Introduction
Macrophage migration inhibitory factor (MIF), a
cytokine first identified in 1966, is expressed fairly ubiquitously
and has both extracellular and intracellular roles (reviewed in
ref. 1). MIF has been implicated
in auto-immune and infectious disease, and cancer (reviewed in ref.
1). The causative role of MIF in
cancer progression was initially linked to its increased expression
by a variety of cancer cells, including prostate, colon,
hepatocellular, lung, ovarian, in addition to melanoma,
glioblastoma and neuroblastoma (2). In these cancers, MIF overexpression
has been associated with a concomitant increase in: a) tumor
invasion/migration, b) metastasis and c) angiogenesis. More recent
data have additionally identified the role of the host MIF in
regulating tumor growth. In support of the latter, the role of MIF
in regulating tumor angiogenesis in the B16–F10 melanoma model has
recently been investigated (3). A
combination either of B16–F10 or of shRNA targeting of MIF in the
same melanoma cells with a MIF knockout (−/−) genetic background,
resulted in significant reduction of tumor growth (by 47%), when
compared to wild-type mice. Furthermore, reduced growth of CT26
colon tumors (by 75%) in MIF−/− mice was reported
(4). Finally, host
macrophage-produced MIF was shown to polarize tumor-associated
macrophages (TAMs) towards an immunosuppressive M2 phenotype,
whereas MIF inhibition or gene loss (MIF−/−) reversed
TAM functionalities to M1-type (5). These studies, in conjunction with the
previous reports, establish MIF as a promising anticancer drug
target.
An increase of MIF levels positively correlates with
a poor prognosis in cancer (6–8).
Anti-MIF neutralizing antibodies (Abs), MIF-directed siRNA/shRNA or
antisense oligonucleotides and compounds hindering MIF secretion
have been tested in vitro and in preclinical models with
notable results (9,10). A more attractive approach to
decrease MIF upregulation is the utilization of small molecule MIF
inhibitors, which advantageously block the activity of both cancer
cell- and host cell-secreted MIF. ISO-1, the ‘gold standard’
inhibitor of MIF, was designed to selectively ligate the
tautomerase catalytic site of MIF which is known to neutralize its
pro-inflammatory activity (1,11,12).
In vitro MIF reduction by ISO-1, has been reported to
effectively reduce cancer cell proliferation, migration, and
invasion of the human lung adenocarcinoma A549 (10,13)
decrease the proliferation and invasiveness of prostate cancer
DU-145 cells (9), restore contact
inhibition of proliferation of LN 229 and LN -18 glioblastoma cells
(14), reduce cell migration and
invasion of HS683 glioma cells (15), and suppress the proliferation of
the murine colorectal cancer cells CT-26 (16). Previous studies have also addressed
the role of ISO-1 in prostate and colorectal cancer in vivo
(9,16). In both mouse models, ISO-1
treatment resulted in significant reduction of the tumor volume or
weight, despite the lack of an optimal dosing regimen.
In our search for more effective MIF inhibitors, we
herein identify ISO-66, as a potent inhibitor of MIF. We show that
ISO-66 enhances the cytotoxicity of human lymphocytes in
vitro and when administered to mice with established tumors
(melanoma and of the colon), is effective in suppressing tumor
growth in vivo. We believe ISO-66 offers promise as a
MIF-reducing therapy in cancer and other MIF-implicated
diseases.
Materials and methods
Preparation of ISO-66
All solvents were HPL C-grade from Fisher
Scientific. Silica gel (Selecto Scientific, 32–63 μm average
particle size) was used for flash column chromatography (FCC).
Aluminum-backed Silica Gel 60 with a 254-nm fluorescent indicator
TLC plates were used. Spots on TLC plates were visualized under a
short wavelength UV lamp or stained with I2 vapor. NMR
spectra were collected on a Jeol Eclipse 400 spectrometer at 400
MHz for 1H NMR spectra and 100 MHz for the
13C NMR spectra. Coupling constants are reported in
Hertz (Hz), and chemical shifts are reported in parts per million
(ppm) relative to deuterated solvent peak. The coupling constants
(J) are measured in Hertz (Hz) and assigned as s (singlet),
d (doublet), t (triplet), m (multiplet) and br (broad).
High-resolution mass spectra were carried out at the Mass
Spectrometry Facility at the Hunter College of the City University
of New York.
To a solution of 3-fluoro-4-hydroxybenzaldoxime (1
g; 6.45 mmol) in anhydrous DMF (120 ml) was added NCS (1.03 g; 7.74
mmol). The reaction mixture was stirred for 5 h at room temperature
affording the chloro oxime. To this solution, 4-penten-2-one (1.0
ml; 9.7 mmol) (oxidize 4-penten-2-ol with PCC in DCM) was added,
followed by the dropwise addition of triethylamine (1.34 ml; 9.68
mmol) in DMF (12 ml). The reaction mixture was stirred under
N2 at room temperature for 48 h. The solvent was removed
in vacuo and the residue was taken up in EtOAc. The EtOAc
solution was washed with 0.5 NHCl, water, brine, and dried with
anhydrous MgSO4. The filtrate was evaporated to dryness
and the residue was purified by FCC (hexane:EtOAc 4:3) to yield
ISO-66 as a pale yellow solid (0.6 g; 39%). 1H NMR (500
MHz, Methanol-d4) δ 7.40 (d, 1H), 7.30 (d, 1H), 6.96 (m,
1H), 5.05 (m, 1H), 3.53 (m, 1H), 3.03 (m, 2H), 2.86 (m, 1H), 2.21
(s, 3H); 13C NMR (125 MHz, Methanol-d4) δ
31.43, 42.08, 79.24, 116.07, 116.23, 119.85, 123.54, 125.70,
149.46, 152.76, 154.68, 158.84, 209.48; HR-MS(ES) m/z (M+H)
238.0871 (found); 238.0873 (calculated). Compound KF III 53Y, the
prodrug of ISO-66, was prepared from the ISO-acid chloride with
bis-trimethylsilyl malonate via methods reported in the literature
(17,18). KF III 53Y has better solubility
than ISO-66, but undergoes fast decarboxlyation to form ISO-66 upon
formulation in buffer.
MIF tautomerase inhibition assay
MIF tautomerase activity was measured using a
UV-Visible spectrophotometer (Shimadzu, UV1600U). A fresh stock
solution of L-dopachrome methyl ester was prepared at 2.4 mM
through oxidation of L-3,4-dihydroxyphenylalanine methyl
ester with sodium periodate. One μl of MIF solution (800–900 μg/ml)
and 1 μl of a dimethyl sulfoxide (DMSO) solution with various
concentrations of the MIF inhibitor were added into a plastic
cuvette (10 mm, 1.5 ml) containing 0.7 ml assay buffer (50 mM
potassium phosphate, pH 7.2). Then, activated L-dopachrome
methyl ester solution (0.3 ml) was added to the assay buffer
mixture. Activity was determined at room temperature and
spectrometric measurements were made every 5 sec at λ = 475 nm for
total 20 sec, by monitoring the rate of decolorization of
L-dopachrome in comparison to a standard solution.
Crystallization and X-ray data
collection
Recombinant human MIF was expressed in E.
coli and purified as described (19). Briefly, cells were lysed using a
French Pressure Cell, the lysate was clarified by centrifugation,
and the supernatant liquid was filtered with a 0.22-μm filter. The
filtered supernatant was purified by ion-exchange in 20 mM Tris (pH
7.5), 20 mM NaCl using a DEAE and an SP column connected in series.
MIF is found in the flow-through. Flow-through fractions were
collected, and fractions containing pure MIF were pooled and
concentrated.
A stock solution of 1.2 mM MIF in 20 mM Tris (pH
7.5), 20 mM NaCl, and a stock of 0.10 M KF III 53Y (the
carboxylated derivative/prodrug, of ISO-66) in DMSO were used to
prepare a solution of 1.1 mM MIF, 10 mM KF III 53Y, 18 mM Tris (pH
7.5), 18 mM NaCl, 10% DMSO. The KF III 53Y spontaneously
decarboxylated non-enzymatically, forming ISO-66. Crystallization
was performed using the hanging-drop vapor diffusion method. Two μl
of the MIF-ISO-66 complex was mixed with an equal volume of
reservoir comprised of 2 M
(NH4)2SO4, 0.1 M Tris (pH 7.5), 3%
isopropanol. Crystals grew in three to five weeks. X-ray
diffraction data were collected from a single crystal at station
X29 of the National Synchrotron Light Source at Brookhaven National
Laboratory. Crystal dimensions were ~0.40×0.25×0.23 mm. Immediately
prior to mounting the crystal, it was soaked briefly in
cryoprotective solution containing 2.25 M NaCl, 2 M
(NH4)2SO4, 50 mM Tris (pH 7.5). An
ω-sweep of 100° of data were collected with 1.0809 Å radiation.
Data were processed using HKL2000. Data collection statistics are
presented in Table I.
 | Table ICrystallographic statistics. |
Table I
Crystallographic statistics.
| Integration and
scaling | |
|---|
| Space group |
P3121 |
| Unit cell | a = b = 95.46 Å, c
= 104.55 Å, α = β = 90°, γ = 120° |
| Resolution, Å
(highest shell) | 1.55
(1.61–1.55) |
| Unique
reflections | 79,934 (7,885) |
| Completeness,
% | 99.6 (99.2) |
| Redundancy | 5.8 (5.3) |
| Avg. I/Avg. σ | 33.2 (2.25) |
|
Rmerge | 0.058 (0.534) |
|
| Refinement | |
|
| R (working) | 19.9% |
| R-free | 22.0% |
| RMS deviations from
idealitya | |
| Bond lengths
(Å) | 0.010 |
| Bond angles
(°) | 1.273 |
|
| Average
B-factorsb, Å2 | |
|
| Overall | 25.577 |
| Protein (number of
residues) | 24.061 (114×3
chains) |
| Water | 35.923 (281) |
| Chloride | 34.078 (6) |
| Sulfate | 28.648 (1) |
| Inhibitors | 39.299 (4) |
Structure determination
The program AMoRe was used for molecular
replacement. Protein coordinates from the complex of MIF with
OXIM-11 (20) was used as the
search model. Refinement was performed primarily using the program
CNS (21). Refmac5 was also used
for refinement. Occupancy refinement was performed using CNS.
Refined occupancies were normalized so that the sum of the
occupancies of corresponding atoms was 1.0. A TLS model was
employed in the Refmac5 refinement. The initial TLS model was found
using the TLSMD web server. Manual manipulation of the model was
performed using O and COOT. The structure was deposited into the
RCSB Protein Data Bank. Crystallographic statistics are presented
in Table I.
Cell lines and culture conditions
The human FM3 (melanoma), HCT-116 (colorectal
carcinoma), K562 (leukemia), Daudi (Burkitt’s lymphoma), and the
murine B16-F1 (melanoma), CT-26.WT (colorectal carcinoma), YAC-1
(lymphoma), WE HI 164 (fibrosarcoma) cell lines, peripheral blood
mononuclear cells (PBMCs) and their subpopulations and mouse spleen
cells were cultured in RP MI-1640, supplemented with 10%
heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, 10 mM
Hepes, 50 μM β-mercaptoethanol, 5 μg/ml gentamycin, 10 U/ml
penicillin and 10 U/ml streptomycin (all from Lonza, Cologne,
Germany) (thereafter referred to as complete medium), at 37°C, in a
humidified 5% CO2 incubator. ISO-66 was dissolved in
DMSO at 50 mM, aliquoted and stored at −20°C. Serial dilutions of
stock solutions were freshly prepared prior to their use.
Cell isolation and separation
Buffy coats were collected from 7 healthy
blood-donors. Prior to blood withdrawal, individuals gave their
informed consent according to the regulations approved by the 2nd
Peripheral Blood Transfusion Unit and Haemorophilic Centre,
‘Laikon’ General Hospital Institutional Review Board, Athens,
Greece.
PBMCs were isolated by centrifugation over
Ficoll-Histopaque (Lonza) density gradient, resuspended in complete
medium or cryopreserved in FBS-10% DMSO for later use. Purified
natural killer (NK; CD56+) cells were obtained using an
immunomagnetic isolation procedure. Briefly, 5–10×106
PBMCs were incubated for 15 min at 4°C with 20 μl of anti-CD56
monoclonal Ab conjugated to magnetic beads (Miltenyi Biotec,
Auburn, CA, USA). CD56+ cells were isolated by positive
selection on an MS column (Miltenyi), according to the
manufacturer’s instructions. Purity of the isolated populations was
tested by flow cytometry (FACSCalibur, Becton-Dickinson, Mountain
View, CA, USA), using FITC- and PE -conjugated anti-CD3 and
anti-CD56 monoclonal Abs (BD Pharmingen, San Diego, CA, USA). In
all cases, purity of the NK population was >90%.
For lymphokine-activated killer (LAK) cell
generation, PBMCs were seeded in 24-well plates (1×106
cells/ml; 2 ml/well) and cultured for 5 days in complete medium
supplemented with 1,000 IU/ml recombinant human interleukin (IL)-2
(Proleukin, Roche, CA, USA). Culture medium containing IL-2 was
renewed every other day. On day 5, LAK cells were harvested and
tested as described.
Monocytes were isolated by seeding PBMCs in 6-well
plates (5×106/ml; 3 ml/well) and let adhere for 2 h at
37°C. The non-adherent cell fraction (comprising >80%
CD3+ cells as assessed by flow cytometry) was collected
and cryopreserved for later use in T cell stimulation cultures.
Proliferation assay
FM3, HCT-116, B16-F1 and CT-26.WT cancer cells were
seeded into 96-well U-bottom microplates (Costar, Cambridge, MA,
USA; 20–25×103 cells/ml; 200 μl/well) and pre-incubated
for 24 h to adhere. ISO-66 was serially diluted at various
concentrations (1 mM-0.04 μM), added to the wells and incubated for
72 h. All cultures were set in triplicates, whereas cultures set in
complete medium or containing an equivalent amount of DMSO (2% v/v)
or doxorubicin (0.5 μM; Sigma-Aldrich, Japan) were used as
controls. For the last 18 h, 3[H]-thymidine (Amersham
Pharmacia Biotech, Amersham, Bucks, UK) was added and inhibition of
proliferation was determined as described (22).
Stimulation of PBMCs, NK, LAK and T cells
with ISO-66
PBMCs were seeded in 48-well plates
(1×106 cells/ml; 1 ml/well), ISO-66 was added at final
concentrations of 1 mM-1 μM and cells were cultured for 3 days.
PBMCs cultured in plain complete medium served as controls. NK
cells were isolated by magnetic beads and used as effector cells
(E) versus K562 (NK-sensitive) targets (T), in standard
51Cr release assays at an E:T ratio of 10:1. Similarly
cultured LAK cells were also used as effectors versus K562 and
Daudi (LAK-sensitive) targets at an E:T ratio of 50:1.
T cells were stimulated with a pool of tumor
antigenic peptides, extracted from the cell surface of FM3 and
HCT-116 [acid wash extract, AWE (25)], as previously described (23). In brief, on day 0 monocytes were
irradiated at 30 Gy, washed and co-incubated with autologous T
cells, at a monocyte:lymphocyte ratio of 1:5. AWE extracted from
20×106 FM3 or HCT-116 cells and ISO-66 at a final
concentration of 10 μM, were added to each well. Cultures set
without ISO-66 served as controls. On day 5, lymphocytes were
restimulated with autologous irradiated monocytes and FM3- or
HCT-116-AWE. On days 2, 5, 7 and 9, IL-2 (40 IU/ml) and ISO-66 (10
μM) were added to the culture medium. T cells were harvested on day
11 and tested for their cytotoxic activity versus FM3, HCT-116 and
Daudi targets, using standard 51Cr-release assays, at an
E:T ratio of 50:1.
In vivo melanoma and colon cancer mouse
models
Female mice, 6–8 weeks of age, 15–20 g of weight, of
the strains C57BL/6 and Balb/C were purchased from Harlan
Laboratories (Udine, Italy). All mice were maintained under
conditions and protocols in accordance with Law 2015/27/2.1992,
Presidential Decree 160/3.5.1991 and the Directive 86/609/EE
C/24.11.1986 of the Council of Europe on Animal Welfare. The study
was approved by the Ethics and Biosafety Committee, Subcommittee on
Ethics, Department of Biology, University of Athens and all
experiments were conducted following the guidelines of the
aforementioned committee.
B16-F1 (syngeneic to C57BL/6 mice) and CT-26.WT
(syngeneic to Balb/C mice) were expanded to sufficient numbers in
complete medium. On day 0, C57BL/6 and Balb/C mice were
subcutaneously (s.c.) inoculated with 1×105 B16-F1 or
1×106 CT-26.WT (in 250 μl PBS), respectively, and mice
of each strain were randomly assigned to 5 groups (8 mice/group).
On days 12–14, when tumors were palpable, and for 20 consecutive
days, mice received intraperitoneally (i.p.) PBS or DMSO (control
groups), or ISO-66 (all diluted in 300 μl PBS) once daily. ISO-66
was administered at 180 μg/dose/animal. DMSO was administered at a
final concentration of 4% v/v, equivalent to the amount present in
ISO-66 injections. Tumor growth rate was recorded every 2–3 days by
measuring the major and minor axes of the tumors formed with a
digital caliper. Measurements were transformed into tumor volume
using the formula: tumor volume (cm3) = major axis ×
minor axis2 × 0.5. On days 34 (for C57BL/6) and 40 (for
Balb/C), when mean tumor volumes of the control groups exceeded
1.5–2 cm3, animals were euthanized and spleens were
aseptically excised from 3 mice/group. Splenocytes were isolated
from individually homogenized spleens and immediately tested for
their cytotoxicity versus B16-F1, CT-26.WT, YAC-1 and WE HI-164
targets in standard 51Cr-release assays.
Cytotoxicity assay
Target cells (T) were 51Cr-labelled
according to Skopeliti et al (24) and co-cultured with the effectors
(E) at the indicated E:T ratios. After 18 h at 37°C, 5%
CO2, 100 μl of each supernatant was removed and isotope
was measured in a γ-counter (1275 Minigamma, LKB Wallac, Turku,
Finland). Targets were incubated with 3 NHCl and in complete medium
to determine maximal and spontaneous isotope release, respectively.
All cultures were set in triplicates. Percentage of specific
cytotoxicity was calculated according to the formula: (cpm
experimental - cpm spontaneous)/(cpm maximal - cpm spontaneous) ×
100.
Statistical analysis
Data were analyzed by the Student’s t-test and
statistical significance was presumed at significance level of 5%
(p<0.05). Tumor size among groups was compared with Wilcoxon’s
signed rank test.
Results
ISO-66 as an inhibitor of MIF
We have identified ISO-66, a novel analog of ISO-1,
as a potent inhibitor of MIF tautomerase activity (Fig. 1). The rationale for the design of
ISO-66 is based on our previous experience with ISO-1 design and
structure activity relationship (SAR) studies. For instance, the
ester functional group in ISO-1 has been shown to play an important
role in binding to MIF, as evident by SAR studies and MIF: ISO-1
co-crystal structure (11).
However, the methyl ester may hydrolyze and this could limit future
application of ISO-1. We hypothesized that the methyl ester could
be replaced by a ketone group and came up with the structure of
ISO-66. ISO-66 was synthesized in three steps and characterized by
NMR and mass spectrometry. ISO-66 was found to inhibit MIF
tautomerase activity with an IC50 of 1.5±0.4 μM compared
to 18.2±.8 μM for ISO-1. It is superior to ISO-1 in both its
potency and stability profile. To improve the solubility of ISO-66,
the β-keto carboxylate was also synthesized (referred to as KF III
53Y/prodrug of ISO-66); however, it was found by mass spectrometry
that KF III 53Y spontaneously decarboxylates to form ISO-66. This
finding was also supported by structural studies, in which MIF was
co-crystallized with KF III 53Y.
Structure of MIF-bound ISO-66
ISO-66 was found bound in the MIF tautomerase active
sites, with the meta-fluoro-para-hydroxyphenyl ring
buried in the innermost part of the cleft, as expected. What was
unexpected, however, was that the five-membered ring of the
inhibitor had been opened. The bond between the nitrogen and oxygen
was cleaved, resulting in free imine and hydroxyl groups (Fig. 2). An open ring structure was seen
in all three active sites. Fig. 2
shows the electron density for the inhibitor in active site C, in
which no active site residues participate in crystal contacts,
clearly revealing the absence of the five-membered ring. The
electron density is also shown for active site B, in which the
density is better defined due to the crystal contacts. The presence
of the modified (ring-opened) inhibitor in the active site
demonstrates that it is itself a potential inhibitor of MIF’s
physiological activity.
Both non-polar and polar interactions occur between
the modified inhibitor and MIF. The
m-fluoro-p-hydroxyphenyl ring is buried in the
hydrophobic cavity of the protein. The hydroxyl group shares a
hydrogen bond with Asn 97 at the base of the cavity. The fluorine
atom has Van der Waals contacts with the side chain of Met 101 and
the β-methylene group of His 62. Electron density for the fluorine
atom is only found on one side of the ring (Fig. 2), demonstrating that the ring does
not stochastically flip between the two states 180° apart. Rotating
the ring by 180° would cause steric conflict between the Tyr 95 and
the electronegative fluorine, which would point toward the Tyr 95
aromatic π-orbital if the inhibitor’s aromatic ring was in such an
orientation. Lys 32 donates a strong hydrogen bond to the hydroxyl
generated by the ring-opening reaction. The nitrogen that was
formerly of the five-membered ring accepts a hydrogen bond from the
main chain amide of Ile 64.
In active site A, however, there was excess electron
density around the methylene and terminal methyl ketone group of
the inhibitor (Fig. 3A). It is
very interesting that this excess density can be explained by the
intact, ring-closed form of the inhibitor, having partial occupancy
(Fig. 3B). Both the ring-opened
and ring-closed forms were modeled into the electron density,
having occupancies of 0.88 and 0.12, respectively. The low
occupancy of the intact inhibitor has no or little effect on the
electron density in the phenyl portion where the B-factors for the
modified inhibitor are lower than those of the intact form.
However, the higher B-factors on the methyl ketone side of the
modified inhibitor yields deterioration of the electron density
there, where we instead see the intact form. The intact inhibitor
also accounts for the extra breadth of electron density located at
the five-membered ring, as shown in Fig. 3.
Both the intact and modified forms of ISO-66
interact with MIF in active site A. The interactions of the
modified compound with MIF in active site A are similar to those in
the other two active sites. The hydrogen bond between the nitrogen
of the ring-opened inhibitor and the Ile 64 backbone amide is
stretched (3.46 Å) relative to the other two active sites. The
binding interactions of intact ISO-66 with MIF are as follows: Lys
32 donates a hydrogen bond to the oxygen atom of the five-membered
ring and is also within hydrogen bonding distance to the ring
nitrogen. The phenyl ring is positioned in the MIF hydrophobic
pocket, but does not fit as deep into the cavity as the phenyl ring
of the modified inhibitor. The hydroxyl group of the phenyl ring of
the intact inhibitor ISO-66 shares a hydrogen bond with Asn 97.
However, this is not as strong as the hydrogen bond between Asn 97
and the modified inhibitor (Fig.
3). This presents the structural view of how ISO-66 binds to
its target MIF, providing a means for it to have the antitumor
effects described in this report. Furthermore, this suggests the
open-ring form we discovered through the crystallographic analysis
could also be a potential chemotherapeutic lead compound, which may
be investigated in future studies.
ISO-66 does not affect cancer cell
proliferation in vitro
To assess whether ISO-66 is directly cytotoxic to
cancer cells, we tested its effect in inhibiting the proliferation
of the human and mouse melanoma (FM3 and B16-F1) and colon cancer
cells (HCT-116 and CT-26.WT). In all experiments, the equivalent
amount of DMSO (2% v/v) present in the highest ISO-66 concentration
tested (1 mM), and the chemotherapeutic drug doxorubicin (0.5 μM)
were used as controls. Our results showed that low concentrations
of ISO-66 did not affect the proliferation of cancer cells, whereas
at the highest ISO-66 concentration tested, an analogous inhibition
of proliferation (~50%) was also caused by the equivalent amount of
DMSO (Fig. 4).
ISO-66 enhances NK and LAK cell
cytotoxicity
For assessing the ability of ISO-66 to induce
cytolytic PBMC responses in vitro, we incubated normal
donor-derived PBMCs with 1 mM-1 μM ISO-66 for 3 days and then we
isolated the CD56+ cells from the cultures. Our
titration experiment (Fig. 5A)
showed that NK cell cytotoxicity versus K562 cell targets was
equally enhanced by 16% upon incubation with 10 or 100 μM ISO-66.
We further generated LAK cells from PBMCs of healthy donors and
cultured them with ISO-66 for 3 days. In the cytotoxicity test, we
observed that ISO-66 increased LAK cell cytotoxicity against K562
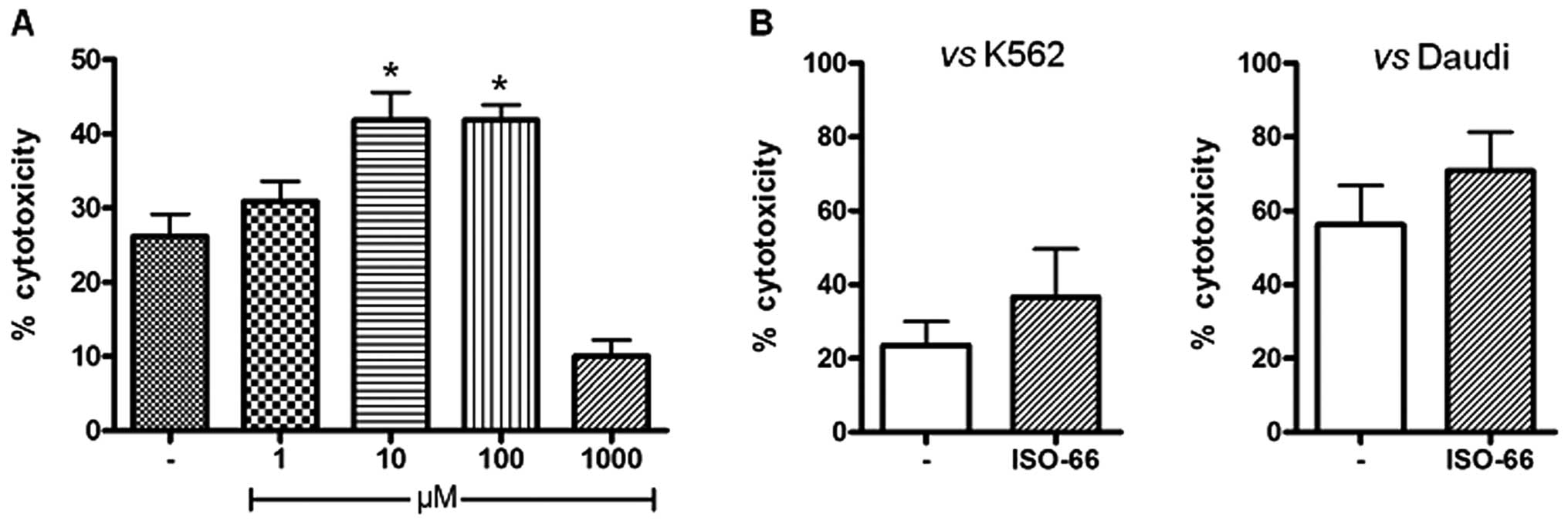
(36.5% for ISO-66, compared to 23.5% of the control; Fig. 5B), which was further enhanced when
Daudi cells were used as targets (70.7% for ISO-66, compared to
56.2% of the control; Fig. 5B).
However, for both targets, this increase in LAK cell cytotoxicity
did not reach statistical significance.
ISO-66 enhances T cell cytotoxicity
We next sought to investigate whether ISO-66 is able
to enhance T cell cytotoxicity. As specific antigen source for T
cell stimulation, we used a pool of tumor antigenic peptides,
detached from MHC molecules present on the surface of melanoma
(FM3) and colon cancer (HCT-116) cells. In the presence of
autologous irradiated monocytes as antigen-presenting cells, this
extract (AWE ) has been already used by us (23) and others (25) to generate in vitro
tumor-reactive T cell lines. On day 5, T cells were restimulated
with autologous AWE -loaded monocytes. To expand AWE -reactive T
cells, low dose IL-2 (40 IU/ml) was used. ISO-66 was added to the
cultures every other day, while cultures in plain medium served as
controls. On day 11, AWE -reactive T cells cultured in the presence
of ISO-66 were able to recognize and lyse more efficiently the
targets used for AWE preparation (Fig.
6). Specifically, FM3-AWE -stimulated T cells killed FM3
targets (58.4%) and this cytotoxicity was enhanced in the presence
of the MIF inhibitor (69.0%). The same T cells marginally lysed
allogeneic HCT-116 colon cancer targets (<25%), but showed some
LAK cytotoxicity (36.1% against Daudi cells), which was higher when
ISO-66 was added in the cultures (49.5%; Fig. 6A). Accordingly, HCT-116-AWE
-stimulated T cells killed HCT-116 targets (44.6%) and ISO-66
increased this percentage to 68.5%. HCT-116-AWE -stimulated T cells
did not lyse FM3 targets (<25%) and similarly to FM3-AWE
-stimulated T cells, they exhibited increased LAK activity as
detected by their cytotoxicity against Daudi (45.0% for ISO-66,
compared to 33.9% of the control; Fig.
6B).
Although, the enhancement of cytotoxicity induced by
ISO-66 was in all cases not statistically significant compared to
unstimulated cells, we cannot rule out the possibility that
additional T cell stimulations could have resulted in a more
pronounced effect. Taken as a whole, our results show that ISO-66
increases the specific and non-specific cytotoxic responses of
activated human T cells in vitro.
ISO-66 retards melanoma and colon tumor
growth in vivo
To test the anticancer activity of ISO-66 in
vivo, mice were implanted with syngeneic melanoma and colon
cancer cells. We specifically selected these two mouse models in
order to be able to compare our results on ISO-66 with already
reported data, in which MIF shRN A and ISO-1 were used to block MIF
in similar in vivo melanoma and colon cancer models,
respectively (3,16).
Following titration experiments (26), on day 0, C57BL6 and Balb/C mice
received s.c. 1×105 B16-F1 or 1×106 CT-26. WT
cells (melanoma and colon carcinoma syngeneic cells, respectively).
By days 12–14, all mice had developed palpable tumors and were
further administered i.p. PBS, DMSO or ISO-66 for 20 consecutive
days. The dose of 180 μg ISO-66/animal/day (3.6 mg/mouse) was based
on previous reports on the anticancer activity of ISO-1, where,
although at different time intervals, 3.2 mg (16) and 4 mg (9) per animal were i.p. administered to
tumor-bearing mice, without reported toxicity. In contrast to these
studies, in our protocols, we administered ISO-66 daily and for 20
consecutive days, to significantly, if not completely, inactivate
MIF constantly produced both by the host and by melanoma (27) or colon cancer cells (28). Tumor growth was monitored until day
34 (for melanoma) or day 40 (for colon cancer). As shown in
Fig. 7A, tumor size (expressed in
cm3) in melanoma-inoculated mice treated with ISO-66,
showed a significantly slower increase as compared to controls
(i.e., mice receiving PBS or DMSO). On the 34th day post-tumor cell
inoculation, control mice were euthanized for ethical reasons
(tumor size ≥2 cm3), whereas the average tumor volume
recorded for animals treated with ISO-66 was 1.2 cm3.
This tumor reduction of ~45% was statistically significant
(p<0.01, compared to controls).
For the colon cancer model (Fig. 7B), by day 40 post-inoculation, both
control groups (i.e., mice receiving PBS or DMSO) exhibited a
similar rapid tumor development (≥1.5 cm3). The
therapeutic administration of the MIF inhibitor reduced tumor
growth rates in all animals of the ISO-66-treated group.
Specifically, the average tumor volume recorded for ISO-66 was 0.55
cm3, and this ca. 60% tumor volume reduction was
statistically significant compared to controls (p<0.001).
The in vivo antitumor responses induced
upon treatment with ISO-66 are mediated by T cells
To verify whether the in vivo reduction of
tumor growth was associated with increased antitumor immune
responses induced by ISO-66, three mice from each group that
developed the smaller tumors were sacrificed on day 34 for melanoma
and on day 40 for the colon cancer model. Without additional ex
vivo stimulation, their splenocytes were used as effectors in
51Cr-release assays against the murine NK-sensitive
targets YAC-1, the LAK-sensitive WE HI-164, and the syngeneic cells
B16-F1 and CT-26.WT. Splenocytes from ISO-66-treated
melanoma-inoculated mice were more efficient in killing B16-F1
targets than PBS- or DMSO-treated mice (41.5 versus 11.7 and 11.9%,
respectively; p<0.01 compared to PBS; Fig. 8A). The same splenocytes did not
lyse the non-syngeneic CT-26.WT colon targets (10.8%), or YAC-1
(13.0%), but showed some cytotoxicity against WE HI-164 (29.8%
compared to 12.8% of the control; p<0.05). These results
indicate that the MIF inhibitor ISO-66 increased the cytotoxicity
of LAK cells and, most importantly, induced the in vivo
expansion of melanoma-reactive T cells.
Accordingly, spleen cells from mice bearing colon
cancer cells when treated with ISO-66, lysed the syngeneic CT-26.WT
targets (39.4% compared to 27.6% of the PBS group; Fig. 8B), as well as the LAK-sensitive WE
HI-164 (20% compared to 11.9% of the PBS group). Marginal
cytotoxicity against the melanoma B16-F1 and YAC-1 was recorded.
Thus, from the results obtained from both cancer models, we observe
that ISO-66 enhances tumor-reactive (T cell-mediated) and
non-specific (LAK cell-mediated) immune responses leading to
decreased cancer cell growth.
Discussion
MIF is a pleiotropic cytokine broadly studied in
sepsis and many infectious and auto-immune diseases (1). Recently, a causative role in cancer
progression has been attributed to MIF, as it has been reported to
inactivate p53 (29), enhance
angiogenesis (30), promote
metastasis (31) and impair both
innate and adaptive immune responses (5,32–34).
Therefore, inactivating MIF provides an alternative therapeutic
option in anticancer treatment (11). Small-molecule inhibitors of host-
and cancer cell-derived MIF have been used to block its activity
(35). The most prominent MIF
inhibitor, ISO-1, is known to inhibit the
proliferation/invasiveness of cancer cell lines in vitro
(9,10,13–16,36)
and tumor growth and vascularization in vivo (9,16).
In this study, using the structure of ISO-1 as a
scaffold, we designed a novel, more potent and more stable MIF
inhibitor. In our initial in vitro studies, we observed that
ISO-66 did not affect human and mouse cancer cell proliferation,
even at concentrations as high as 1 mM. On the contrary, ISO-66
induced the cytotoxic potential of distinct effector cell
populations with antitumor activity, namely NK and LAK cells and
cytotoxic T cells (CTLs) (37).
Specifically, NK cells purified from human PBMCs cultured with
ISO-66 showed increased killing of tumor targets (K562) and LAK
cells generated with high concentrations of IL-2, when stimulated
with ISO-66 efficiently lysed both K562 and Daudi targets. Most
importantly, in the presence of ISO-66, tumor antigen-reactive CTLs
generated in vitro, recognized and effectively killed the
relative tumor-antigen expressing targets. Our results are in
agreement with previous reports showing that MIF hampers anticancer
immunity by inhibiting tumor-specific CTL and NK cell activity
(33,34). The mechanisms responsible for this
CTL deficiency, probably include MIF-induced downregulation of
receptors involved in tumor cell recognition (34) and/or excess T cell activation,
leading to cell death (33). MIF
is also known to impair the ability of NK cells to release
cytolytic perforin granules (32).
Additionally, it has been suggested that MIF’s effect is optimally
exerted on activated cells, whereas normal functions of resting
cells evolve independently of MIF (9). Our results are in agreement with this
scenario, as NK, IL-2- activated LAK cells and antigen-stimulated
CTLs, increased their cytotoxicity in the presence of ISO-66.
Although ISO-1 has been reported to inhibit proinflammatory
cytokine and IFN-γ secretion by human PBMCs or subpopulations
thereof (38,39), the enhanced functional responses of
NK, LAK and CTLs as observed in our in vitro system, do not
support such an ISO-66-induced suppressive effect. More likely,
ISO-66 acts by blocking the activity of MIF produced by host
macrophages, possibly also T cells, allowing effectors to exert
their lytic functionalities.
Translating our in vitro observations in
vivo, the therapeutic anticancer activity of ISO-66 was tested
in C57BL/6 and Balb/C mice inoculated with syngeneic melanoma
B16-F1 and colon CT-26.WT tumor cells, respectively. In line with
similar doses of ISO-1 given by others (9,16),
we observed that injecting a total of 3.6 mg of ISO-66 per mouse
was well-tolerated and did not cause any adverse effects. Further,
it led to a statistically significant retardation of tumor growth
in both models (45% for melanoma and ca. 60% for colon carcinoma,
compared to controls). Although the therapeutic protocol of the
in vivo ISO-66 administration used in our study differs from
other regimens used to date in terms of intervals between
injections, our results are comparable with previously reported
data on the antitumor activity both of ISO-1 and anti-MIF Abs.
Specifically, He et al (16) recorded a 25% inhibition of CT-26
colon tumors upon treatment with either ISO-1 or anti-MIF Abs
administered twice per week, whereas Ogawa et al (40) reported a much higher inhibition
(55%) of the same tumor upon a more frequent (every other day)
treatment with anti-MIF Abs. These data support our rationale of
maximizing MIF inactivation by injecting mice with ISO-66 daily for
20 consecutive days. Of greater importance are the recent elegant
studies of Girard et al (3)
and Choi et al (4) in which
MIF−/− mice inoculated with B16-F10 melanoma and CT26
colon cancer cells, respectively, exhibited a 47% and 75% tumor
reduction compared to wild-type animals. Melanoma growth in
wild-type mice given daily ISO-66 (Fig. 7A) resembles that observed in
MIF−/− mice (3).
Similarly, colon tumor growth in our model (Fig. 7B), greatly coincides the growth
recorded in MIF−/− mice (4). Therefore, we can propose that
repetitive, daily dosing of ISO-66 effectively reduces the
cancer-promoting effects of MIF.
This was further confirmed upon analyzing the ex
vivo cytotoxicity of spleen cells from mice administered ISO-66
against the inoculated tumors. We used splenocytes from 3 animals
per group with minimal tumor load, as these most likely had
developed the highest percentages of cytotoxic effectors. Our
results revealed that splenocytes from mice therapeutically
administered ISO-66 exhibited increased specific lysis only of the
syngeneic cancer cells, both for melanoma and colon cancer.
Although we also observed a non-negligible percentage of
non-specific cytotoxic responses enhanced by ISO-66, these
accounted for <50% of the syngeneic tumor cell killing. To our
knowledge, our results for the first time suggest that successful
MIF inactivation in vivo by ISO-66 leads to restoration of
the impaired tumor-reactive lymphocyte responses in cancer-bearing
mice. Due to ethical reasons (Guidelines of Ethics and Biosafety
Committee) we could not follow murine tumor growth for a prolonged
period, but based on our results we could safely speculate that
ISO-66 administration would also lead to prolonged animal
survival.
There are several advantages to using small
molecules, such as ISO-1 or ISO-66, to block MIF activity instead
of neutralizing Abs against MIF or its receptor (9,12,16,41).
Small-molecule inhibitors can be developed and synthesized via a
less complex and expensive process, are non-immunogenic upon
repetitive administration, are more mobile in reaching their target
and do not usually require intravenous injection after formulation
optimization [(12,42), and the present report]. Moreover,
although administered at low concentrations, they can still
effectively and specifically target MIF irrespective of its origin,
i.e., whether deriving from host and/or cancer cells. Indeed, in
our study 9 mg/kg of ISO-66 was shown to be non-toxic even after
frequent administration and sufficient to enhance in vivo
tumor-reactive immune responses leading to the reduction of
tumor-cell expansion. In support of our last assumption,
Yaddanapudi et al (5) most
recently reported that the suicide inhibitor of MIF, 4-IPP,
significantly slowed the rate of B16 tumor growth, but to
intracellularly block MIF secretion by TAMs, 4-IPP was given at
doses 10-fold higher (80 mg/kg) than ISO-66 in our models, raising
specificity (35) and toxicity
issues, if such high concentrations were to be adapted for clinical
use. In another study, the drug ibudilast has been used in humans
for two decades in the Far East for bronchial asthma and
post-stroke complication. It is an allosteric inhibitor of MIF at
clinically relevant concentrations, but its antitumor effect has
yet to be studied (44).
Taken as a whole, our results in conjunction with
accumulated evidence from other cancer studies, suggest that
selective MIF inactivation upon treatment with ISO-66 restores the
lytic ability of tumor-reactive CTLs and NK cells, which become
immunocompetent. These cells can further efficiently kill tumor
cells, thereby reducing the size of the tumors. Therefore, anti-MIF
therapies using improved small molecule inhibitors selectively
targeting MIF, such as ISO-66, may provide new treatment options
with the potential to complement currently applied anticancer
strategies.
Acknowledgements
The authors would like to thank Pinelopi Samara and
Efthymis Paronis for their assistance in animal handling. This
study was partly supported by Grant 05 NON EU -404, funded by the
GSRT, Hellenic Ministry of Development (to O.E.T and Y.A.A), R01
AI065029 (to E.L.) and EU FP7 Capacities REGP OT-CT-2011-284460,
INsPiRE.
References
|
1
|
Al-Abed Y and VanPatten S: MIF as a
disease target: ISO-1 as a proof-of-concept therapeutic. Future Med
Chem. 3:45–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bach JP, Deuster O, Balzer-Geldsetzer M,
Meyer B, Dodel R and Bacher M: The role of macrophage inhibitory
factor in tumorigenesis and central nervous system tumors. Cancer.
115:2031–2040. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Girard E, Strathdee C, Trueblood E and
Queva C: Macrophage migration inhibitory factor produced by the
tumour stroma but not by tumour cells regulates angiogenesis in the
B16 F10 melanoma model. Br J Cancer. 107:1498–1505. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choi S, Kim HR, Leng L, Kang I, Jorgensen
WL, Cho CS, Bucala R and Kim WU: Role of macrophage migration
inhibitory factor in the regulatory T cell response of
tumor-bearing mice. J Immunol. 189:3905–3913. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yaddanapudi K, Putty K, Rendon BE, Lamont
GJ, Faughn JD, Satoskar A, Lasnik A, Eaton JW and Mitchell RA:
Control of tumor-associated macrophage alternative activation by
macrophage migration inhibitory factor. J Immunol. 190:2984–2993.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Krockenberger M, Kranke P, Hausler S,
Engel JB, Horn E, Nurnberger K, Wischhusen J, Dietl J and Honig A:
Macrophage migration-inhibitory factor levels in serum of patients
with ovarian cancer correlates with poor prognosis. Anticancer Res.
32:5233–5238. 2012.PubMed/NCBI
|
|
7
|
Kindt N, Lechien J, Decaestecker C,
Rodriguez A, Chantrain G, Remmelink M, Laurent G, Gabius HJ and
Saussez S: Expression of macrophage migration-inhibitory factor is
correlated with progression in oral cavity carcinomas. Anticancer
Res. 32:4499–4505. 2012.PubMed/NCBI
|
|
8
|
Wang XB, Tian XY, Li Y, Li B and Li Z:
Elevated expression of macrophage migration inhibitory factor
correlates with tumor recurrence and poor prognosis of patients
with gliomas. J Neurooncol. 106:43–51. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meyer-Siegler KL, Iczkowski KA, Leng L,
Bucala R and Vera PL: Inhibition of macrophage migration inhibitory
factor or its receptor (CD74) attenuates growth and invasion of DU
145 prostate cancer cells. J Immunol. 177:8730–8739. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rendon BE, Roger T, Teneng I, Zhao M,
Al-Abed Y, Calandra T and Mitchell RA: Regulation of human lung
adenocarcinoma cell migration and invasion by macrophage migration
inhibitory factor. J Biol Chem. 282:29910–29918. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lubetsky JB, Dios A, Han J, Aljabari B,
Ruzsicska B, Mitchell R, Lolis E and Al-Abed Y: The tautomerase
active site of macrophage migration inhibitory factor is a
potential target for discovery of novel anti-inflammatory agents. J
Biol Chem. 277:24976–24982. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Al-Abed Y, Dabideen D, Aljabari B, Valster
A, Messmer D, Ochani M, Tanovic M, Ochani K, Bacher M, Nicoletti F,
Metz C, Pavlov VA, Miller EJ and Tracey KJ: ISO-1 binding to the
tautomerase active site of MIF inhibits its pro-inflammatory
activity and increases survival in severe sepsis. J Biol Chem.
280:36541–36544. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Winner M, Meier J, Zierow S, Rendon BE,
Crichlow GV, Riggs R, Bucala R, Leng L, Smith N, Lolis E, Trent JO
and Mitchell RA: A novel, macrophage migration inhibitory factor
suicide substrate inhibits motility and growth of lung cancer
cells. Cancer Res. 68:7253–7257. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schrader J, Deuster O, Rinn B, Schulz M,
Kautz A, Dodel R, Meyer B, Al-Abed Y, Balakrishnan K, Reese JP and
Bacher M: Restoration of contact inhibition in human glioblastoma
cell lines after MIF knockdown. BMC Cancer. 9:4642009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Piette C, Deprez M, Roger T, Noel A,
Foidart JM and Munaut C: The dexamethasone-induced inhibition of
proliferation, migration, and invasion in glioma cell lines is
antagonized by macrophage migration inhibitory factor (MIF) and can
be enhanced by specific MIF inhibitors. J Biol Chem.
284:32483–32492. 2009. View Article : Google Scholar
|
|
16
|
He XX, Chen K, Yang J, Li XY, Gan HY, Liu
CY, Coleman TR and Al-Abed Y: Macrophage migration inhibitory
factor promotes colorectal cancer. Mol Med. 15:1–10.
2009.PubMed/NCBI
|
|
17
|
Barnick JWFK, Van Der Baan JL and
Bickelhaupt F: Convenient direct method for the preparation of
keto-acids. Synthesis. 79:787–788. 1979. View Article : Google Scholar
|
|
18
|
Rathkea MW and Nowaka MA: Synthesis of
beta-keto acids and methyl ketones using bis(trimethylsilyl)
malonate and triethylamine in the presence of lithium or magnesium
Halides. Synthesis Communications. 15:1039–1049. 1985. View Article : Google Scholar
|
|
19
|
Sun HW, Swope M, Cinquina C, Bedarkar S,
Bernhagen J, Bucala R and Lolis E: The subunit structure of human
macrophage migration inhibitory factor: evidence for a trimer.
Protein Eng. 9:631–635. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Crichlow GV, Cheng KF, Dabideen D, Ochani
M, Aljabari B, Pavlov VA, Miller EJ, Lolis E and Al-Abed Y:
Alternative chemical modifications reverse the binding orientation
of a pharmacophore scaffold in the active site of macrophage
migration inhibitory factor. J Biol Chem. 282:23089–23095. 2007.
View Article : Google Scholar
|
|
21
|
Brunger AT, Adams PD, Clore GM, DeLano WL,
Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu
NS, Read RJ, Rice LM, Simonson T and Warren GL: Crystallography
& NMR system: A new software suite for macromolecular structure
determination. Acta Crystallogr D Biol Crystallogr. 54:905–921.
1998.
|
|
22
|
Argyropoulou A, Samara P, Tsitsilonis O
and Skaltsa H: Polar constituents of Marrubium thessalum
Boiss. & Heldr (Lamiaceae) and their cytotoxic/cytostatic
activity. Phytother Res. 26:1800–1806. 2012.
|
|
23
|
Baxevanis CN, Voutsas IF, Tsitsilonis OE,
Gritzapis AD, Sotiriadou R and Papamichail M: Tumor-specific
CD4+ T lymphocytes from cancer patients are required for
optimal induction of cytotoxic T cells against the autologous
tumor. J Immunol. 164:3902–3912. 2000.PubMed/NCBI
|
|
24
|
Skopeliti M, Voutsas IF, Klimentzou P,
Tsiatas ML, Beck A, Bamias A, Moraki M, Livaniou E, Neagu M,
Voelter W and Tsitsilonis OE: The immunologically active site of
prothymosin alpha is located at the carboxy-terminus of the
polypeptide. Evaluation of its in vitro effects in cancer patients.
Cancer Immunol Immunother. 55:1247–1257. 2006. View Article : Google Scholar
|
|
25
|
Nair SK, Boczkowski D, Snyder D and Gilboa
E: Antigen-presenting cells pulsed with unfractionated
tumor-derived peptides are potent tumor vaccines. Eur J Immunol.
27:589–597. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ioannou K, Kavrochorianou N, Bega C,
Thyphronitis G, Haralambous S and Tsitsilonis O: The C-Terminal
decapeptide of prothymosin α induces a TH1-Type immune response in
vitro and retards tumor growth in vivo. In: 8th Joint Conference of
the International Cytokine Society and the International Society
for Interferon and Cytokine Research Elsevier; Chicago, IL. pp.
17–20. 2010
|
|
27
|
Shimizu T, Abe R, Nakamura H, Ohkawara A,
Suzuki M and Nishihira J: High expression of macrophage migration
inhibitory factor in human melanoma cells and its role in tumor
cell growth and angiogenesis. Biochem Biophys Res Commun.
264:751–758. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dessein AF, Stechly L, Jonckheere N,
Dumont P, Monte D, Leteurtre E, Truant S, Pruvot FR, Figeac M,
Hebbar M, Lecellier CH, Lesuffleur T, Dessein R, Grard G, Dejonghe
MJ, de Launoit Y, Furuichi Y, Prevost G, Porchet N, Gespach C and
Huet G: Autocrine induction of invasive and metastatic phenotypes
by the MIF-CXCR4 axis in drug-resistant human colon cancer cells.
Cancer Res. 70:4644–4654. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hudson JD, Shoaibi MA, Maestro R, Carnero
A, Hannon GJ and Beach DH: A proinflammatory cytokine inhibits p53
tumor suppressor activity. J Exp Med. 190:1375–1382. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun B, Nishihira J, Suzuki M, Fukushima N,
Ishibashi T, Kondo M, Sato Y and Todo S: Induction of macrophage
migration inhibitory factor by lysophosphatidic acid: relevance to
tumor growth and angiogenesis. Int J Mol Med. 12:633–641.
2003.PubMed/NCBI
|
|
31
|
Sun B, Nishihira J, Yoshiki T, Kondo M,
Sato Y, Sasaki F and Todo S: Macrophage migration inhibitory factor
promotes tumor invasion and metastasis via the Rho-dependent
pathway. Clin Cancer Res. 11:1050–1058. 2005.PubMed/NCBI
|
|
32
|
Apte RS, Sinha D, Mayhew E, Wistow GJ and
Niederkorn JY: Cutting edge: role of macrophage migration
inhibitory factor in inhibiting NK cell activity and preserving
immune privilege. J Immunol. 160:5693–5696. 1998.PubMed/NCBI
|
|
33
|
Yan X, Orentas RJ and Johnson BD:
Tumor-derived macrophage migration inhibitory factor (MIF) inhibits
T lymphocyte activation. Cytokine. 33:188–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Krockenberger M, Dombrowski Y, Weidler C,
Ossadnik M, Honig A, Hausler S, Voigt H, Becker JC, Leng L, Steinle
A, Weller M, Bucala R, Dietl J and Wischhusen J: Macrophage
migration inhibitory factor contributes to the immune escape of
ovarian cancer by down-regulating NKG2D. J Immunol. 180:7338–7348.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xu L, Li Y, Sun H, Zhen X, Qiao C, Tian S
and Hou T: Current developments of macrophage migration inhibitory
factor (MIF) inhibitors. Drug Discov Today. 18:592–600. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Binsky I, Lantner F, Grabovsky V, Harpaz
N, Shvidel L, Berrebi A, Goldenberg DM, Leng L, Bucala R, Alon R,
Haran M and Shachar I: TAp63 regulates VLA-4 expression and chronic
lymphocytic leukemia cell migration to the bone marrow in a
CD74-dependent manner. J Immunol. 184:4761–4769. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ioannou K, Samara P, Livaniou E,
Derhovanessian E and Tsitsilonis OE: Prothymosin alpha: a
ubiquitous polypeptide with potential use in cancer diagnosis and
therapy. Cancer Immunol Immunother. 61:599–614. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
West PW, Parker LC, Ward JR and Sabroe I:
Differential and cell-type specific regulation of responses to
Toll-like receptor agonists by ISO-1. Immunology. 125:101–110.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chuang CC, Chuang YC, Chang WT, Chen CC,
Hor LI, Huang AM, Choi PC, Wang CY, Tseng PC and Lin CF: Macrophage
migration inhibitory factor regulates interleukin-6 production by
facilitating nuclear factor-kappa B activation during Vibrio
vulnificus infection. BMC Immunol. 11:502010. View Article : Google Scholar
|
|
40
|
Ogawa H, Nishihira J, Sato Y, Kondo M,
Takahashi N, Oshima T and Todo S: An antibody for macrophage
migration inhibitory factor suppresses tumour growth and inhibits
tumour-associated angiogenesis. Cytokine. 12:309–314. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Takahashi K, Koga K, Linge HM, Zhang Y,
Lin X, Metz CN, Al-Abed Y, Ojamaa K and Miller EJ: Macrophage CD74
contributes to MIF-induced pulmonary inflammation. Respir Res.
10:332009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Imai K and Takaoka A: Comparing antibody
and small-molecule therapies for cancer. Nat Rev Cancer. 6:714–727.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Engh RA and Huber R: Accurate bond and
angle parameters for X-ray protein structure refinement. Acta
Cryst. 47:392–400. 1991. View Article : Google Scholar
|
|
44
|
Cho Y, Crichlow GV, Vermeire JJ, Leng L,
Du X, Hodsdon ME, Bucala R, Cappello M, Gross M, Gaeta F, Johnson K
and Lolis EJ: Allosteric inhibition of macrophage migration
inhibitory factor revealed by ibudilast. Proc Natl Acad Sci USA.
107:11313–11318. 2010. View Article : Google Scholar : PubMed/NCBI
|