Introduction
Pancreatic cancer is a devastating disease with the
worst prognosis among all the major human malignancies. Long-term
prognosis remains poor with a 5-year survival rate of <5% after
the initial diagnosis (1). One of
the major hallmarks of pancreatic cancer is its extensive local
tumor invasion and drug resistance, but the molecular events
underlying this remain mysterious (2).
The alterations of the chromatin structure by
histone acetylases (HATs) and histone deacetylases (HDACs) are
involved in the regulation of gene transcription and also in the
process of tumorigenesis. The deacetylation of lysine residues by
HDACs can lead to chromatin compaction and gene inactivation
(3). Recently studies showed that
elevated levels of HDAC3 expression and activity caused epigenetic
alterations associated with malignancies (4,5).
However, the role of HDAC3 in pancreatic cancer has not been well
elucidated.
In this study, the potential effects of HDAC3 on
pancreatic cancer were investigated. We found that HDAC3 was
overexpressed in pancreatic cancer as compared to paired
paracancerous tissues. Consistently, all of the eight pancreatic
cancer cell lines had higher level of HDAC3 relative to human
pancreatic ductal epithelial cells (HPDE). Further function
analysis revealed that high HDAC3 expression could promote cancer
cell proliferation, migration and invasion, and may increase drug
resistance. Moreover, the functional involvement of HDAC3 was
partially correlated with post-induction repression of P53, P27 and
Bax gene transcription, acting via H3K9 deacetylation.
Materials and methods
Patients and tissue samples
Four cases of pancreatic cancer and paired
paracancerous fresh tissues were obtained from Department of
General Surgery, First People’s Hospital, School of Medicine,
Shanghai Jiao Tong University. For the use of all clinical
materials for research purposes, prior written informed consent
from all the patients and approval from the Ethics Committees of
the First People’s Hospital, School of Medicine, Shanghai Jiao Tong
University were obtained.
Immunofluorescence
The experimental steps were performed as previous
described (6). Pancreatic cancer
tissue specimens stored at −80°C were moved to −20°C to equilibrate
to the temperature of the cryostat. Sections were cut of the 7–8-μm
thickness, placed on slides and dried overnight at room
temperature. Slides were then fixed by immersion in cold acetone
(−20°C) for 5 min, air dried at room temperature and exposed to
microwave for 15 min. The prepared frozen tissue sections were
either subjected to immunostaining or stored at −20°C.
The frozen tissue slices were re-fixed with
paraformaldehyde at room temperature for 10 min. After four rinses,
the slices were blocked with 5% BSA and 5% goat serum for 60 min at
37°C. Properly diluted primary antibody against HDAC3 (Abcam,
dilution 1:2,500) was applied to frozen sections at 4°C overnight,
followed by four rinses. The slides were stained with Dylight
546-conjugated goat anti-rabbit antibody (Jackson, dilution 1:100)
for 1 h at room temperature. Finally, slides were stained with DAPI
(Sigma) for 15 min and mounted in Antifade medium (Beyotime).
Cancer cell lines
Human pancreatic ductal epithelial cells (HPDE),
pancreatic cancer cells BxPC-3, PANC-1, AsPC-1, HPAC, CFPAC-1,
HS-766T, HPAF-II and SW1990 were obtained from Shanghai Institute
for Life Science, Chinese Academy of Sciences. PANC-1 and HPAC were
grown in 5% CO2 saturated humidity, at 37°C and cultured
in DMEM (Gibco, USA) supplemented with 2 mmol/l glutamine and 10%
FBS (Gibco) and subcultured by harvesting with trypsin-EDTA. HPDE,
BxPC-3, AsPC-1, CFPAC-1, HS-766T, HPAF-II and SW1990 cells were
cultured in RPMI-1640 (Gibco) supplemented with 10% FBS.
Knockdown and overexpression of the HDAC3
gene
Human HDAC3 cDNA (NM_003883) was amplified by RT-PCR
with the RNA extracted from PANC-1 cells, and then cloned into pLV4
vector. HDAC3 were searched for suitable siRNA target sequences,
three siRNA sequences were designed, synthesized, and confirmed by
sequencing, respectively. Preliminary experiments picked out the
most efficient siRNA sequences of knockdown from the above three
candidate siRNAs. siRNA sequences: 5′-GCCUCAUCGCCUGGCAUUGdTdT-3′
(sense), 5′-CAAUGCCAGGCGAUGAGGCdTdT-3′ (antisense); with a negative
control siRNA sequence: 5′-UUCUCCGAAC GUGUCACGUdTdT-3′ (sense),
5′-ACGUGACACGUUCGGA GAAdTdT-3′ (antisense). Lentivirus particles
were produced by co-transfecting expression vector pLV4-HDAC3 cDNA,
pLV4-HDAC3 shRNA or pLV4-vector with viral particle packaging
helper vector into 293T cells. Titer of viral particles was
determined by limited serial dilution. PANC-1 cells or HPAC cells
were infected with the lentivirus with the pLV4-HDAC3 cDNA,
pLV4-HDAC3 shRNA or pLV4-vector. The efficiency of knockdown or
overexpression of HDAC3 was determined by western blot
analysis.
Cell proliferation assay and cytotoxicity
studies
Cell proliferation was determined from three
separate experiments using MTT
(3-(4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide)
assays (7). Briefly, the medium
was removed and replaced by 200 μl fresh medium with 500 μg/ml MTT
per well. The cells were incubated for 4 h. The medium was then
removed and 200 μl of DMSO was added to each well. The absorbance
value of each well was determined spectrophotometrically at 570 nm
on a Microplate ELISA Reader (Bio-Tek Instruments). The cell growth
inhibitory effect of 72-h gemcitabine exposure was studied as
described previously (8). The 50%
inhibitory concentration (IC50) of cell growth for each
cell line was determined by non-linear least squares curve fitting
(GraphPad PRISM, Intuitive Software for Science). The experiment
was performed in triplicate. Each experiment was repeated three
times.
In vitro invasion assay and migration
assay
MilliCell (12-mm diameter with 8-μm pores) chambers
(Millipore, Bedford, MA, USA) were pre-coated with Matrigel (BD,
Bedford, MA, USA) on the upper side. A total of 1×105
pancreatic cancer cells were added to the upper compartment in
medium supplemented with 0.1% serum, and the chambers were placed
into 24-well plates with medium containing 10% serum. After 24 h at
37°C, cells on the upper side of the membrane were wiped off,
invaded cells on the lower membrane surface were fixed and stained
with DAPI (Sigma). Invasive activity was quantified by counting 10
high-power fields (HPFs, ×200) per chamber. Mean values were
obtained from at least three individual chambers for each
experimental point per assay. The migration assay is the same with
invasion assay excepting no Matrigel was used and the permeating
time for cells was 16 h.
Reverse transcription qPCR analysis
Total RNA was extracted from the cells with RNAiso
(Takara) according to the manufacturer’s protocol. For mRNA
detection, reverse transcription was performed according to the
protocol of RevertAid™ First Strand cDNA Synthesis kits
(Fermentas); qPCR was performed with SYBR premix Ex Taq (Takara) on
an Applied Biosystems 7500 Real-Time PCR system supplied with
analytical software (Applied Biosystems, USA). GAPDH mRNA was used
to normalize RNA inputs. Primers used were as follows: P27
[5′-GCAACCAATGGATCTCCT CCT-3′ (sense) and
5′-GGGGAGAAAAACACCCCGAA-3′ (antisense)], P53
[(5′-TGACACGCTTCCCTGGATTG-3′ (sense) and 5′-TCCGGGGACAGCATCAAATC-3′
(antisense)], Bax [(5′-GTGGTTGGGTGAGACTCCTC-3′ (sense) and 5′-GCA
GGGTAGATGAATCGGGG-3′ (antisense)], Bim [(5′-TTGAT
TCTTGCAGCCACCCT-3′ (sense) and 5′-CGCAGGCTGC AATTGTCTAC-3′
(antisense)], GAPDH [(5′-CTCTGCTCCT CCTGTTCGAC-3′ (sense) and
5′-GCGCCCAATACGACCAA ATC-3′ (antisense)]. Data were processed by
using the 2−ΔΔCt method. The results are represented as
the means ± SE of three independent experiments.
Western blotting
Cells were lysed with lysis buffer (Beyotime)
containing PMSF. Lysates was centrifuged at 12,000 g at 4°C for 10
min, and the supernatant was collected and preserved at −80°C for
later use. Protein concentrations were determined using BCA Protein
Assay kit (Beyotime). Proteins were subjected to one-dimensional
SDS-PAGE and transferred to PVDF membrane (Millipore) by using a
transfer apparatus according to the manufacturer’s protocols
(Bio-Rad, Richmond, CA, USA). Membranes were blocked with non-fat
dry milk in TBST buffer (10 mM Tris-HCl, pH 8.0, and 150 mM NaCl)
containing 0.1% Tween-20, washed in the same buffer and probed with
the following antibodies: anti-HDAC3 (Abcam), anti-histone H3
(Millipore Biotechnology), anti-acetyl-histone H3 (Millipore
Biotechnology) and anti-β-actin antibodies (Cell Signaling
Technology) at 4°C overnight. Then the membranes were washed in
TBST buffer and incubated with respective secondary antibody. The
infrared fluorescence image was obtained using Odyssey infrared
imaging system (Li-Cor Bioscience). Data were obtained from at
least three independent experiments.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed
using the Upstate Biotechnology ChIP kit and following a modified
protocol from the manufacturer. Briefly, tissues was fixed in 1%
formaldehyde and cell lysates were sheared by sonication in 1% SDS
lysis buffer to generate chromatin fragments with an average length
of 200–1,000 bp. The chromatin was then immunoprecipitated
overnight at 4°C with antibodies specific to HDAC3 (Abcam), AcH3,
AcH3K9 and AcH3K14 (Millipore Biotechnology) or an equivalent
amount of control IgG. Protein-DNA-antibody complexes were
precipitated with protein A-agarose beads for 2 h at 4°C. Input or
DNA in the complex was subjected to quantitative PCR with specific
primer for P27 promoter [(5′-GGCTCACAAGTTAGAGA CAA-3′ (sense) and
5′-GCAGAAGGAATTAGCAAGTG-3′ (antisense)], P53 promoter
[(5′-CACAGGAACAGACGACAA-3′ (sense) and 5′-TGGACACGGCTAAGTAGA-3′
(antisense)], Bax promoter [(5′-TCTTACTATTGGTTGCTCTAGG-3′ (sense)
and 5′-AGGTCTCGGTTCTGTCTG-3′ (antisense)], Bim promoter
[(5′-CTTAGAAGAATGGTGGAGTTG-3′ (sense) and
5′-CATAGACAAGTGTTCAGATGG-3′ (antisense)].
Statistical analysis
All statistical analyses were performed using SPSS
10.0. Data are expressed as mean ± SEM. The statistical correlation
of data between groups was analyzed by one-way analysis of variance
(ANOVA) and Student’s t-test as appropriate. In vitro cell
growth assay was tested using factorial design ANOVA. P-values of
<0.05 were considered statistically significant.
Results
HDAC3 protein is overexpressed in human
pancreatic cancer
Four samples of pancreatic cancer tissues and paired
paracancerous tissues were collected. Protein levels of HDAC3 were
detected by western blot analysis. In comparison with paired
paracancerous tissues, HDAC3 protein expression was significant
higher in pancreatic cancer tissues. Next, we detected protein
levels of histone H3 and acetylated histone H3 (AcH3). While total
levels of histone H3 were similar, levels of AcH3 were
significantly higher at paracarcinoma tissues as compared to tumor
tissues (Fig. 1A). The results
suggested that HDAC3 overexpression could lead to decreased level
of AcH3 in pancreatic cancer tissues. In addition,
immunofluorescence results confirmed that HDAC3 protein expression
was higher in pancreatic cancer (Fig.
1B). Furthermore, we compared the expression of HDAC3 between
human normal pancreatic ductal epithelial cell (HPDE) and
pancreatic cancer cell lines. Interestingly, consistently higher
expression level of HDAC3 was found in all the eight pancreatic
cancer cell lines relative to the HPDE (Fig. 1C). Taken together, it is suggested
that upregulation of HDAC3 is a frequent event in human pancreatic
cancer.
HDAC3 promotes pancreatic cancer cell
proliferation, migration and invasion, and increases drug
resistance in vitro
In order to investigate the biological function
involved in pancreatic cancer, we knocked down or overexpressed
HDAC3 gene expression in PANC-1 and HPAC cells through
lentivirus-mediated constructs. In addition, TSA, an inhibitor of
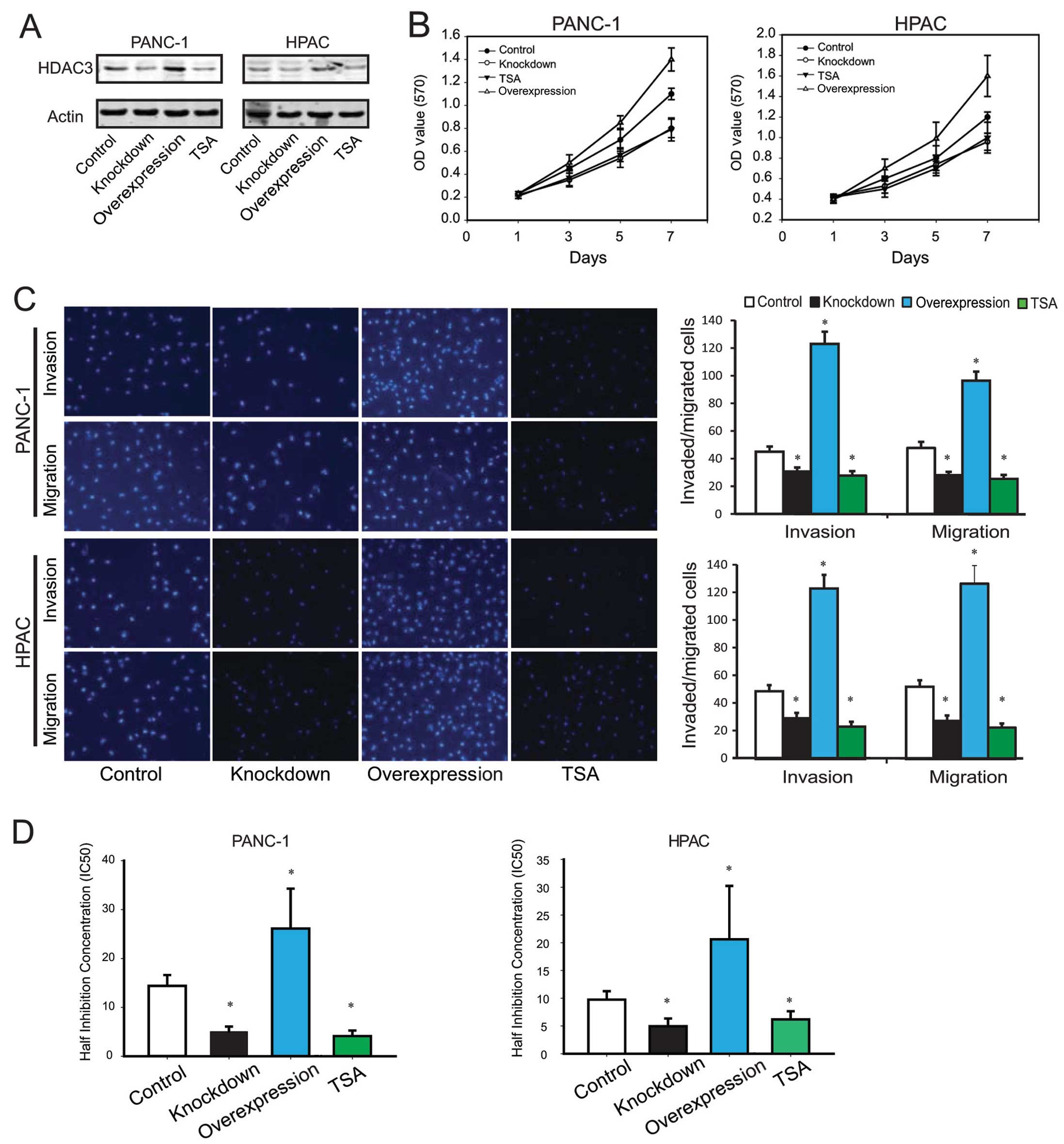
HDACs, was used to inhibit HDAC3 activity. As shown in Fig. 2A, comparing to the control, cells
transfected with shRNA-HDAC3 or treatment with TSA had decreased
levels of HDAC3 protein, and HDAC3 overexpression was able to
upregulate HDAC3 protein level significantly.
To detect the effect of HDAC3 expression on
pancreatic cell proliferation, we investigated the proliferative
activities by MTT assays. As a result, it was found that HDAC3
downregulation significantly reduced the proliferative activities
of pancreatic cancer cell lines, while HDAC3 upregulation increased
the cell proliferation (Fig.
2B).
Cell migration and invasion are important processes
of tumor development and metastasis. Therefore, we evaluated the
effects of HDAC3 on the migration and invasion of PANC-1 and HPAC
by the Transwell assay. The results revealed that
migration/invasion capability of PANC-1 and HPAC cells were
significantly decreased by HDAC3 knockdown or treatment with TSA,
and increased by HDAC3 upregulation (Fig. 2C).
To screen the effect of HDAC3 on drug resistance, we
investigated the impact gemcitabine on the proliferation of PANC-1
and HPAC cells, and calculated the half inhibition concentration
(IC50). The results are shown in Fig. 2D, compared to control, HDAC3
knockdown or TSA treatment decreased IC50 of gemcitabine
(PANC-1, 5.04±1.04/4.11±1.14 vs. 14.40±2.21 nmol/l; HPAC,
4.93±1.41/6.17±1.48 vs. 9.74±1.52 nmol/l), and HDAC3 overexpression
increased IC50 significantly (PANC-1, 26.13±8.16 vs.
14.40±2.21 nmol/l; HPAC, 20.62±9.61 vs. 9.74±1.52 nmol/l).
The recruitment of high level of HDAC3 to
P27, P53 and Bax gene promoter mediate transcription
suppression
Elevated levels of HDACs expression among
malignancies caused transcriptional repression of a diverse set of
genes, involved in tumor progression (5). Previous reports suggested that P27,
P53, Bax and Bim played important roles in pancreatic cancer
progression. The P53 tumor suppressor gene encodes a nuclear
protein that plays a crucial role in cell cycle regulation and
major early events in pancreatic cancer (9). P27 (Kip1/CDKN1B) is a member of the
Cip/Kip family of cyclin-dependent kinase inhibitors, which can
induce cell cycle arrest and serve as tumor suppressors. Moreover,
P27 and P53 provide independent prognostic or predictive
information in pancreatic cancer (10). Bax and Bim belong to the BCL-2
family forming hetero- or homodimers and acting as anti- or
pro-apoptotic regulators that are involved in a wide variety of
cellular activities. Bax (11) and
Bim (12) function as an apoptotic
activator regulated apoptosis signaling in pancreatic cancer. To
investigate the possible mechanisms of HDAC3-mediated biological
function in pancreatic cancer, we analysed the effects of HDAC3
expression on P27, P53, Bax and Bim mRNA. The results revealed that
expression of P27, P53 and Bax mRNA was upregulated by HDAC3
knockdown, and significantly downregulated by HDAC3 overexpression.
However, no changes of the expression level of Bim gene mRNA was
observed (Fig. 3A). Therefore, we
hypothesized that recruitment of high level of HDAC3 to the above
gene promoters in pancreatic cancer led to transcription
suppression. The ChIP result revealed that, compared to paired
paracancerous tissues, higher level of HDAC3 was recruited to P27,
P53 and Bax promoter in pancreatic cancer. In addition, there was
no statistically significant difference for Bim gene. Accordingly,
the recruitment of AcH3 was significantly diminished because of the
higher level of HDAC3 (Fig.
3B).
Specifically, HDAC3 negatively regulates the
transcription of genes by directly interacting with co-repressors
recruited to target gene promoters by various transcription factors
(13). The silencing mediator of
retinoid and thyroid receptor (SMRT) is a nuclear receptor
corepressor that binds and enhances the HDAC activity of HDAC3
(14). ChIP experiments with SMRT
antibodies indicated increased recruitment in the P27, P53 and Bax
promoter region in pancreatic cancer (Fig. 3B), thus confirming a functional
involvement of HDAC3 in the post-induction repression of gene
transcription.
Furthermore, the recruitments of HDAC3 and SMRT to
the promoters of P27, P53 and Bax gene were significantly
diminished by knockdown of HDAC3 or TSA administration, and
increased by HDAC3 overexpression. Inversely, HDAC3 knockdown or
treatment with TSA could increase the recruitment of AcH3, and
HDAC3 overexpression resulted in decreased level of AcH3
recruitment (Fig. 3C).
H3K9 deacetylation is directly correlated
with the recruitment of HDAC3 to P27, P53 and Bax gene
promoters
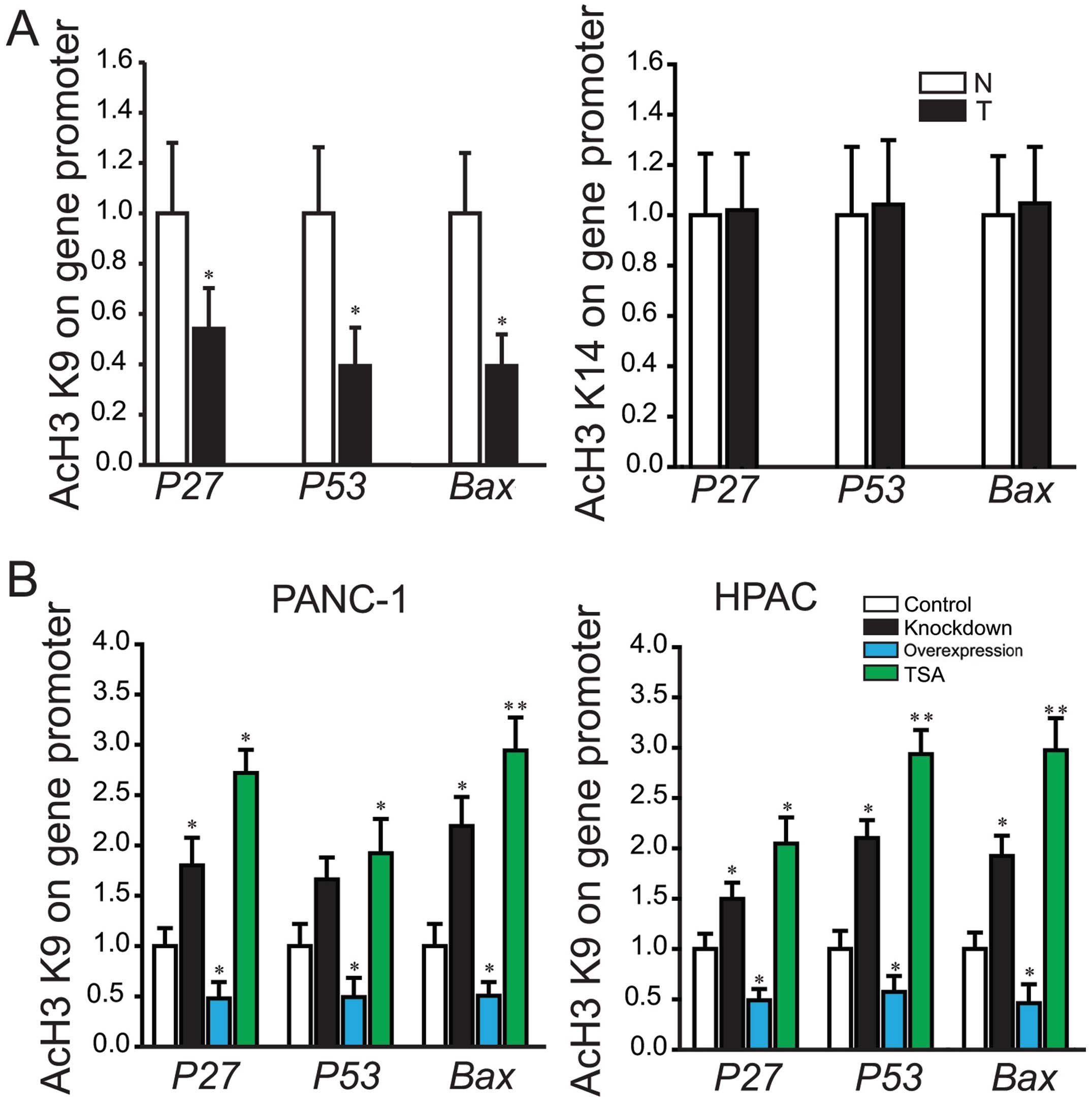
Genome-wide analyses of histone acetylation have
demonstrated that the acetylation of individual lysines in histone
H3 and H4 tails and more specifically histone H3 acetylation at
lysines K9 (H3K9) and K14 (H3K14) correlates with the active state
of gene transcription (15,16).
To further understand the involvement of histone acetylation in
regulating gene expression, the modulation of histone H3K9 and
H3K14 acetylation at the P27, P53 and Bax promoters were examined.
Compared to the constitutive levels detected in paired
paracancerous tissues, decreased H3K9 acetylation by 2–3-fold was
detected in pancreatic cancer tissues (Fig. 4A). However, for H3K14 acetylation,
no significant difference was detected (Fig. 4A). These results indicated H3K9
deacetylation correlates directly with the recruitment of HDAC3 to
the above gene promoters, thus providing a mechanism to maintain
controlled expression of genes.
Furthermore, ChIP assay was carried out to determine
the levels of H3K9 acetylation at the gene promoter in PANC-1 and
HPAC with stable transfection of HDAC3 or siRNA-HDAC3. The
recruitment levels of H3K9 acetylation were significantly
diminished by HDAC3 overexpression, and increased by knockdown of
HDAC3 or treatment with TSA (Fig.
4B).
Taken together, these results indicated that
recruitment of HDAC3 to P27, P53 and Bax gene promoters led to
histone H3K9 deacetylation and post-inductional inhibition of gene
transcription in pancreatic cancer.
Discussion
Within tumor cells, aberrant deacetylation of
histones due to enhanced HDACs activity results in conformational
changes within the nucleosome that lead to transcriptional
repression of genes involved in differentiation and negative
regulation of cell proliferation, invasion and metastasis (Fig. 5). Increased expressions of HDAC3
have been found closely associated with many malignancies (5). In several studies analyzing patient
cancer samples, overexpression of HDAC3 was found in hepatocellular
(17), lung (18) and prostate carcinomas (19) and in most of the cases HDAC3
upregulation associates with poor prognosis. Also it was proposed
to serve as a candidate biomarker (20). However, the contributions of HDAC3
to pancreatic cancer remain incompletely understood.
In the present study, we firstly analyzed HDAC3
expression in pancreatic cancer and paired paracancerous tissues as
a control. The result showed higher HDAC3 expression in pancreatic
cancer tissues. Consistently, all the eight pancreatic cancer cell
lines had high level of HDAC3. In addition, our data suggested that
elevated expression of HDAC3 in pancreatic cancer led to reduced
level of histone H3 acetylation. To the best of our knowledge, this
is the first report that demonstrates higher level of HDAC3
expression in pancreatic cancer.
Although we demonstrated that HDAC3 functioned as a
stimulus in pancreatic cancer, the function of HDAC3 in tumor is
still very controversial. Various researchers have viewed HDAC3 as
an oncogenic protein. SiRNA-mediated knock-down of HDAC3 in HeLa
cells resulted in inhibition of cell proliferation (21). Inhibition of HDAC3 in glioma cell
lines could suppress proliferation and tumor sphere formation,
induce G0/G1 arrest and apoptosis, and suppress the migration of
glioma cells in comparison with controls (22). However, it has also been reported
as a tumor suppressor protein (23). To further understand the biologic
function of HDAC3, pancreatic cancer cell lines PANC-1 and HPAC
with stable overexpression and knockdown HDAC3 gene were
constructed by lentivirus-mediated methods. Our data showed that
knock-down of HDAC3 expression could suppress pancreatic cancer
cell proliferation, migration and invasion, and reduce
drug-resistance of gemcitabine, consistent with TSA administration,
an inhibitor of HDAC(s). Accordingly, HDAC3 overexpression promoted
cell proliferation, migration and invasion, and increased
drug-resistance. Our study revealed that HDAC3 may function as an
oncogenic protein in pancreatic cancer cells, which was in
agreement with previous findings in other cell types (22,24).
Together, these results suggested that the role of HDAC3 in
pancreatic cancer may mainly lie in tumor growth, invasion and
metastasis.
Previous studies suggested that P27, P53, Bax and
Bim played major roles in pancreatic cancer progression, which
could be regulated by HDAC and/or histone deacetylase inhibitors
(HDACi). HDACi activated P53, and entinostat-induced cytotoxic
effects partially depended on P53 in colon cancer cell lines
(25). Valproic acid and TSA
affect acetylation status of p53 and induce apoptosis in
ERG-positive prostate cancer cells (26). In human lung cancer cell lines,
HDAC inactivation results in the induction of apoptosis via p53 and
Bax activation (27). P27, which
inhibits CDK4- and CDK2-containing complexes, was induced by
vorinostat and/or TSA, in leukemia cells (28) and breast cancer cells (29). In addition, HDACi could upregulate
pro-apoptotic proteins of Bcl-2 family, such as Bax, Bim, Bmf, Bak
and Bik (30,31), which mediated the intrinsic
apoptosis pathway. Moreover, HDAC3 played a role in cell cycle
processes and DNA damage response (32). Therefore, we hypothesized the
functional involvement of HDAC3 in pancreatic cancer were possibly
correlated with the post-induction repression of gene
transcription, including P27, P53, Bax and Bim. Firstly, our
results showed that P27, P53 and Bax mRNA expressions were
upregulated by HDAC3 knockdown, and downregulated significantly by
HDAC3 overexpression. Further ChIP analysis revealed that, compared
to paracancerous tissues, higher level of HDAC3 and HDAC3
corepressors SMRT were recruited to P27, P53 and Bax promoter in
pancreatic cancer, leading to the decreased AcH3 at gene promoter.
In regulation of Bim gene expression, other mechanisms may also
exist. Together, the above data suggest that the functional
involvements of HDAC3 in pancreatic cancer were possible partially
associated with the transcription repression of P27, P53 and Bax
genes. However, the detail mechanisms of HDAC3 regulating histone
modifications and key factors in pancreatic cancer still need
further exploration.
Furthermore, histone H3 acetylation at lysines K9
(H3K9) and K14 (H3K14) were directly correlated with the active
state of gene transcription (16).
In order to identify which lysines acetylation/deacetylation of
histone H3 was responsible for regulating genes expression, ChIP
assays with H3K9 and H3K14 antibodies were performed. The results
indicated that inhibition of P27, P53 and Bax gene transcription in
pancreatic cancer were related to inhibition of histone H3K9
acetylation in the gene promoters. Knockdown of HDAC3 expression or
treatment with TSA resulted in increase of H3K9 acetylation,
whereas the levels of H3K14 acetylation were unaffected.
Accordingly, HDAC3 overexpression led to decrease of H3K9
acetylation. Taken together, the functional involvement of HDAC3 in
the post-induction repression of regulated gene transcription, were
directly correlated to histone H3K9 deacetylation.
In conclusion, our findings reveal strong expression
of HDAC3 in patients with pancreatic cancer, and demonstrate that
the functional involvement of HDAC3 is partially related to
post-induction repression of P53, P27 and Bax gene transcription,
acting via H3K9 deacetylation. HDAC3 participates in the
pathogenesis and progression of pancreatic cancer, which might be a
pivotal epigenetic target against this devastating disease.
Acknowledgements
This study was supported by the Natural Science
Foundation of China (grant nos. 81101846, 81171887, 91229117 and
31101016), Program of Shanghai Subject Chief Scientist (grant no.
12XD1404200), Shanghai International Science and Technology
Cooperation Project (grant no. 12410709000) and Shanghai Science
and Technology Committee (grant no. 11DZ1922002).
References
|
1
|
Ducreux M, Boige V, Goere D, Deutsch E,
Ezra P, Elias D and Malka D: The multidisciplinary management of
gastrointestinal cancer. Pancreatic cancer: from pathogenesis to
cure. Best Pract Res Clin Gastroenterol. 21:997–1014. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ghaneh P, Costello E and Neoptolemos JP:
Biology and management of pancreatic cancer. Postgrad Med J.
84:478–497. 2008. View Article : Google Scholar
|
|
3
|
Chueh AC, Togel L, Mariadason J and Tse
JW: Mechanisms of HDAC inhibitor-regulated gene expression in
cancer cells. Antioxid Redox Signal. March 27–2014.[Epub ahead of
print].
|
|
4
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 124:30–39.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barneda-Zahonero B and Parra M: Histone
deacetylases and cancer. Mol Oncol. 6:579–589. 2012. View Article : Google Scholar
|
|
6
|
Huang X, Ji G, Wu Y, Wan B and Yu L:
LAMA4, highly expressed in human hepatocellular carcinoma from
Chinese patients, is a novel marker of tumor invasion and
metastasis. J Cancer Res Clin Oncol. 134:705–714. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xue Y, Ren H, Xiao W, Chu Z, Lee JJ and
Mao L: Antitumor activity of AZ64 via G2/M arrest in non-small cell
lung cancer. Int J Oncol. 41:1798–1808. 2012.PubMed/NCBI
|
|
8
|
Giovannetti E, Del TM, Mey V, Funel N,
Nannizzi S, Ricci S, Orlandini C, Boggi U, Campani D, Del CM,
Iannopollo M, Bevilacqua G, Mosca F and Danesi R: Transcription
analysis of human equilibrative nucleoside transporter-1 predicts
survival in pancreas cancer patients treated with gemcitabine.
Cancer Res. 66:3928–3935. 2006. View Article : Google Scholar
|
|
9
|
Bardeesy N, Aguirre AJ, Chu GC, Cheng KH,
Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U,
Hanahan D, Redston MS, Chin L and Depinho RA: Both p16(Ink4a) and
the p19(Arf)-p53 pathway constrain progression of pancreatic
adenocarcinoma in the mouse. Proc Natl Acad Sci USA. 103:5947–5952.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ansari D, Rosendahl A, Elebro J and
Andersson R: Systematic review of immunohistochemical biomarkers to
identify prognostic subgroups of patients with pancreatic cancer.
Br J Surg. 98:1041–1055. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hamada S, Masamune A, Miura S, Satoh K and
Shimosegawa T: MiR-365 induces gemcitabine resistance in pancreatic
cancer cells by targeting the adaptor protein SHC1 and
pro-apoptotic regulator BAX. Cell Signal. 26:179–185. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen Z, Chen LY, Dai HY, Wang P, Gao S and
Wang K: miR-301a promotes pancreatic cancer cell proliferation by
directly inhibiting Bim expression. J Cell Biochem. 113:3229–3235.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Genin P, Lin R, Hiscott J and Civas A:
Recruitment of histone deacetylase 3 to the interferon-A gene
promoters attenuates interferon expression. PLoS One. 7:e383362012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Karagianni P and Wong J: HDAC3: taking the
SMRT-N-CoRrect road to repression. Oncogene. 26:5439–5449. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pokholok DK, Harbison CT, Levine S, Cole
M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer
E, Zeitlinger J, Lewitter F, Gifford DK and Young RA: Genome-wide
map of nucleosome acetylation and methylation in yeast. Cell.
122:517–527. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guenther MG, Levine SS, Boyer LA, Jaenisch
R and Young RA: A chromatin landmark and transcription initiation
at most promoters in human cells. Cell. 130:77–88. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu C, Liu L, Shan J, Shen J, Xu Y, Zhang
Q, Yang Z, Wu L, Xia F, Bie P, Cui Y, Zhang X, Bian X and Qian C:
Histone deacetylase 3 participates in self-renewal of liver cancer
stem cells through histone modification. Cancer Lett. 339:60–69.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Minamiya Y, Ono T, Saito H, Takahashi N,
Ito M, Motoyama S and Ogawa J: Strong expression of HDAC3
correlates with a poor prognosis in patients with adenocarcinoma of
the lung. Tumour Biol. 31:533–539. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weichert W, Roske A, Gekeler V, Beckers T,
Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M
and Kristiansen G: Histone deacetylases 1, 2 and 3 are highly
expressed in prostate cancer and HDAC2 expression is associated
with shorter PSA relapse time after radical prostatectomy. Br J
Cancer. 98:604–610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu LM, Yang Z, Zhou L, Zhang F, Xie HY,
Feng XW, Wu J and Zheng SS: Identification of histone deacetylase 3
as a biomarker for tumor recurrence following liver transplantation
in HBV-associated hepatocellular carcinoma. PLoS One. 5:e144602010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Glaser KB, Li J, Staver MJ, Wei RQ, Albert
DH and Davidsen SK: Role of class I and class II histone
deacetylases in carcinoma cells using siRNA. Biochem Biophys Res
Commun. 310:529–536. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu J, Wan H, Xue C, Jiang T, Qian C and
Zhang Y: Histone deacetylase 3 implicated in the pathogenesis of
children glioma by promoting glioma cell proliferation and
migration. Brain Res. 1520:15–22. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Campos B, Bermejo JL, Han L, Felsberg J,
Ahmadi R, Grabe N, Reifenberger G, Unterberg A and Herold-Mende C:
Expression of nuclear receptor corepressors and class I histone
deacetylases in astrocytic gliomas. Cancer Sci. 102:387–392. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Spurling CC, Godman CA, Noonan EJ,
Rasmussen TP, Rosenberg DW and Giardina C: HDAC3 overexpression and
colon cancer cell proliferation and differentiation. Mol Carcinog.
47:137–147. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sonnemann J, Marx C, Becker S, Wittig S,
Palani CD, Kramer OH and Beck JF: p53-dependent and p53-independent
anticancer effects of different histone deacetylase inhibitors. Br
J Cancer. 110:656–667. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fortson WS, Kayarthodi S, Fujimura Y, Xu
H, Matthews R, Grizzle WE, Rao VN, Bhat GK and Reddy ES: Histone
deacetylase inhibitors, valproic acid and trichostatin-A induce
apoptosis and affect acetylation status of p53 in ERG-positive
prostate cancer cells. Int J Oncol. 39:111–119. 2011.
|
|
27
|
Jung KH, Noh JH, Kim JK, Eun JW, Bae HJ,
Xie HJ, Chang YG, Kim MG, Park H, Lee JY and Nam SW: HDAC2
overexpression confers oncogenic potential to human lung cancer
cells by deregulating expression of apoptosis and cell cycle
proteins. J Cell Biochem. 113:2167–2177. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nimmanapalli R, Fuino L, Stobaugh C,
Richon V and Bhalla K: Cotreatment with the histone deacetylase
inhibitor suberoylanilide hydroxamic acid (SAHA) enhances
imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia
cells. Blood. 101:3236–3239. 2003. View Article : Google Scholar
|
|
29
|
Huang L and Pardee AB: Suberoylanilide
hydroxamic acid as a potential therapeutic agent for human breast
cancer treatment. Mol Med. 6:849–866. 2000.PubMed/NCBI
|
|
30
|
Zhao Y, Tan J, Zhuang L, Jiang X, Liu ET
and Yu Q: Inhibitors of histone deacetylases target the Rb-E2F1
pathway for apoptosis induction through activation of proapoptotic
protein Bim. Proc Natl Acad Sci USA. 102:16090–16095. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu W, Ngo L, Perez G, Dokmanovic M and
Marks PA: Intrinsic apoptotic and thioredoxin pathways in human
prostate cancer cell response to histone deacetylase inhibitor.
Proc Natl Acad Sci USA. 103:15540–15545. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Reichert N, Choukrallah MA and Matthias P:
Multiple roles of class I HDACs in proliferation, differentiation,
and development. Cell Mol Life Sci. 69:2173–2187. 2012. View Article : Google Scholar : PubMed/NCBI
|