with Crystal Violet Staining Solution. The experiments were
performed in triplicate and the number of colonies containing
>50 cells were microscopically counted to calculate the colony
formation rate as number of colonies/number of cells × 100%.
Statistical analysis
Each set of experiments was repeated at least three
times with similar results. Results are expressed as means with
standard deviations (SD). Statistical evaluation was carried out by
one-way analysis of variance (ANOVA). P-values of <0.05 were
considered statistically significant.
Results
Establishment of a HepG2 cell line stably
expressing the HBx gene
Lentiviral vectors have become one of the most
powerful gene transfer vehicles, they provide several advantages
that include being able to transduce a wide range of cell types
irrespective of their division status, leading to prolonged
transgene expression, lack of pre-existing immunity and less
genotoxic (34,35). Our study used lentiviral vectors to
establish a HepG2 cell line stably expressing HBx gene in
vitro. The GFP positive cells were 95–98% by fluorescence
microscope (Fig. 1A). Stable
expression the HBx mRNA and protein was verified by RT-PCR and
western blot, respectively (Fig.
1B). A HepG2 cell line stably transduced with a lentivirus
expressing the HBx gene was successfully constructed.
HBx targets to COXIII leading to changes
of mitochondrial biogenesis and morphology in HepG2 cells
We proceeded with fluorescent staining experiments
by a combination of anti-flag antibody, anti-COXIII antibody and a
mitochondria-specific staining agent, Mito Tracker Red, then
observed cells by confocal microscopy. As indicated in Fig. 2, the staining of HBx protein (green
color) together with COXIII protein (blue color) in mitochondria
(red color) was observed (white) in HepG2-HBx cells, which
suggested that HBx protein co-localized with inner mitochondrial
membrane protein COXIII in HepG2 cells, further verifying our
previous studies (12,13). Moreover, the localization of HBx
was both in the nucleus and the cytoplasm, with a fraction of
cytosolic HBx in the mitochondria.
Since COXIII is an essential component of the
mitochondrial respiratory chain, the co-localization of HBx with
COXIII would induce an alteration in the normal mitochondrial
function and morphology in HepG2-HBx cells. We found that the
protein expression of COXIII was increased in HepG2-HBx cells,
while the RNA level did not change as shown in Fig. 3A and B. We then examined the COX
activities. Correlating with their upregulated proteins level, they
were significantly increased in HepG2-HBx cells compared with
HepG2-control and HepG2-mock cells (Fig. 3C). Next, we measured mitochondrial
membrane potencial (ΔΨm) of cells in the three groups using the
fluorochrome TMRM, HepG2-HBx cells displayed the highest ΔΨm
(Fig. 3D). Increased COXIII
protein expression level, upregulation of COX activities as well as
the highest ΔΨm demonstrated that HBx promoted mitochondrial
function in HepG2 cells. To assess the changes of mitochondrial
morphology accompanied by the alteration of mitochondrial
biogenesis in HepG2-HBx cells, we observed the mitochondria, nuclei
and other intracellular membrane structures by TEM. Results showed
that only a detectable slight swelling of mitochondria in HepG2-HBx
cells was found compared with the other two groups (Fig. 3E). Thus, our studies demonstrated
that HBx co-localized with inner mitochondrial protein COXIII in
HepG2 cells, leading to the changes of mitochondrial biogenesis and
morphology.
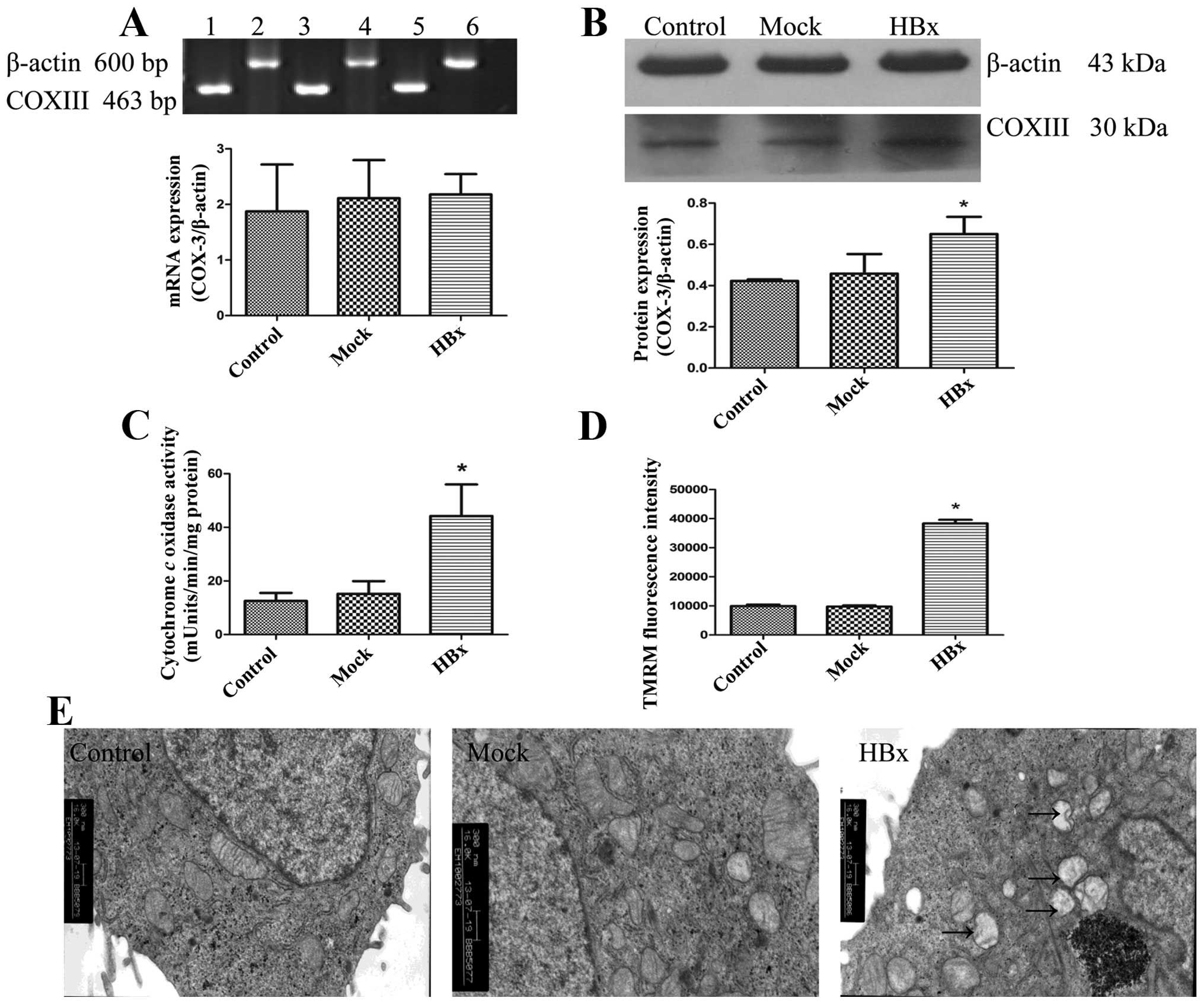 | Figure 3HBx leads to changes of mitochondrial
biogenesis and morphology in HepG2 cells. HBx, Mock and Control
represent HepG2-HBx, HepG2-mock and HepG2 cells, respectively. (A)
Expression of COXIII mRNA was detected using RT-PCR. Lane 1 and 2,
3 and 4–6 represent HepG2, HepG2-mock and HepG2-HBx cells,
respectively. (B) Levels of COXIII protein were detected using
western blotting by antibody specific against COXIII protein and
β-actin. (C) Changes of cytochrome c oxidase (COX) activity
were determinated using the Cytochrome c Oxidase Assay kit.
(D) Mitochondrial membrane potencial was measured by using the
fluorescence dye, TMRM, followed by flow cytometry. (E)
Morphological changes of mitochondria were examined by electronic
microscope (original magnification, ×16,000). Arrows show the
slight swelling in mitochondria. Values are means ± SD of at least
three independent experiments; *P<0.05. |
ROS from mitochondria induce the
cyclooxygenase-2 gene expression in HepG2-HBx cells
Mitochondria are major ROS generators, thus changes
in mitochondrial function and morphology by HBx would be expected
to cause ROS generation in cells (36). We evaluated the ROS level by flow
cytometry with the DihyDroeThidium (DHE) probe. As shown in
Fig. 4A, the ROS generation was
the strongest in HepG2-HBx cells compared to the others. Recent
studies have shown that ROS from mitochondria is necessary for
COX-2 induction (28,32). We then analyzed whether COX-2 gene
expression increased with the ROS generation. Fig. 4B and C show that expression of both
COX-2 mRNA and protein was increased in HepG2-HBx cells. To further
confirm the notion that ROS from mitochondria induced
cyclooxygenase-2 gene expression in HepG2-HBx cells, we treated
cells with the antioxidant, N-acetyl cysteine (NAC) (10 mM) for 12
h. As can be seen from Fig. 4D,
the induction of COX-2 protein expression by HBx was decreased in
HepG2-HBx cells with NAC, but still higher than the other two
groups. Therefore, it was clear that ROS from mitochondria induce
COX-2 gene expression in HepG2-HBx cells, resulting in inflammatory
injury.
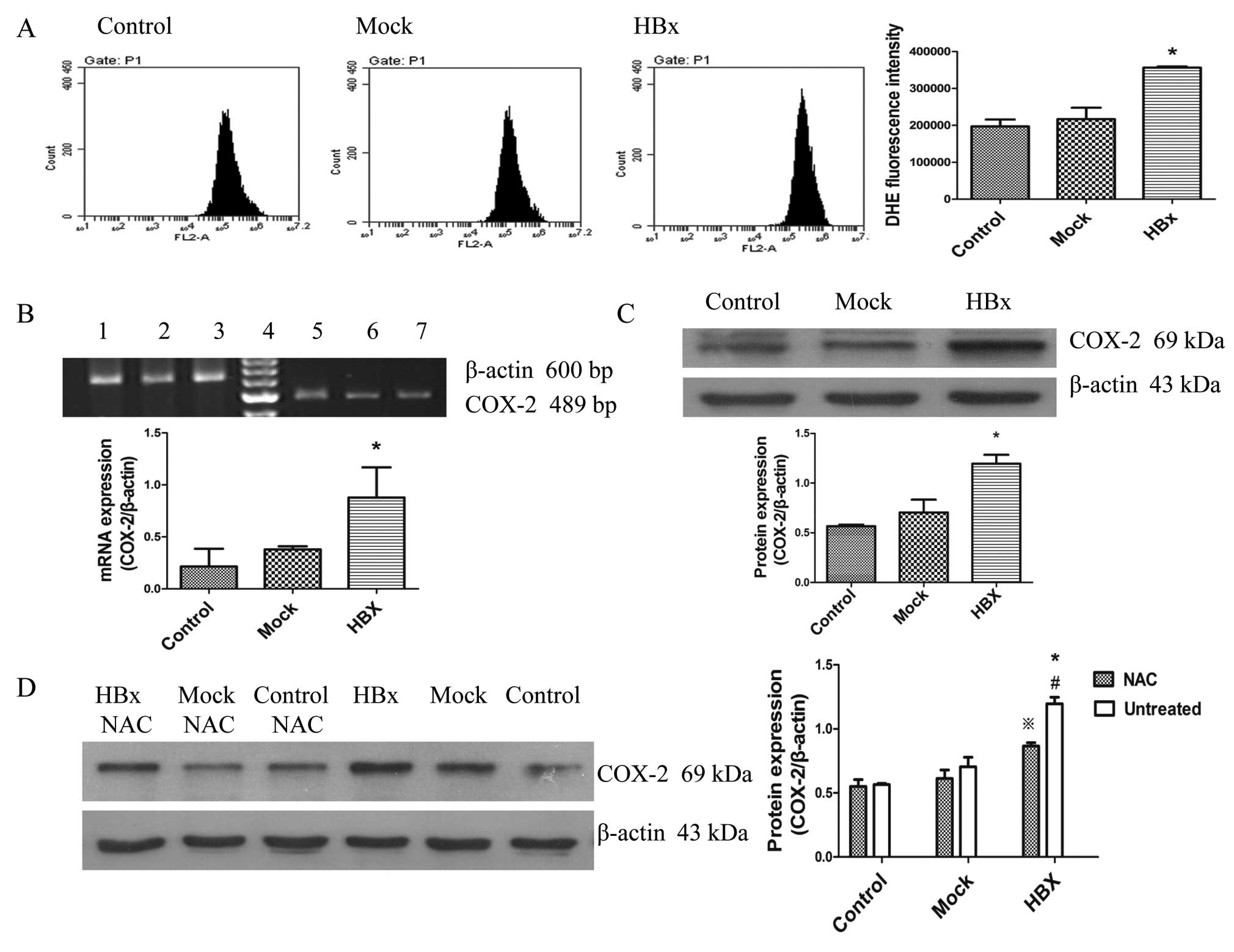 | Figure 4HBx induces ROS generation in HepG2
cells and ROS plays a key role in HBx-induced COX-2 expression.
HBx, Mock and Control represent HepG2-HBx, HepG2-mock and HepG2
cells, respectively. (A) HBx increased ROS production in HepG2-HBx
cells. Intracellular ROS levels were determined by using the
fluorescence dye DHE, followed by flow cytometry. (B) Levels of
COX-2 mRNA in cells were detected using RT-PCR. Lane 1 and 5, 2 and
3, 6 and 7 represent HepG2-HBx, HepG2-mock and HepG2 cells,
respectively while D is a marker. (C) Levels of COX-2 protein were
detected using western blotting by antibody specific against COX-2
protein and β-actin. (D) Western blot analysis for COX-2 protein
levels in HepG2-HBx, HepG2-mock and HepG2 cells. Cells in the left
three groups were treated with NAC (10 mM) for 12 h. Cells in the
right three groups were treated at the same conditions without NAC.
Values are means ± SD for at least three independent experiments.
*P<0.05 compared with HepG2 and HepG2-mock cells,
**P<0.05 compared with HepG2-HBx cells with NAC.
#P<0.05 compared with HepG2 and HepG2-mock cells with
NAC. |
COX-2 promotes the proliferation ability
of HepG2 cells
Substantial research in recent years has
demonstrated that HBx promoted cell proliferation associating with
HCC in different models and conditions (37,38).
To observe the effects of HBx on cell proliferation, we performed
the cell viability assay and plate colony formation assay. As shown
in Fig. 5A and B, HepG2-HBx cells
increased the proliferation rate and formed more colonies,
indicating that HBx promoted the proliferation of HepG2 cells.
Previous studies have pointed out that COX-2 stimulates
proliferation, angiogenesis, invasiveness and inhibits apoptosis to
promote the growth of HCC cells (39). We considered that the upregulation
of COX-2 contributed to the proliferation mediated by HBx. Further
studies were carried out by treating cells with the selective COX-2
inhibitor NS-398 (50 μmol/l) for 72 h. The proliferation of
HepG2-HBx cells were significantly suppressed following the
inhibition of COX-2 protein expression (Fig. 5C and D). Of note, our data strongly
suggest that HBx induced cell proliferation through
inflammation.
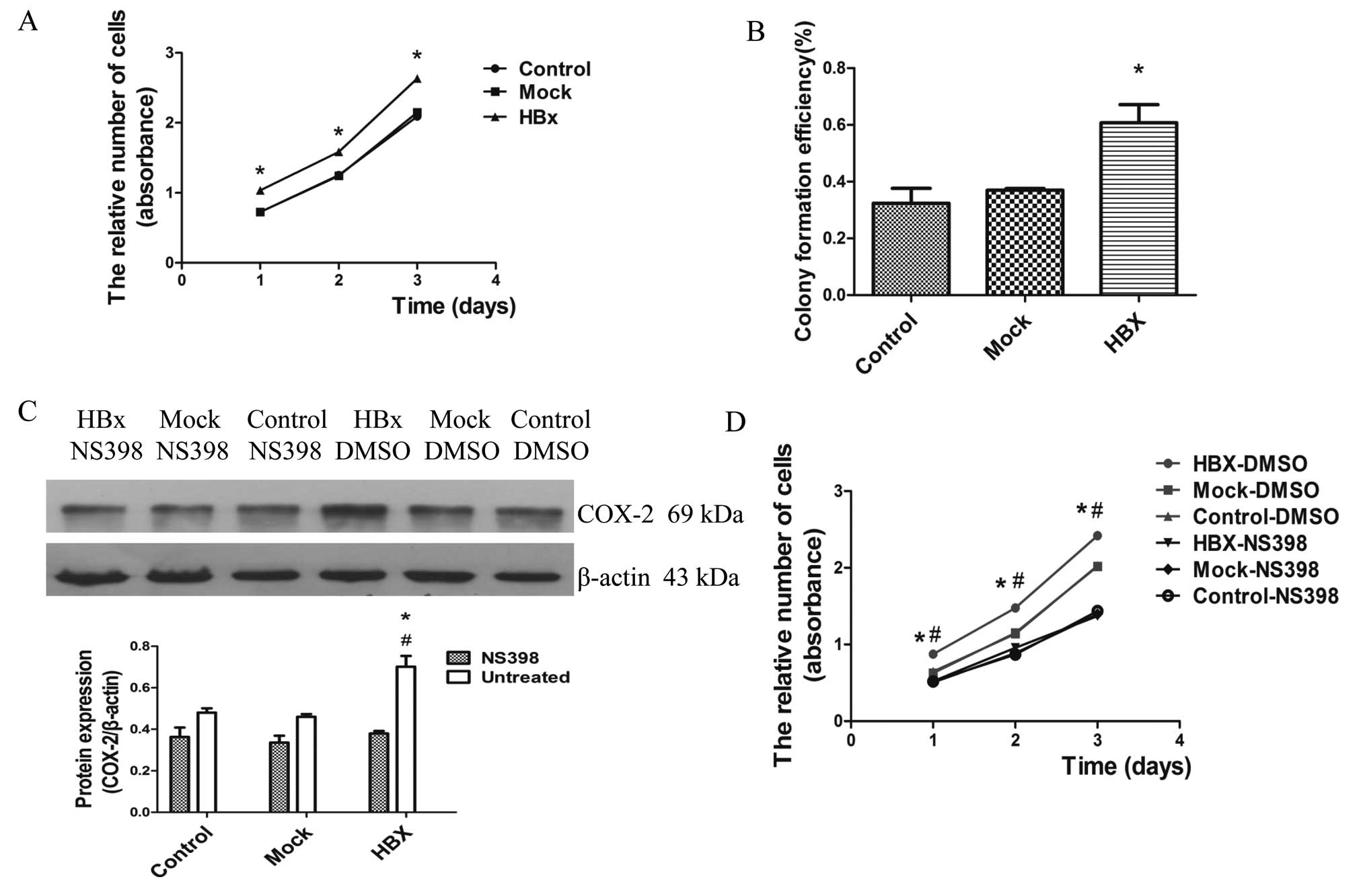 | Figure 5HBx promotes the proliferation
ability of HepG2 cells through upregulation of COX-2. HBx, Mock and
Control represent HepG2-HBx, HepG2-mock and HepG2 cells,
respectively. (A) After incubation for 1, 2 and 3 days, cell
proliferation was examined by cell viability assay with
cell-counting kit-8 (CCK-8). (B) Colony formation efficiency was
detected by plate clone formation assay. After incubation for 2
weeks, the cells were stained with Crystal Violet staining solution
and microscopically counted. (C) Western blot analysis for COX-2
protein levels in HepG2-HBx, HepG2-mock and HepG2 cells. Cells in
the left three groups were treated with COX-2 inhibitor NS-398 (50
μmol/l) for 72 h. Cells in the right three groups were treated with
DMSO. (D) Cells were treated with NS-398 (50 μmol/l) and DMSO as
controls for 72 h, respectively. Following incubation for 1, 2 and
3 days, cell proliferation was examined by Cell viability assay
with cell-counting kit-8 (CCK-8). *P<0.05 compared
with HepG2 and HepG2-mock cells, #P<0.05 compared
with HepG2-HBx cells with NS398. |
Discussion
Hepatocellular carcinoma (HCC) is one of the most
common malignant diseases and has the fourth highest cancer
mortality rate worldwide. During recent years evidence has
accumulated that HBx contributes to the development of
HBV-associated HCC (3). The
subcellular localization of HBx is primarily cytoplasmic, with a
small fraction in the nucleus. In addition, a fraction of cytosolic
HBx localizes to the outer mitochondrial membrane in different cell
lines, affecting mitochondrial physiology, metabolism and other
relevant functions (7–11,40).
In this study, we successfully constructed a HepG2 cell line stably
expressing the HBx gene by lentivirus vectors which can better
simulate the progress of chronic hepatitis B virus infection. Then
we confirmed the co-localization of HBx with the inner
mitochondrial membrane protein COXIII by confocal microscopy in
HepG2 cells (Fig. 2), further
verifying our previous findings that HBx can also localize to the
inner mitochondrial membrane (12,13).
The new localization of HBx will give new insight into the role of
HBx associated with HCC.
COX plays a pivotal role in the generation and
maintenance of mitochondrial membrane potential (ΔΨm) and ATP
(14,15). In this study, to clarify the impact
of the co-localization of HBx with COXIII on mitochondrial relevant
functions and morphology, we first demonstrated that HBx regulated
COXIII gene function at the post-transcription level as the protein
expression was increased in HepG2-HBx cells, while the RNA level
did not change as shown in Fig. 3A and
B. The effect of cytochrome oxidase changed with the alteration
of subunit III (19,41). We then found COX activities were
significantly increased in HepG2-HBx cells, indicating that COXIII
functions in the process of growth and differentiation,
transcription, signal transduction by supporting more energy
available for cells (16–18). Next, a higher ΔΨm was detected in
HepG2-HBx cells. TEM results showed a detectable slight swelling in
mitochondria of HepG2-HBx cells. Together with the previous
findings, we demonstrated that HBx promotes mitochondrial
biogenesis in HepG2 cells. Since mitochondria are the key
organelles which regulate apoptosis, cellular energetics and signal
transduction pathways (42), the
alteration of mitochondrial function and morphology may destroy
cellular homeostasis.
Several investigators have demonstrated that COX
encoded gene expression changes are associated with the development
of tumors mainly through increased mitochondrial ROS (20–22,43).
A similar increasing trend was observed in our results of
mitochondrial ROS as shown in Fig.
4A. HBx increases the level of mitochondrial ROS which is
related to hepatocellular carcinogenesis (44,45),
but the mechanism remains unclear. Increasing evidence has exposed
the relationship between the two main inflammatory mediators, ROS
and COX-2 (31). We found that
COX-2 gene expression increased with the ROS generation (Fig. 4D). When cells were treated with the
antioxidant NAC (10 mM) for 12 h, the induction of COX-2 protein
expression by HBx was decreased in HepG2-HBx cells, consistent with
previous studies that ROS from mitochondria is necessary for COX-2
induction (28,32). Thus, our work proved that HBx
promoted the inflammation in HepG2 cells by generation of
mitochondria ROS and led to induction of COX-2, that may contribute
to HCC development (23–25).
Research in recent years has demonstrated that
inflammation caused by COX-2 stimulates proliferation and inhibits
apoptosis to promote the growth of HBx transfected HCC cells
(29,39,46).
We found HBx promoted cell proliferation by cell viability and
plate colony formation assays (Fig. 5A
and B). The addition of the selective COX-2 inhibitor NS-398
(50 μmol/l) to cells for 72 h significantly suppressed the
proliferation of HepG2-HBx cells followed by the inhibition of
COX-2 protein expression (Fig. 5C and
D). These results strongly indicated that COX-2 mediated HBx
promotion of HepG2 cell growth.
All the data above are consistent with the finding
that HBx induced cell proliferation through HBx/COXIII/COX-2
pathway. First, we successfully constructed a HepG2 cell line
stably expressing the HBx gene by lentivirus vectors. Then we
confirmed the co-localization of HBx with the inner mitochondrial
membrane protein COXIII leading to the alteration of mitochondrial
function and morphology. The upregulation of COX-2 caused by HBx in
HepG2 cells through generation of mitochondria ROS contributed to
cell growth. Such a mechanism provides deeper insights into the
molecular mechanism of HBV-associated HCC.
Acknowledgements
This study was supported by the National Natural
Science grant 81300321 from the Foundation of China and the Key
Clinical Specialty Discipline Construction Program of Fujian, P.R.
China (Min Wei Ke Jiao 2012 No. 49). We thank Professor M.J.
Bouchard for the PcDNA3.1-X plasmid.
References
|
1
|
Tian Y, Yang W, Song J, Wu Y and Ni B:
Hepatitis B virus X protein-induced aberrant epigenetic
modifications contributing to human hepatocellular carcinoma
pathogenesis. Mol Cell Biol. 33:2810–2816. 2013. View Article : Google Scholar
|
|
2
|
Xu C, Zhou W, Wang Y and Qiao L: Hepatitis
B virus-induced hepatocellular carcinoma. Cancer Lett. 345:216–222.
2013. View Article : Google Scholar
|
|
3
|
Motavaf M, Safari S, Saffari JM and
Alavian SM: Hepatitis B virus-induced hepatocellular carcinoma: the
role of the virus x protein. Acta Virol. 57:389–396. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ng SA and Lee C: Hepatitis B virus X gene
and hepatocarcinogenesis. J Gastroenterol. 46:974–990. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Assrir N, Soussan P, Kremsdorf D and
Rossignol JM: Role of the hepatitis B virus proteins in pro- and
anti-apoptotic processes. Front Biosci (Landmark Ed). 15:12–24.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kew MC: Hepatitis B virus x protein in the
pathogenesis of hepatitis B virus-induced hepatocellular carcinoma.
J Gastroenterol Hepatol. 26:144–152. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rawat S, Clippinger AJ and Bouchard MJ:
Modulation of apoptotic signaling by the hepatitis B virus X
protein. Viruses. 4:2945–2972. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma J, Sun T, Park S, Shen G and Liu J: The
role of hepatitis B virus X protein is related to its differential
intracellular localization. Acta Biochim Biophys Sin (Shanghai).
43:583–588. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shoshan-Barmatz V, Israelson A, Brdiczka D
and Sheu SS: The voltage-dependent anion channel (VDAC): function
in intracellular signalling, cell life and cell death. Curr Pharm
Des. 12:2249–2270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rahmani Z, Huh KW, Lasher R and Siddiqui
A: Hepatitis B virus X protein colocalizes to mitochondria with a
human voltage-dependent anion channel, HVDAC3, and alters its
transmembrane potential. J Virol. 74:2840–2846. 2000. View Article : Google Scholar
|
|
11
|
Clippinger AJ and Bouchard MJ: Hepatitis B
virus HBx protein localizes to mitochondria in primary rat
hepatocytes and modulates mitochondrial membrane potential. J
Virol. 82:6798–6811. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang XZ, Li D, Tao QM, Lin N and Chen ZX:
A novel hepatitis B virus X-interactive protein: cytochrome C
oxidase III. J Gastroenterol Hepatol. 21:711–715. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li D, Wang XZ, Yu JP, Chen ZX, Huang YH
and Tao QM: Cytochrome C oxidase III interacts with hepatitis B
virus X protein in vivo by yeast two-hybrid system. World J
Gastroenterol. 10:2805–2808. 2004.PubMed/NCBI
|
|
14
|
Mkaouar-Rebai E, Ellouze E, Chamkha I,
Kammoun F, Triki C and Fakhfakh F: Molecular-clinical correlation
in a family with a novel heteroplasmic Leigh syndrome missense
mutation in the mitochondrial cytochrome c oxidase III gene. J
Child Neurol. 26:12–20. 2011. View Article : Google Scholar
|
|
15
|
Bauerfeld CP, Rastogi R, Pirockinaite G,
et al: TLR4-mediated AKT activation is MyD88/TRIF dependent and
critical for induction of oxidative phosphorylation and
mitochondrial transcription factor A in murine macrophages. J
Immunol. 188:2847–2857. 2012. View Article : Google Scholar
|
|
16
|
Liang L, Qu L and Ding Y: Protein and mRNA
characterization in human colorectal carcinoma cell lines with
different metastatic potentials. Cancer Invest. 25:427–434. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bafna S, Singh AP, Moniaux N, Eudy JD,
Meza JL and Batra SK: MUC4, a multifunctional transmembrane
glycoprotein, induces oncogenic transformation of NIH3T3 mouse
fibroblast cells. Cancer Res. 68:9231–9238. 2008. View Article : Google Scholar
|
|
18
|
Wu H, Rao GN, Dai B and Singh P: Autocrine
gastrins in colon cancer cells up-regulate cytochrome c oxidase Vb
and down-regulate efflux of cytochrome c and activation of
caspase-3. J Biol Chem. 275:32491–32498. 2000. View Article : Google Scholar
|
|
19
|
Soto IC, Fontanesi F, Valledor M, Horn D,
Singh R and Barrientos A: Synthesis of cytochrome c oxidase subunit
1 is translationally downregulated in the absence of functional
F1F0-ATP synthase. Biochim Biophys Acta. 1793:1776–1786. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Athar M, Chaudhury NK, Hussain ME and
Varshney R: Hoechst 33342 induced reactive oxygen species and
impaired expression of cytochrome c oxidase subunit 1 leading to
cell death in irradiated human cancer cells. Mol Cell Biochem.
352:281–292. 2011. View Article : Google Scholar
|
|
21
|
Bernstein C, Facista A, Nguyen H, et al:
Cancer and age related colonic crypt deficiencies in cytochrome c
oxidase I. World J Gastrointest Oncol. 2:429–442. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gutierrez-Gonzalez L, Graham TA,
Rodriguez-Justo M, et al: The clonal origins of dysplasia from
intestinal metaplasia in the human stomach. Gastroenterology.
140:1251–1260. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Szabo G and Lippai D: Molecular hepatic
carcinogenesis: impact of inflammation. Dig Dis. 30:243–248. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nishida N, Arizumi T, Takita M, et al:
Reactive oxygen species induce epigenetic instability through the
formation of 8-hydroxydeoxyguanosine in human hepatocarcinogenesis.
Dig Dis. 31:459–466. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Castello G, Costantini S and Scala S:
Targeting the inflammation in HCV-associated hepatocellular
carcinoma: a role in the prevention and treatment. J Transl Med.
8:1092010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hino K, Hara Y and Nishina S:
Mitochondrial reactive oxygen species as a mystery voice in
hepatitis C. Hepatol Res. 44:123–132. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Indo HP, Inanami O, Koumura T, et al:
Roles of mitochondria-generated reactive oxygen species on
X-ray-induced apoptosis in a human hepatocellular carcinoma cell
line, HLE. Free Radic Res. 46:1029–1043. 2012. View Article : Google Scholar
|
|
28
|
Lim W, Kwon SH, Cho H, et al: HBx
targeting to mitochondria and ROS generation are necessary but
insufficient for HBV-induced cyclooxygenase-2 expression. J Mol Med
(Berl). 88:359–369. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng AS, Chan HL, Leung WK, et al:
Expression of HBx and COX-2 in chronic hepatitis B, cirrhosis and
hepatocellular carcinoma: implication of HBx in upregulation of
COX-2. Mod Pathol. 17:1169–1179. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bu X and Zhao C: The association between
cyclooxygenase-2 1195 G/A polymorphism and hepatocellular
carcinoma: evidence from a meta-analysis. Tumour Biol.
34:1479–1484. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hernanz R, Briones AM, Salaices M and
Alonso MJ: New roles for old pathways? A circuitous relationship
between reactive oxygen species and cyclo-oxygenase in
hypertension. Clin Sci (Lond). 126:111–121. 2014. View Article : Google Scholar
|
|
32
|
Kiritoshi S, Nishikawa T, Sonoda K, et al:
Reactive oxygen species from mitochondria induce cyclooxygenase-2
gene expression in human mesangial cells: potential role in
diabetic nephropathy. Diabetes. 52:2570–2577. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nybo K: GFP imaging in fixed cells.
Biotechniques. 52:359–360. 2012. View Article : Google Scholar
|
|
34
|
Hu B, Tai A and Wang P: Immunization
delivered by lentiviral vectors for cancer and infectious diseases.
Immunol Rev. 239:45–61. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liechtenstein T, Perez-Janices N and
Escors D: Lentiviral vectors for cancer immunotherapy and clinical
applications. Cancers (Basel). 5:815–837. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee YI, Hwang JM, Im JH, et al: Human
hepatitis B virus-X protein alters mitochondrial function and
physiology in human liver cells. J Biol Chem. 279:15460–15471.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tang R, Kong F, Hu L, et al: Role of
hepatitis B virus X protein in regulating LIM and SH3 protein 1
(LASP-1) expression to mediate proliferation and migration of
hepatoma cells. Virol J. 9:1632012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Khattar E, Mukherji A and Kumar V: Akt
augments the oncogenic potential of the HBx protein of hepatitis B
virus by phosphorylation. FEBS J. 279:1220–1230. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cheng AS, Yu J, Lai PB, Chan HL and Sung
JJ: COX-2 mediates hepatitis B virus X protein abrogation of
p53-induced apoptosis. Biochem Biophys Res Commun. 374:175–180.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huh KW and Siddiqui A: Characterization of
the mitochondrial association of hepatitis B virus X protein, HBx.
Mitochondrion. 1:349–359. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hosler JP: The influence of subunit III of
cytochrome c oxidase on the D pathway, the proton exit pathway and
mechanism-based inactivation in subunit I. Biochim Biophys Acta.
1655:332–339. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Salvioli S, Bonafe M, Capri M, Monti D and
Franceschi C: Mitochondria, aging and longevity - a new
perspective. FEBS Lett. 492:9–13. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ray AM, Zuhlke KA, Levin AM, Douglas JA,
Cooney KA and Petros JA: Sequence variation in the mitochondrial
gene cytochrome c oxidase subunit I and prostate cancer in African
American men. Prostate. 69:956–960. 2009. View Article : Google Scholar
|
|
44
|
Ha HL and Yu DY: HBx-induced reactive
oxygen species activates hepatocellular carcinogenesis via
dysregulation of PTEN/Akt pathway. World J Gastroenterol.
16:4932–4937. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lee YI, Hwang JM, Im JH, et al: Human
hepatitis B virus-X protein alters mitochondrial function and
physiology in human liver cells. J Biol Chem. 279:15460–15471.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bae SH, Jung ES, Park YM, et al:
Expression of cyclooxygenase-2 (COX-2) in hepatocellular carcinoma
and growth inhibition of hepatoma cell lines by a COX-2 inhibitor,
NS-398. Clin Cancer Res. 7:1410–1418. 2001.PubMed/NCBI
|
















