Introduction
Histone deacetylase inhibitors (HDIs) unfold
specific effects against cancer cells. Their pleiotropic mode of
action is not fully understood but includes induction of cell cycle
arrest, direct activation of apoptotic pathways and reactivation of
epigenetically silenced tumor suppressor genes (1,2).
However, many genes are downregulated by HDI treatment (3). Often, this downregulation is mediated
via many non-histone target proteins of histone deacetylases
(HDACs) (4) such as TP53 or RUNX3
(5,6).
Despite the high clinical potential of HDIs against
hematological malignancies, their effectiveness against solid
tumors has not been proven yet (7,8).
Different HDIs were shown to induce CDKN1A (encoding the
cell cycle kinase inhibitor p21) expression and cell death in
colorectal carcinoma cell lines (9,10).
In a mouse model of colorectal cancer, inhibition of HDAC2 led to a
significant reduction in adenoma formation (11). Yet, clinically proven benefits of
HDI treatment are observed in individual cases only (7). However, solid cancer types in
clinical studies are diverse and so are the genetic aberrations in
these tumors. Hence, we sought to analyze HDI’s properties in one
specific type of cancer cells, i.e., colorectal carcinoma cell
lines characterized by constitutively active Wnt signaling.
Wnt signaling controls many cellular processes
during neoplastic transformation (12) and thus represents an important
therapeutic target. In a Wnt activated cell, the protein β-catenin,
the central player of the pathway, accumulates in the cytoplasm and
enters the nucleus. Nuclear β-catenin associates with transcription
factors of the TCF/LEF family and recruits transcriptional
co-activators and chromatin remodeling complexes which in concert
drive the expression of target genes (13). Wnt target genes include
CCND1, MMP7, VEGF, MYC, survivin
and also genes involved in feed-back regulation such as
TCF7L2 or LEF1. In many epithelial cancers, the Wnt
pathway is constitutively active due to mutations in different
components of the pathway as for instance almost every colorectal
carcinoma harbors either an APC or a β-catenin
mutation (14).
The mode of action of HDIs in respect to Wnt
signaling is not well defined. HDIs were shown to downregulate the
expression of single endogenous Wnt target genes, e.g.,
CCND1, VEGF and survivin (15,16).
In other studies activation of Wnt signaling due to HDI treatment
could be demonstrated (17). We
assessed the effects of HDI treatment in colorectal carcinoma cell
lines to further elucidate the mode of HDI action.
Materials and methods
Cell culture and drug treatment
Colorectal carcinoma cell lines SW480 and HCT116
were obtained from the American Type Culture Collection (LGC
Promochem, Wesel, Germany). Cells were maintained in Dulbecco’s
modified Eagle’s medium (DMEM) including 10% fetal bovine serum
(FBS), sodium pyruvate, L-glutamine and non-essential amino acids
(PAN Biotech, Aidenbach, Germany) at 37°C in a 7.5% CO2
humidified chamber. Cells were treated with 1 μM trichostatin A
(TSA; Sigma, Munich, Germany) or 5 μM suberoylanilide hydroxamic
acid (SAHA; Cayman Chemicals, Ann Arbor, MI, USA) dissolved in
ethanol and 1 mM valproic acid (VPA; Sigma) dissolved in PBS.
Vehicle treatment with either ethanol or PBS served as internal
control. The proteasome inhibitors MG132 and Epoxomicin (both from
Calbiochem, Darmstadt, Germany) were dissolved in DMSO.
Cellular assays
Cell growth and viability were evaluated by using
bromodeoxyuridine incorporation (BrdU) and methyl thiazol
tetrazolium bromide reduction (MTT) assays, respectively. Cells
were plated in triplicates in 96-well plates. After treatment with
HDIs and control substances for 24, 30 and 48 h or 24 and 48 h
after siRNA treatment, MTT or BrdU assay (BrdU cell proliferation
ELISA; Roche, Mannheim, Germany) was carried out. The mean
absorbance values of HDI-treated cells were normalized to the mean
absorbance values of vehicle-treated cells. To detect apoptotic
cells, the Apo-One homogeneous Caspase-3/7 assay (Promega, Madison,
WI, USA) was carried out according to the manufacturer’s
instructions.
For cell cycle analysis, cells were trypsinized
after 24 h HDI treatment or 48 h after siRNA transfection. The cell
cycle was measured via propidium iodide incorporation on a LSRII
flow cytometer and analyzed with FACS DiVa software (Becton
Dickinson, Franklin Lakes, NJ, USA). Data evaluation was performed
with FlowJo software (TreeStar Inc., Ashland, OR, USA).
HDAC knockdown - siRNA transfection
HDAC knockdown was performed with siGENOME SMARTpool
siRNA from Dharmacon for the genes HDAC6 (M-003499-00), HDAC10
(M-004072-00) and HDAC11 (M-004258-00). Transfection was carried
out with DharmaFECT1 (Dharmacon, Lafayette CO, USA) at a ratio of
1:40 in HCT116 and 1:50 in SW480 cells according to the
manufacturer’s instructions. Experiments were performed 24 to 72 h
after siRNA transfection. Specific target knockdown was measured on
mRNA level by quantitative real-time PCR after 24 or 48 h.
Reverse transcription real-time PCR
RNA isolation was carried out with the NucleoSpin
RNA II kit (Macherey-Nagel, Düren, Germany), cDNA synthesis was
performed with RevertAID H Minus First Strand cDNA Synthesis kit
(Fermentas, St. Leon-Rot, Germany). The ABsolute QPCR SYBR-Green
ROX Mix (Abgene, Epsom, UK) was used for quantitative real-time
PCRs on a GeneAmp 5700 Sequence detection system (Applied
Biosystems, Foster City, CA, USA). Primer sequences are stated in
Table I. Real-time PCR data was
analyzed using the ΔΔCt method whereby the relative expression
change of a treated sample over a control sample is: fold change =
2−ΔΔCt.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Primer name | Sequence 5′→3′ |
|---|
| LEF1 |
| Forward |
CTTGGTGAACGAGTCTGAAATC |
| Reverse |
GTGTTCTCTGGCCTTGTCGTG |
| TCF7L2 |
| Forward |
GGAGCCTCCAGAGTAGACAAG |
| Reverse |
CCACTGGCACTTTGTTAGAGAC |
| Cyclin D1 |
| Forward |
GCCTGAACCTGAGGAGCCCCA |
| Reverse |
GTCACACTTGATCACTCTGG |
| c-Myc |
| Forward |
TCCTGAGACAGATCAGCAACAACCG |
| Reverse |
TCCTCTGGCGCTCCAAGACGTT |
| VEGF |
| Forward |
CATGACAGCGCCCCTTCCTGG |
| Reverse |
TGTGAGGACATAGGTCCTTTTAGGCTG |
| MMP7 |
| Forward |
TGTTGCAGAATACTCACTATTTCC |
| Reverse |
GATCCACTGTAATATGCGGTAAG |
| GAPDH |
| Forward |
GCTCTCCAGAACATCATCCCTGCC |
| Reverse |
AGGCCATGCCAGTGAGCTTCC |
Western blot analysis
Total cell lysates were made from HCT116 or SW480
cells after treatment with HDIs for 3, 6 and 16 or 20 h. After SDS
gel electrophoresis, western blot analysis was used to determine
the relative protein levels of TCF7L2 and β-catenin in total cell
lysates. GAPDH was used as an internal control. The antibodies were
as follows: TCF7L2 (Cell Signaling Technology, Danvers, MA, USA)
1:2,500 in 5% BSA/TBST; β-catenin (BD Biosciences, Franklin Lakes,
NJ, USA) 1:10,000 in 0.5% SlimFast/TBST; GAPDH (Abcam) 1:30,000 in
5% milk/TBST. Band intensities were determined using AIDA software
(Raytest, Straubenhardt, Germany). The band intensity (BI) of the
analyzed protein was referred to the BI of GAPDH to give the
normalized BI. The normalized BI of control treated cells was set
to 100%.
Immunoprecipitation
For cytosolic and nuclear fractionation of cells,
HCT116 cells were grown in 10-cm dishes and HDI treated for 16 h.
Cells were washed with ice-cold PBS, scraped into a microcentrifuge
tube and centrifuged for 5 min at 1,600 rpm. The cell pellet was
resuspended in 100 μl membrane lysis buffer [10 mM HEPES, pH 8.0;
10 mM KCl; 1.5 mM MgCl2; 1 mM DTT; 1X Complete Protease
Inhibitor Cocktail (Roche)] and incubated for 15 min on ice. NP-40
was added to a final concentration of 1%. After vortexing for 10
sec, the sample was centrifuged for 3 min at 13,000 rpm. The
supernatant is the cytoplasmic fraction. The pellet containing the
nuclei was washed 3 times with 500 μl membrane lysis buffer and
centrifuged for 3 min. Then the pellet was resuspended in 100 μl
ice cold nuclear envelope lysis buffer [20 mM HEPES, pH 8.0; 1.5 mM
MgCl2; 25% glycerol; 0.42 M NaCl; 0.2 mM EDTA; 1 mM DTT;
1X Complete Protease Inhibitor Cocktail (Roche)] and vortexed for
30 sec. After rotation at 4°C for 30 min, samples were centrifuged
for 15 min at 13,000 rpm. The protein concentration of the
supernatant was determined by Bradford assay. Concentrations of
HDI- and vehicle-treated samples were adjusted to 3 μg/μl. Lysates
were used immediately or stored in aliquots at −80°C until use.
For immunoprecipitation, protein G coupled magnetic
beads (Dynabeads, Invitrogen) were used according to the
manufacturer’s instructions. In brief, 50 μl of beads were loaded
with 5 μg of antibody. After washing, the loaded beads were
incubated with cellular lysates for 10 to 20 min. After several
washing steps, the beads were resuspended in 5X SDS loading buffer
and heated to 70°C for 10 min.
To test the acetylation status of β-catenin,
immunoprecipitations were carried out with an anti-β-catenin
antibody (BD Biosciences) followed by western blot analysis with an
anti-β-catenin antibody or an anti-acetylated β-catenin antibody
(Cell Signaling Technology).
Statistical analysis
Statistical calculations were carried out with the
GraphPad QuickCalcs online calculator (www.graphpad.com/quickcalcs) and STATISTICA 8 software
(StatSoft, Hamburg, Germany) using different t-tests as indicated
in the text. A p-value of <0.05 was considered to indicate
statistical significance. Significant results are indicated by
*p<0.05; **p<0.01 or
***p<0.001 in the figures.
Results
The HDIs SAHA and TSA attenuate
proliferation and induce apoptosis in Wnt-activated colorectal
carcinoma cell lines
We analyzed the effects of the HDIs SAHA, TSA and
VPA on the growth of the colorectal carcinoma cell lines HCT116 and
SW480. Both cell lines possess an activated Wnt signaling pathway
due to mutations in β-catenin or APC, respectively.
SAHA and TSA belong to the chemical class of hydroxamic acids;
whereas VPA is a short-chain fatty acid. Treatment with hydroxamic
acids led to a strong reduction in the number of viable cells,
while VPA had no, or only little effect on viability even after 48
h as measured by MTT assay (Fig.
1A). Furthermore, a BrdU assay revealed a pronounced decrease
in the cell proliferation rate of SAHA- or TSA-treated cells as
opposed to VPA-treated cells (Fig.
1B). To gain further insights into the underlying mechanisms we
carried out an apoptosis detection assay. After treatment with
hydroxamic acids for 24 and 30 h, the number of apoptotic cells
strongly increased (Fig. 1C).
After 48 h, the relative apoptotic rate dropped due to the low
number of living cells. In contrast, VPA treatment induced no
significant changes in the apoptosis rate. These findings were
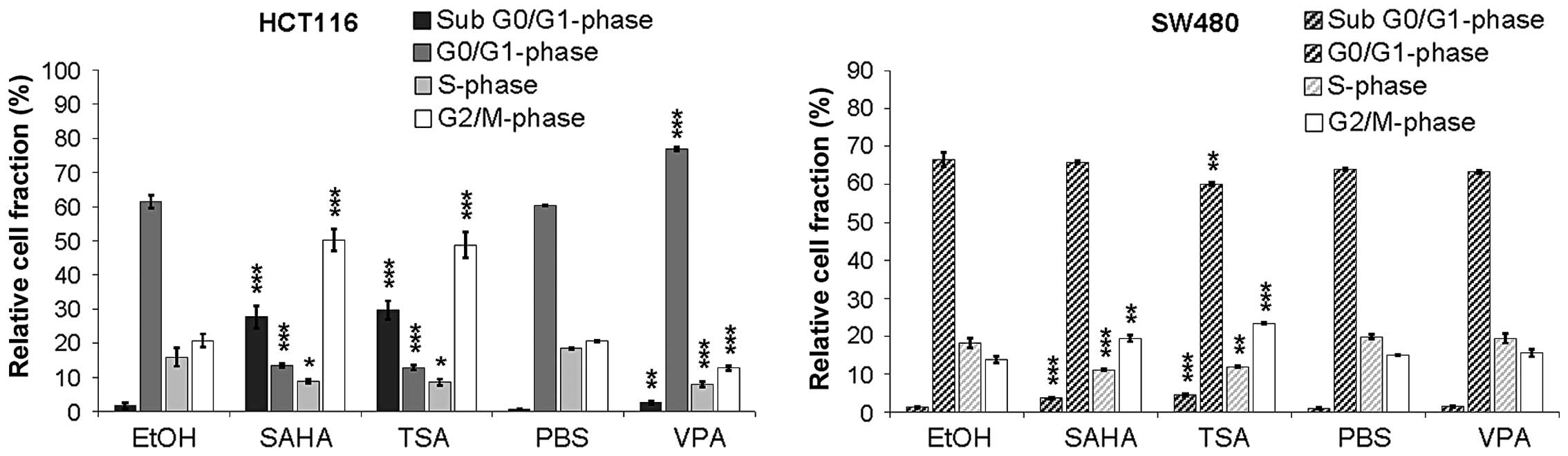
further underlined by flow cytometric cell cycle analysis revealing
an increase in the apoptotic cell fraction and an induction of a
G2/M-phase cell cycle arrest in response to TSA and SAHA (Fig. 2). In contrast, treatment with VPA
led to little, or no changes in the cell cycle.
The HDIs SAHA and TSA repress Wnt target
genes in colorectal carcinoma cells
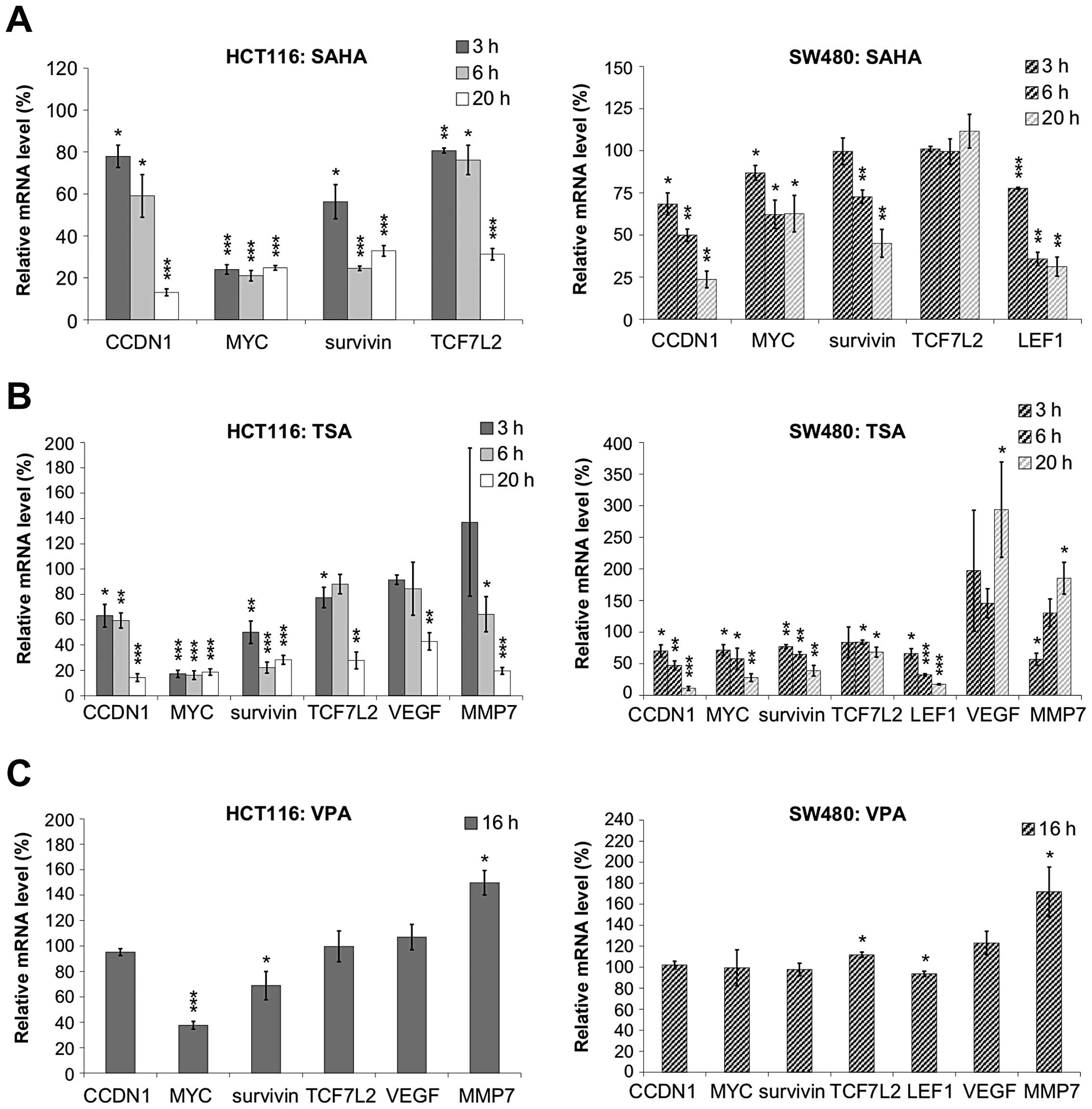
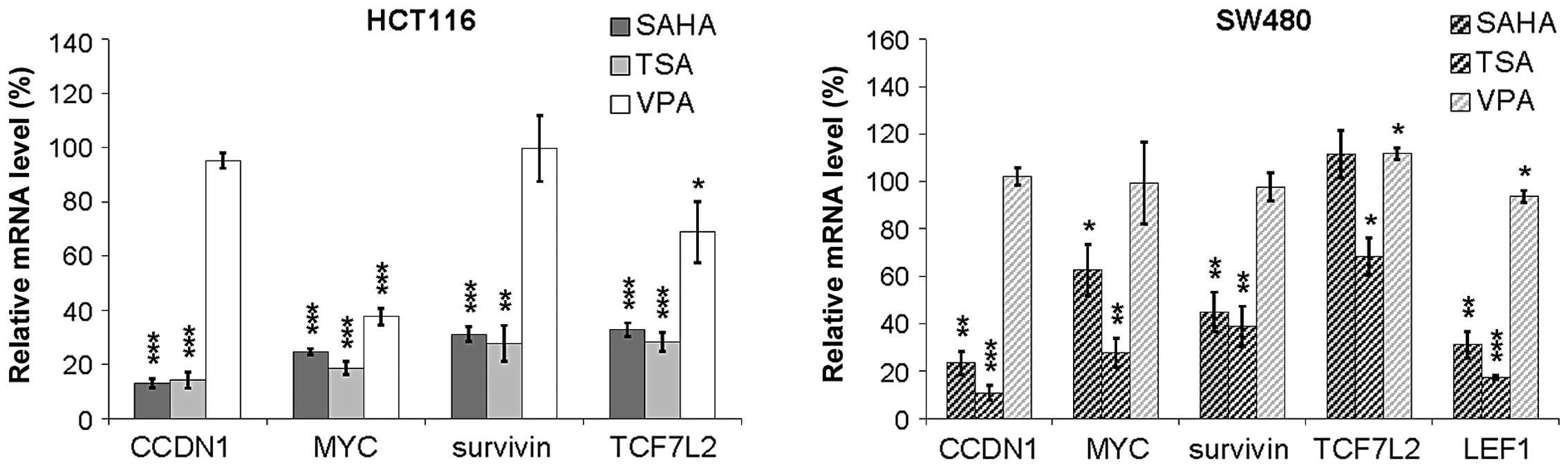
In order to explore how HDIs influence Wnt regulated
genes we measured the expression of endogenous Wnt target genes by
quantitative real-time PCR after treatment with HDIs (Fig. 3). Analyzed Wnt target genes
included MYC, CCDN1 (encoding cyclin D1),
VEGF, MMP7, survivin and the two transcription
factors LEF1 and TCF7L2 which are integral parts of
the pathway. We observed downregulation of all analyzed genes upon
20 h treatment with hydroxamic acids whereas VPA treatment left the
expression of most genes unchanged (Fig. 4). In HCT116 cells, the expression
of CCDN1, MYC, VEGF, MMP7,
survivin and TCF7L2 was significantly downregulated
after 16 to 20 h exposure to SAHA or TSA, but not to VPA. In SW480
cells, the expression levels of CCDN1, MYC,
survivin and LEF1 were significantly downregulated by
SAHA or TSA, but not by VPA treatment. Only MYC expression, which
is known to be regulated by a variety of mechanisms (18), was significantly downregulated by
VPA treatment in HCT116.
β-catenin acetylation status remains
unchanged in response to TSA treatment
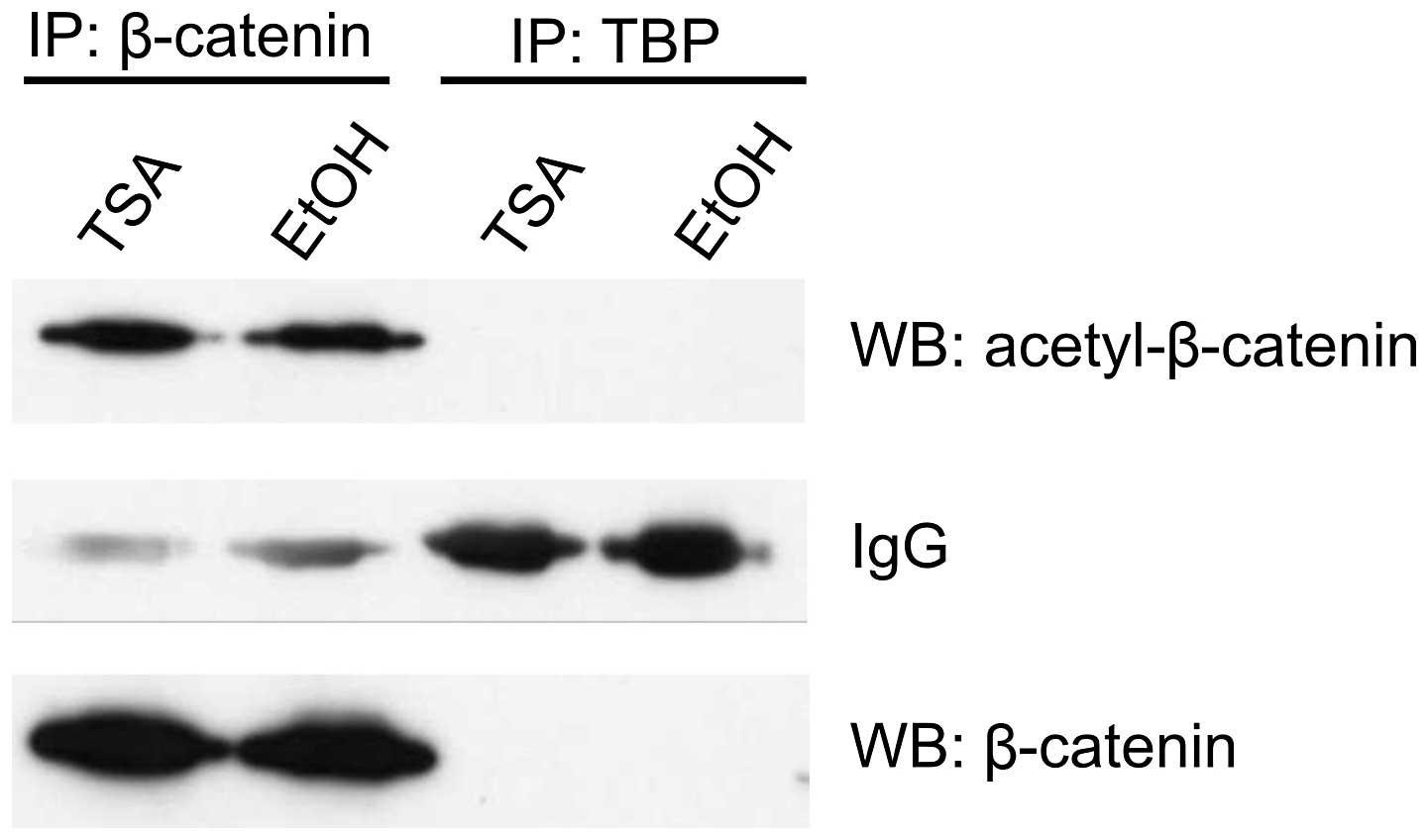
Based on recent findings that the β-catenin protein
can be acetylated, we questioned if a modification of this central
player of the Wnt pathway may be involved in the transcriptional
downregulation of target genes in response to HDI treatment.
Immunoprecipitation experiments with anti-acetylated β-catenin
antibody and nuclear lysates from HCT116 cells after 16 h TSA
treatment revealed no difference in the amount of precipitated
acetylated β-catenin between TSA- and vehicle-treated cells
(Fig. 5). Similarly, there was no
change in the total amount of β-catenin protein in total cell
lysates.
The HDIs SAHA and TSA induce TCF7L2
depletion
We investigated if TCF7L2 might be a non-histone
target of HDACs by analyzing the relative TCF7L2 protein level in
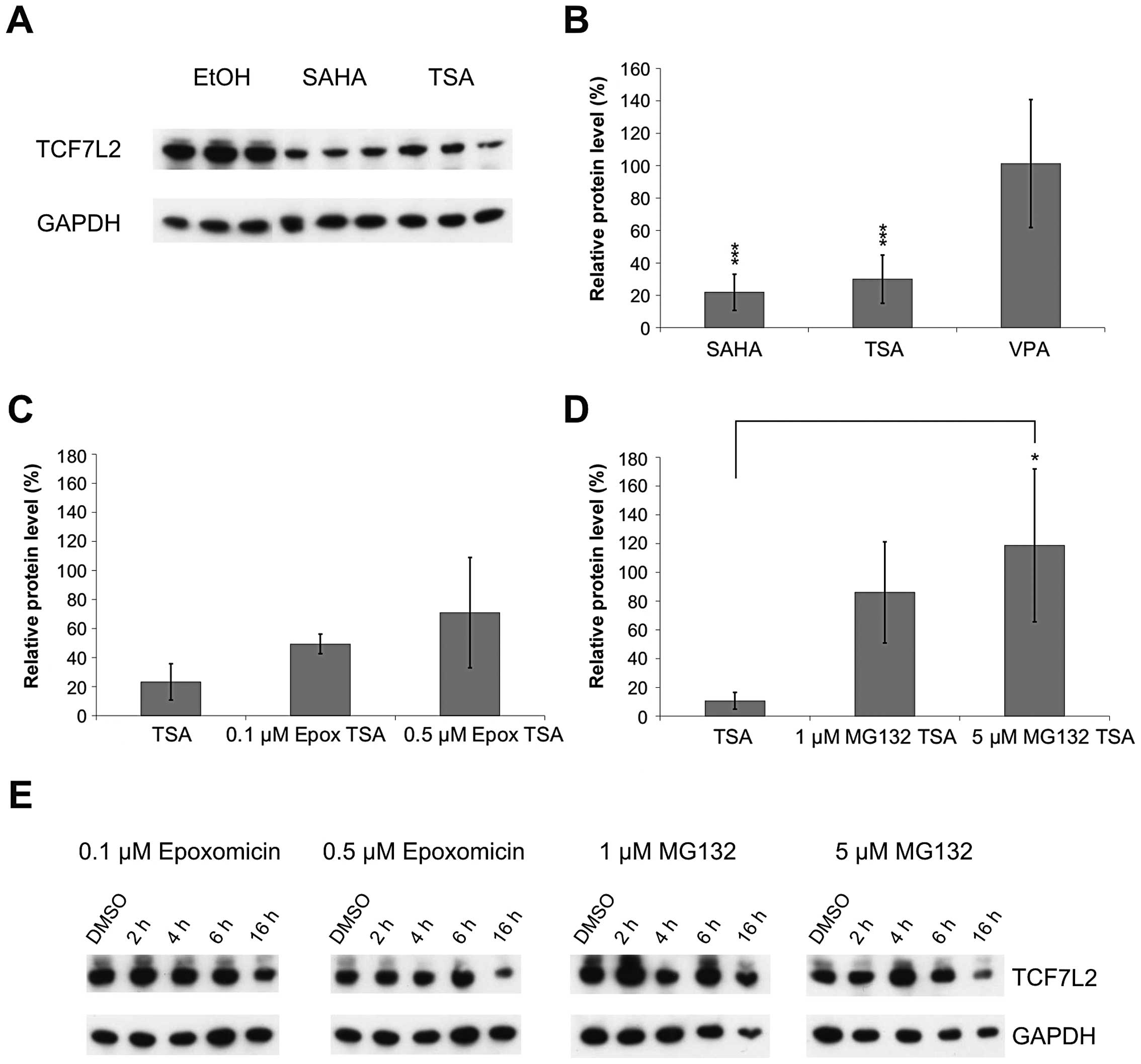
total HCT116 cell lysates after HDI treatment. We found a strong
depletion of TCF7L2 protein after 20 h SAHA and TSA, but not after
VPA treatment (p<0.001) (Fig.
6). This TCF7L2 depletion could not be attributed to
transcriptional downregulation alone, as, according to the N-end
rule (19), the estimated
half-life of the TCF7L2 protein is 30 h. Hence, TCF7L2 depletion
might be partly caused by proteasomal degradation. In this case,
co-treatment with proteasome inhibitors should attenuate the effect
of the HDI treatment. We treated HCT116 cells with TSA for 16 h and
added one of the proteasome inhibitors Epoxomicin or MG132 4 or 6 h
before the end of the treatment, respectively. As control we
treated the cells with the proteasome inhibitors alone.
Co-treatment with HDIs and proteasome inhibitors indeed attenuated
the downregulation of TCF7L2 protein level (Fig. 6C and D). Treatment with proteasome
inhibitors alone did not alter the TCF7L2 level substantially
(Fig. 6E), which showed that
TCF7L2 was not subjected to constant proteasomal degradation under
normal conditions. Thus we demonstrated that HDIs of the
hydroxamate class induce a strong proteasome-dependent depletion of
TCF7L2.
Knockdown of single HDACs cannot mimic
the HDI-induced attenuation of cell proliferation
The previous experiments showed that the hydroxamic
acids SAHA and TSA, which inhibit all known HDACs, induced
apoptotic cell death of colon carcinoma cells and attenuation of
Wnt signaling. In contrast, treatment with VPA, which inhibits only
some HDAC classes, led to little, or no change, in the colon
carcinoma cell lines. We reasoned that the different effects of the
HDIs might be caused by the differences in their HDAC inhibition
profiles. Therefore, we carried out siRNA experiments to analyze
the effects of the knockdown of HDACs 6, 10 and 11, which are
inhibited by hydroxamic acids but not by VPA, on Wnt signaling and
colon cancer cell proliferation.
We controlled for a proper gene knockdown by
measuring the mRNA expression level of the three HDACs 24 and 48 h
after siRNA transfection. All three HDAC siRNAs reduced the
respective HDAC expression to at least 40% after 48 h treatment. By
using MTT, BrdU, apoptosis assay and flow cytometric analysis, we
found that none of the analyzed HDAC siRNAs induced any significant
changes in proliferation or apoptosis of HCT116 and SW480 cells.
This indicated that the knockdown of a single HDAC is not
sufficient to induce major changes in cell proliferation of the
analyzed colon carcinoma cell lines.
Knockdown of HDAC 6, 10 and 11 affects
Wnt target gene expression
Next we examined whether siRNA mediated HDAC
knockdown had an effect on Wnt target gene expression. Therefore,
we performed quantitative real-time PCR of the endogenous Wnt
target genes CCDN1, MYC, survivin, LEF1
and TCF7L2 48 h after HDAC siRNA transfection.
In HCT116 cells, HDAC 6 or 10 knockdown reduced the
expression of several Wnt target genes, whereas HDAC11 knockdown
had an effect on all analyzed genes (Fig. 7). In SW480 cells, the transfection
with HDAC10 siRNA led to a significant decrease in all analyzed Wnt
target genes, while the siRNAs for HDAC 6 or 11 reduced only the
expression of two of the five analyzed genes (MYC and LEF1 or LEF1
and TCF7L2, respectively) (Fig.
7).
Western blot analysis was performed to determine the
effects of HDAC knockdown on the TCF7L2 protein level. The
knockdown of HDACs 6 or 10 led to a reduction of the TCF7L2 protein
level to approximately 50% of the initial protein level after 72 h
(p<0.001 and p<0.01) (Fig.
8); while HDAC11 knockdown only slightly decreased the TCF7L2
protein level. Thus, HDACs 6 and 10 may be involved in the SAHA and
TSA induced proteasomal degradation of TCF7L2 leading to the
reduced expression of Wnt target genes.
Discussion
Through the regulation of a vast number of different
target genes, active Wnt signaling is implicated in different
aspects of tumorigenesis, including cell proliferation, apoptosis,
angiogenesis and metastasis.
In this study, we observed the rapid induction of
apoptotic colorectal carcinoma cell death and transcriptional
down-regulation of Wnt target genes upon treatment with the
hydroxamic acid HDIs SAHA and TSA. While SAHA and TSA rapidly
induced apoptosis and cell death in colorectal carcinoma cells, we
noted only slight effects upon VPA treatment. Likewise, APC
mutant cells were relatively insensitive to VPA-induced apoptosis
(20). However, VPA has been shown
to be a potent inducer of apoptosis in various cancer models,
including colorectal carcinoma cell lines (21), when cell viability was determined
after 72-h treatment. In contrast, we found that after 24 and 48 h
of treatment HCT116 and SW480 cells were much more sensitive to
hydroxamic acid HDIs than to the fatty acid VPA.
The transcriptional regulation of Wnt target genes
also displayed a differential mode of regulation upon HDI
treatment. While TSA and SAHA downregulated Wnt target genes, VPA
had no effects on the expression of most genes. In several
different cellular contexts, the downregulation of single Wnt
targets such as CCND1 or survivin upon HDI treatment
has been described (22,23). MMP7 expression was reduced
by TSA and butyrate in chondrosarcoma cells (24). In addition, we found the two Wnt
transcription factors TCF7L2 and LEF1 to be
downregulated by hydroxamic acids. However, as all HDIs induced
histone hyperacetylation, the impact of HDIs on the Wnt pathway
must be explained by a mechanism independent of histone
hyperacetylation.
Recently, a growing number of non-histone targets of
HDACs has been described, one of them being p53. Acetylation of p53
increases its binding affinity to target promoters and also its
transactivation during DNA damage response (5). β-catenin also was shown to be a
target of acetylation (25) which
increases its affinity for TCF/LEF factors (26). However, we did not find a change in
the β-catenin acetylation level upon TSA treatment.
Another non-histone target of HDACs is HSP90. HDI
treatment disrupts HSP90 client protein interactions with
subsequent proteasomal degradation of client proteins, e.g.,
Bcr-Abl (27). We analyzed if
TCF7L2 might be regulated in a similar mode as HSP90 client
proteins. Interestingly, TSA and SAHA induced depletion of TCF7L2
which could be attenuated by co-treatment with proteasome
inhibitors. Given that TCF7L2 is the main transcription factor
mediating Wnt target gene expression in colorectal carcinoma cells
(28), TCF7L2 depletion might be
the primary event in the observed Wnt target gene
downregulation.
To elucidate the role of HDACs 6, 10 and 11, which
are inhibited by hydroxamic acids but not by VPA (29), in Wnt signaling, we studied Wnt
target gene expression and TCF7L2 protein level by siRNA
experiments. Single knockdowns of HDACs 6, 10 and 11 reduced the
expression of at least some of the analyzed Wnt target genes. In
addition, knockdown of HDACs 6 or 10 induced a strong reduction in
TCF7L2 protein level, while knockdown of HDAC11 induced a minor
reduction. These results suggest that inhibition of HDACs 6 and 10
leads to the attenuation of Wnt signaling through the depletion of
its main transcription factor TCF7L2. Interestingly, it was
previously published that inhibition of HDACs 6 and 10 induces
hyperacetylation of HSP90 leading to release and degradation of
client proteins like VEGFR (30–32).
Moreover, HSP90 inhibitors were shown to downregulate Wnt target
gene expression (33). Taken
together our data could indicate that TCF7L2 might be an HSP90
client protein, which has not been described yet.
To sum up the mode of HDI action on Wnt signaling,
we suggest the following line of events: HDIs of the hydroxamate
class inhibit the predominantly cytosolic HDACs 6 and 10 which
results in hyperacetylation of a yet unidentified interaction
partner of TCF7L2; thereby the release of TCF7L2 from its
interaction partner is induced and TCF7L2 is subjected to
proteasomal degradation. Concomitantly, Wnt target gene expression
is downregulated and colorectal carcinoma cell death is induced. In
numerous studies, the attenuation of Wnt signaling results in
cancer cell death, e.g., TCF7L2 antisense RNA inhibits
proliferation and in vivo tumor formation of liver cancer
cell lines (34). The
re-introduction of wild-type APC induces apoptosis in
colorectal carcinoma cells (35).
Likewise, a Wnt1 antibody or small molecule inhibitors of Wnt
signaling stimulate apoptosis in different types of human cancer
cells (36,37). Other reports show that active Wnt
signaling inhibits apoptosis in cancer cells (38). Thus, TCF7L2 depletion and
attenuation of Wnt signaling renders cancer cells sensitive to
apoptotic stimuli.
In conclusion, we describe the depletion of TCF7L2
by HDIs of the hydroxamate class, through the inhibition of HDAC6
and HDAC10, leading to an attenuation of Wnt signaling. TCF7L2
degradation might be the primary event in the observed Wnt target
gene downregulation and apoptotic colorectal carcinoma cell death.
These findings provide a molecular rationale for the use of HDIs
against colorectal carcinomas with activated Wnt signaling.
Acknowledgements
S.G. was supported by a predoctorial grant of the
Hans-Bockler Stiftung.
References
|
1
|
Insinga A, Monestiroli S, Ronzoni S, et
al: Inhibitors of histone deacetylases induce tumor-selective
apoptosis through activation of the death receptor pathway. Nat
Med. 11:71–76. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schrump DS: Cytotoxicity mediated by
histone deacetylase inhibitors in cancer cells: mechanisms and
potential clinical implications. Clin Cancer Res. 15:3947–3957.
2009. View Article : Google Scholar
|
|
3
|
Rada-Iglesias A, Enroth S, Ameur A, et al:
Butyrate mediates decrease of histone acetylation centered on
transcription start sites and down-regulation of associated genes.
Genome Res. 17:708–719. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu J and Colburn NH: Histone deacetylase
inhibition down-regulates cyclin D1 transcription by inhibiting
nuclear factor-kappaB/p65 DNA binding. Mol Cancer Res. 3:100–109.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luo J, Li M, Tang Y, Laszkowska M, Roeder
RG and Gu W: Acetylation of p53 augments its site-specific DNA
binding both in vitro and in vivo. Proc Natl Acad Sci USA.
101:2259–2264. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jin YH, Jeon EJ, Li QL, et al:
Transforming growth factor-beta stimulates p300-dependent RUNX3
acetylation, which inhibits ubiquitination-mediated degradation. J
Biol Chem. 279:29409–29417. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Piekarz RL and Bates SE: Epigenetic
modifiers: basic understanding and clinical development. Clin
Cancer Res. 15:3918–3926. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim HJ and Bae SC: Histone deacetylase
inhibitors: molecular mechanisms of action and clinical trials as
anti-cancer drugs. Am J Transl Res. 3:166–179. 2011.PubMed/NCBI
|
|
9
|
Kobayashi H, Tan EM and Fleming SE:
Acetylation of histones associated with the p21WAF1/CIP1 gene by
butyrate is not sufficient for p21WAF1/CIP1 gene transcription in
human colorectal adenocarcinoma cells. Int J Cancer. 109:207–213.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen YX, Fang JY, Zhu HY, Lu R, Cheng ZH
and Qiu DK: Histone acetylation regulates p21WAF1 expression in
human colon cancer cell lines. World J Gastroenterol. 10:2643–2646.
2004.PubMed/NCBI
|
|
11
|
Zhu P, Martin E, Mengwasser J, Schlag P,
Janssen KP and Gottlicher M: Induction of HDAC2 expression upon
loss of APC in colorectal tumorigenesis. Cancer Cell. 5:455–463.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reya T and Clevers H: Wnt signalling in
stem cells and cancer. Nature. 434:843–850. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Willert K and Jones KA: Wnt signaling: is
the party in the nucleus? Genes Dev. 20:1394–1404. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Morin PJ, Sparks AB, Korinek V, et al:
Activation of beta-catenin-Tcf signaling in colon cancer by
mutations in beta-catenin or APC. Science. 275:1787–1790. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lallemand F, Courilleau D, Sabbah M,
Redeuilh G and Mester J: Direct inhibition of the expression of
cyclin D1 gene by sodium butyrate. Biochem Biophys Res Commun.
229:163–169. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim MS, Kwon HJ, Lee YM, et al: Histone
deacetylases induce angiogenesis by negative regulation of tumor
suppressor genes. Nat Med. 7:437–443. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bordonaro M, Lazarova DL, Augenlicht LH
and Sartorelli AC: Cell type- and promoter-dependent modulation of
the Wnt signaling pathway by sodium butyrate. Int J Cancer.
97:42–51. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Heruth DP, Zirnstein GW, Bradley JF and
Rothberg PG: Sodium butyrate causes an increase in the block to
transcriptional elongation in the c-myc gene in SW837 rectal
carcinoma cells. J Biol Chem. 268:20466–20472. 1993.PubMed/NCBI
|
|
19
|
Varshavsky A, Bachmair A and Finley D: The
N-end rule of selective protein turnover: mechanistic aspects and
functional implications. Biochem Soc Trans. 15:815–816.
1987.PubMed/NCBI
|
|
20
|
Huang X and Guo B: Adenomatous polyposis
coli determines sensitivity to histone deacetylase
inhibitor-induced apoptosis in colon cancer cells. Cancer Res.
66:9245–9251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mologni L, Cleris L, Magistroni V, et al:
Valproic acid enhances bosutinib cytotoxicity in colon cancer
cells. Int J Cancer. 124:1990–1996. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jin JS, Tsao TJ, Sun PC, Yu CT and Tzao C:
SAHA inhibits the growth of colon tumors by decreasing histone
deacetylase and the expression of cyclin D1 and survivin. Pathol
Oncol Res. 18:713–720. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hsu YF, Sheu JR, Lin CH, et al:
Trichostatin A and sirtinol suppressed survivin expression through
AMPK and p38MAPK in HT29 colon cancer cells. Biochim Biophys Acta.
1820:104–115. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Young DA, Lakey RL, Pennington CJ, et al:
Histone deacetylase inhibitors modulate metalloproteinase gene
expression in chondrocytes and block cartilage resorption.
Arthritis Res Ther. 7:R503–R512. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wolf D, Rodova M, Miska EA, Calvet JP and
Kouzarides T: Acetylation of beta-catenin by CREB-binding protein
(CBP). J Biol Chem. 277:25562–25567. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Levy L, Wei Y, Labalette C, et al:
Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4
interaction. Mol Cell Biol. 24:3404–3414. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nimmanapalli R, Fuino L, Bali P, et al:
Histone deacetylase inhibitor LAQ824 both lowers expression and
promotes proteasomal degradation of Bcr-Abl and induces apoptosis
of imatinib mesylate-sensitive or -refractory chronic myelogenous
leukemia-blast crisis cells. Cancer Res. 63:5126–5135. 2003.
|
|
28
|
Korinek V, Barker N, Morin PJ, et al:
Constitutive transcriptional activation by a beta-catenin-Tcf
complex in APC−/− colon carcinoma. Science.
275:1784–1787. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bali P, Pranpat M, Bradner J, Balasis M,
et al: Inhibition of histone deacetylase 6 acetylates and disrupts
the chaperone function of heat shock protein 90: a novel basis for
antileukemia activity of histone deacetylase inhibitors. J Biol
Chem. 280:26729–26734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Park JH, Kim SH, Choi MC, et al: Class II
histone deacetylases play pivotal roles in heat shock protein
90-mediated proteasomal degradation of vascular endothelial growth
factor receptors. Biochem Biophys Res Commun. 368:318–322. 2008.
View Article : Google Scholar
|
|
32
|
New M, Olzscha H and La Thongue NB: HDAC
inhibitor-based therapies: Can we interpret the code? Mol Oncol.
6:637–656. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kurashina R, Ohyashiki JH, Kobayashi C, et
al: Anti-proliferative activity of heat shock protein (Hsp) 90
inhibitors via beta-catenin/TCF7L2 pathway in adult T cell leukemia
cells. Cancer Lett. 284:62–70. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiang Y, Zhou XD, Liu YK, et al: Antisense
Tcf inhibits the neoplastic growth of liver cancer cells. J Cancer
Res Clin Oncol. 130:671–678. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morin PJ, Vogelstein B and Kinzler KW:
Apoptosis and APC in colorectal tumorigenesis. Proc Natl Acad Sci
USA. 93:7950–7954. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
He B, You L, Uematsu K, et al: A
monoclonal antibody against Wnt-1 induces apoptosis in human cancer
cells. Neoplasia. 6:7–14. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Minke KS, Staib P, Puetter A, et al: Small
molecule inhibitors of WNT signaling effectively induce apoptosis
in acute myeloid leukemia cells. Eur J Haematol. 82:165–175. 2009.
View Article : Google Scholar
|
|
38
|
Chen S, Guttridge DC, You Z, et al: Wnt-1
signaling inhibits apoptosis by activating beta-catenin/T cell
factor-mediated transcription. J Cell Biol. 152:87–96. 2001.
View Article : Google Scholar : PubMed/NCBI
|