Introduction
Apo2 ligand/tumor necrosis factor (TNF)-related
apoptosis-inducing ligand (Apo2L/TRAIL), a member of the TNF
superfamily, has emerged as a promising candidate for cancer
therapy on the basis of its capacity to induce apoptosis in various
types of cancer cells without significant cytotoxicity toward
normal cells (1–4). Binding of TRAIL to two death
receptors (DRs), TRAIL receptor (TRAIL-R)1/DR4 and TRAIL-R2/DR5
triggers the extrinsic and intrinsic apoptotic pathways (5,6).
However, some cancer cell types such as melanoma, non-small cell
lung cancer and osteosarcoma are resistant to TRAIL, despite
expressing DRs (7,8). Moreover, TRAIL-responsive tumors
acquire resistance rendering TRAIL therapy ineffective. Thus, drugs
that overcome this resistance are urgently needed for effective
TRAIL therapy.
Mitochondria are highly dynamic organelles with a
reticular organization that is regulated by the balance between
fission and fusion. Mitochondrial morphology is critical for cell
function and survival (9,10). The mitochondrial network depends on
the delicate balance between two antagonistic machineries
responsible for fission and fusion of the mitochondrial membrane.
Disruption of mitochondrial fission leads to an extensively
interconnected and collapsed mitochondrial network, while defects
in mitochondrial fusion lead to mitochondrial fragmentation and
loss of mitochondrial DNA (11).
Mitochondrial fission helps to eliminate damaged mitochondria
through autophagy (mitophagy) (12). On the other hand, mitochondrial
fusion facilitates the exchange of mitochondrial DNA and
metabolites required for mitochondrial function (13). Efficient mitochondrial fusion is
important for cell viability, because cells defective in fusion
exhibit reduced growth, decreased mitochondrial membrane potential
(ΔΨm), and defective respiration (14). Mitochondrial fusion and fission in
mammalian cells are controlled by dynamin-related proteins with
GTPase activity, namely mitofusin 1/2 (Mfn1/2), optic atrophy 1 and
dynamin-related protein 1 (Drp1). Mfn1/2, and optic atrophy 1 act
in concert to regulate mitochondrial fusion and cristae
organization, while Drp1 regulates mitochondrial fission (14,15).
Controversial results have been reported on the role
of mitochondrial fission in cancer cell apoptosis. Mitochondrial
fission has been shown to act as both pro-apoptotic and
anti-apoptotic events in cancer cell apoptosis, depending on the
cell types and the apoptotic stimuli applied (16–22).
At present, there is no model that can depict the dual functions of
mitochondrial fission in cancer cell apoptosis. Mitochondrial
division inhibitor-1 (mdivi-1) was recently identified as a potent
inhibitor of Drp1 GTPase in yeast and mammalian cells (23). To gain insight into the role of
mitochondrial morphology dynamics in cancer cell apoptosis, we
examined whether mdivi-1 affected mitochondrial morphology and
TRAIL-induced apoptosis. We found that mdivi-1 killed different
cancer cell types including malignant melanoma, lung cancer and
osteosarcoma cells, while sparing normal cells. mdivi-1 treatment
also sensitized cancer cells to TRAIL-induced apoptosis. This
potentiation of apoptosis by mdivi-1 occurred through activation of
mitochondrial and endoplasmic reticulum (ER) pathways by increasing
mitochondrial oxidative stress, a major cause of these
pathways.
Materials and methods
Reagents
Soluble recombinant human TRAIL and mdivi-1 were
obtained from Enzo Life Sciences (San Diego, CA, USA).
Thapsigargin, rotenone, antimycin A, oligomycin and carbonyl
cyanide p-trifluoromethoxyphenylhydrazone (FCCP) were obtained from
Sigma-Aldrich (St. Louis, MO, USA). The general caspase inhibitor
z-VAD-fluoromethylketone (FMK), caspase-3/7-specific inhibitor
z-DEVD-FMK, caspase-8-specific inhibitor z-IETD-FMK, and
caspase-9-specific inhibitor z-LEHD-FMK were purchased from Merck
Japan. The reagents were dissolved in dimethylsulfoxide and diluted
with Hanks’ balanced salt solution (HBSS) (pH 7.4) to a final
concentration of <0.1% before use.
Cell culture
The human melanoma cell lines, human A549 lung
cancer cells, osteosarcoma cell lines, and human fetal
fibroblast-like lung WI-38–40 cells were obtained from Health
Science Research Resource Bank (Osaka, Japan). These cell lines
were cultured in Dulbecco’s modified Eagle’s medium (DMEM;
Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS;
Sigma-Aldrich) in a 5% CO2 incubator. Normal human
epidermal melanocytes were obtained from Cascade Biologics and
cultured in DermaLife Basal Medium supplemented with DermaLife M
LifeFactors (Kurabo, Osaka, Japan). Cells were harvested by
incubation in 0.25% trypsin-ethylenediamine-tetraacetic acid (Life
Technologies Japan) for 5 min at 37°C.
Cell viability and apoptosis
measurements
Cell viability was measured with WST-8 assay using
the Cell Counting kit (Dojindo, Kumamoto, Japan), a colorimetric
assay based on the formation of a water-soluble formazan product.
Cells (1×103/well) were seeded in 96-well plates and
cultured with the agents to be tested for 72 h at 37°C in a 5%
CO2 incubator. Then 1/10 volume of WST-8 reagent was
added, incubated for 1 h at 37°C and absorbance at 450 nm was
measured using a microplate reader (ARVO MX, Perkin Elmer, Japan).
Apoptotic cell death was quantitatively assessed by double-staining
with fluorescein isothiocyanate (FITC)-conjugated Annexin V and
propidium iodide (PI) as previously described (24). Briefly, cells
(2×105/well) in 24-well plates were incubated with the
agents to be tested for 24 h in 10% FBS-containing medium at 37°C.
Subsequently, the cells were stained with FITC-conjugated Annexin V
and PI using a commercially available kit (Annexin V FITC Apoptosis
Detection kit I; BD Biosciences, Japan). The stained cells were
evaluated in the FACSCalibur and analyzed using CellQuest software
(BD Biosciences). Four cellular subpopulations evaluated: viable
cells (Annexin V−/PI−); early apoptotic cells
(Annexin V+/PI−); late apoptotic cells
(Annexin V+/PI+); and necrotic/damaged cells
(Annexin V−/PI+). Annexin V+ cells
were considered to be apoptotic cells.
Mitochondrial morphology imaging
Mitochondrial morphology was analyzed by staining
with the mitochondria-targeting dye MitoTracker® Red
CMXRos (Life Technologies Japan) followed by observation under a
fluorescence microscope. Briefly, cells (1×104/well) in
DMEM were placed on coverslips (Asahi Glass Co, Tokyo, Japan) and
treated with the agents to be tested for 24 h at 37°C in a 5%
CO2 incubator. After removing the medium, the cells were
washed and stained with 20 nM MitoTracker Red CMXRos and Hoechst
33342 (Dojindo) in HBSS for 1 h at 37°C in the dark in a 5%
CO2 incubator. The cells were then washed with HBSS and
immersed in HBSS to prevent cell damage. Low magnification images
were obtained with a fluorescence microscope (IX71 inverted
microscope, Olympus, Tokyo, Japan) and analyzed using LuminaVision
software (Mitani Corporation, Fukui, Japan). High magnification
images were gained and analyzed with EVOS FL Cell Imaging System
(Life Technologies Japan).
Caspase-3/7 activation and ΔΨm
measurements
Activation of caspase-3/7 and changes in
ΔΨm were simultaneously measured by flow cytometry as
previously described (24).
Briefly, cells (2×105/ml) in 24-well plates were treated
with the agents to be tested for 24 h in 10% FBS/DMEM at 37°C and
then stained with the dual sensor MitoCasp™ kit (Cell Technology,
Mountain View, CA, USA). Caspase-3/7 activation and ΔΨm
were evaluated using FACSCalibur, and the data were analyzed using
CellQuest software.
Caspase-12 activation assay
Activation of caspase-12 in living cells was
measured using the caspase-12 inhibitor ATAD-FMK conjugated to FITC
(FITC-ATAD-FMK) as previously described (24). FITC-ATAD-FMK is cell-permeable and
non-toxic and binds irreversibly to active caspase-12, but not
inactive caspase-12, in apoptotic cells. Briefly, cells
(2×105/ml) in 24-well plates were treated with the
agents to be tested for 24 h in 10% FBS/DMEM at 37°C and then
stained with a CaspGlow™ Fluorescein Active Caspase-12 Staining kit
(BioVision, Mountain View, CA, USA). Fluorescence was determined
using the FL-1 channel of FACSCalibur and analyzed using CellQuest
software.
Determination of surface DR4/DR5
expression
DR4 and DR5 expression on the cell surface was
assessed by flow cytometry as previously described (25). Briefly, cells (5×105/100
μl) were incubated with monoclonal anti-human DR4 and DR5
antibodies or mouse isotype-matched control antibodies (R&D
Systems, Minneapolis, MN, USA) for 30 min at 4°C. The cells were
then centrifuged into a pellet, resuspended in phosphate-buffered
saline (PBS), and then incubated with phycoerythrin-conjugated goat
F(ab’)2 anti-mouse IgG (R&D Systems) for 30 min at
4°C. The fluorescence was measured using the FL-2 channel of
FACSCalibur and analyzed using CellQuest software.
Mitochondrial oxidative stress
measurements
Mitochondrial oxidative stress was measured by
monitoring mitochondrial ROS production using MitoSOX™ Red (Life
Technologies Japan) by flow cytometry and the signals were
calibrated as previously described (26). Briefly, cells (5×105/500
μl) suspended in HBSS were incubated with the agents to be tested
for 4 h at 37°C and then incubated with 5 μM MitoSOX for 15 min at
37°C for loading. The cells were washed, resuspended in HBSS on
ice, centrifuged at 4°C and analyzed for their fluorescence.
Mitochondrial oxidative stress was also assessed by measuring
oxidation of cardiolipin by flow cytometry using the fluorescent
dye 10-N-nonyl acridine orange (NAO, Life Technologies Japan),
which binds to non-oxidized cardiolipin, but not oxidized
cardiolipin, as previously described (26). Briefly, cells (5×105/500
μl) suspended in HBSS were incubated with the agents to be tested
for 4 h at 37°C and then incubated with 100 nM NAO for 15 min at
37°C. The cells were washed and resuspended in HBSS on ice, and
fluorescence was analyzed. Red fluorescence (MitoSOX) and green
fluorescence (NAO) were measured using the FL-2 and FL-1 channel,
respectively, of FACSCalibur and analyzed using CellQuest software.
The data were expressed as F/F0, where
F0 is the fluorescence of unstimulated
cells and F is the fluorescence of stimulated cells.
Drp1 gene silencing by siRNA
Mitochondrial fission was inhibited by
downregulating Drp1 expression using small interfering RNA. Cells
(2.5×105/well) were plated in 6-well plates and
transfected with 20 nM each of Drp1-targeting siRNA or scrambled
control siRNA (Santa Cruz Biotechnology, Santa Cruz, CA, USA) using
Lipofectamine RNA/Max Kit (Life Technologies Japan) according to
the manufacturer’s instructions and cultured for 72 h at 37°C in a
5% CO2 incubator. The downregulation of Drp1 protein
levels was assessed by immunoblotting.
Immunoblotting
The levels of Drp1, pro-caspase and cleaved
caspase-3 proteins were determined using immunoblotting. For the
analysis of Drp1 protein levels, cells (1×106/ml) were
washed with PBS, and lysed with RIPA buffer containing protease
inhibitors. Whole cell lysates (30 μg protein) were subjected to
reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) using a 10% separation gel (ATTO Co., Tokyo, Japan) and
transferred onto polyvinylidene difluoride membranes (Nippon
Millipore, Tokyo, Japan). The membranes were blocked with BlockAce
(Dainippon Sumitomo Pharma, Osaka, Japan) for 1 h at room
temperature, washed with PBS containing 0.1% Tween-20, then
incubated with anti-Drp1 antibody (Santa Cruz Biotechnology)
overnight at 4°C followed by incubation with horseradish peroxidase
(HRP)-conjugated species-specific anti-rabbit Ig (GE Healthcare
Japan, Tokyo, Japan) for 1 h at room temperature. After washing,
immunoreactive proteins were detected using the ECL Prime Kit (GE
Healthcare Japan). For the analysis of caspase-3 protein levels,
cells (1×106/ml) in 10% FBS/DMEM were treated with the
agents to be tested for the indicated times, washed with PBS and
lysed with RIPA buffer. Whole cell lysates (15 μg protein) were
separated by SDS-PAGE using 4–12% gradient gel (Life Technologies
Japan) and transferred onto Immobilon-P membranes (Nippon
Millipore). The membranes were blocked with Blocking-One (Nacalai
Tesque, Tokyo, Japan) overnight at 4°C, and then incubated with
anti-caspase-3 or anti-cleaved caspase-3 antibodies (Cell Signaling
Technology Japan) overnight at 4°C, followed by incubation with
HRP-conjugated secondary antibody (GE Healthcare Japan) for 1 h at
room temperature. After washing in PBS containing 0.1% Tween-20,
immunoreactive bands were visualized with Chemi-Lumi One Super
(Nacalai Tesque). To verify equal loading, the membranes were
re-probed with monoclonal anti-GAPDH (Santa Cruz Biotechnology) or
anti-β-actin antibody (Sigma-Aldrich).
Statistical analysis
Data were analyzed by one-way analysis of variance
followed by the post-hoc Tukey’s test. All values were expressed as
mean ± SE, and P<0.05 was considered to be significant.
Results
Mdivi-1 reduces cell viability and
sensitizes cancer cells to TRAIL cytotoxicity
We examined the ability of mdivi-1 to affect cell
viability in human cancer cells. For this purpose, we utilized
different cancer cell types with different sensitivities to TRAIL:
the human malignant melanoma cell lines A375 and GAK and the human
lung cancer cell line A549. Cells were treated with mdivi-1 and
TRAIL alone or in combination for 24–72 h, and cell viability was
measured using WST-8 assay. In all cell lines tested, mdivi-1 alone
up to 50 μM only modestly decreased cell viability in the initial
24 h. However, mdivi-1 (≥12.5 μM) caused a robust decrease in cell
viability at 72 h in a dose-dependent manner (Fig. 1A–C). GAK cells, which were
relatively sensitive to TRAIL cytotoxicity, were also sensitive to
mdivi-1 (maximum of 40% decrease at 50 μM). Moreover, mdivi-1
markedly sensitized these cells to TRAIL cytotoxicity during the
initial 24 h (data not shown) and this effect became increasing
prominent over another 48 h (Fig.
1A–C). The human osterosarcoma cell lines MG63, HOS and G292
were relatively resistant to TRAIL, because treatment with 100
ng/ml TRAIL for 72 h resulted in only a modest decrease (maximum of
30%) in cell viability. In contrast, mdivi-1 alone dose-dependently
decreased cell viability in all these cell lines. In addition,
mdivi-1 markedly potentiated TRAIL cytotoxicity (Fig. 1D–F). This effect was clearer in
TRAIL-sensitive cells (MG63) than in TRAIL-resistant cells (HOS and
G292). These results show that mdivi-1 decreases cell viability and
sensitizes cancer cells to TRAIL cytotoxicity.
mdivi-1 potentiates DR4/5-mediated
apoptosis in cancer cells
To clarify the mode of cell death potentiated by
mdivi-1, A375 cells were analyzed for Annexin V and propidium
iodide (PI) double staining by flow cytometry. Fig. 2A shows typical results. mdivi-1 up
to 25 μM alone induced minimal increase in Annexin V+
cells but enhanced the increase by TRAIL, indicating that mdivi-1
sensitizes these cells to TRAIL-induced apoptosis. This
sensitization was dose-dependent with a minimal effective dose of
12.5 μM and was clearly observed with TRAIL (≥25 ng/ml) (Fig. 2A and B). In contrast, as previously
reported by others (27),
thapsigargin up to 1 μM was ineffective at inducing apoptosis, and
mdivi-1 did not potentiate the effect (Fig. 2A and C). mdivi-1 also sensitized
the cells to apoptosis induced by the anti-DR5 or DR4 agonist
antibodies (Fig. 2D), suggesting
that the sensitization mainly occurs through increased DR4/DR5
apoptotic signaling. Similarly, mdivi-1 substantially sensitized
A549 cells to TRAIL-induced apoptosis (Fig. 2E). TRAIL up to 100 ng/ml minimally
increased the number of necrotic (Annexin
V−/PI+) cells (<2.5%), and mdivi-1 did not
significantly increase this cell population in either of the two
cell types, indicating that necrotic cell death plays a minor role
in the potentiation effect. In contrast, MG63 cells displayed
robust basal necrotic cell death (11.1±2.4%, n=3), which was
increased by TRAIL up to 16%, while mdivi-1 decreased it.
Interestingly, as basal necrotic cell death increased, apoptosis
was induced to a lesser extent in response to TRAIL and/or mdivi-1,
suggesting a negative effect of necrotic cell death on apoptosis
rate. Collectively, these results show that mdivi-1 potentiates
DR4/5-mediated cell death in different human cancer cell types.
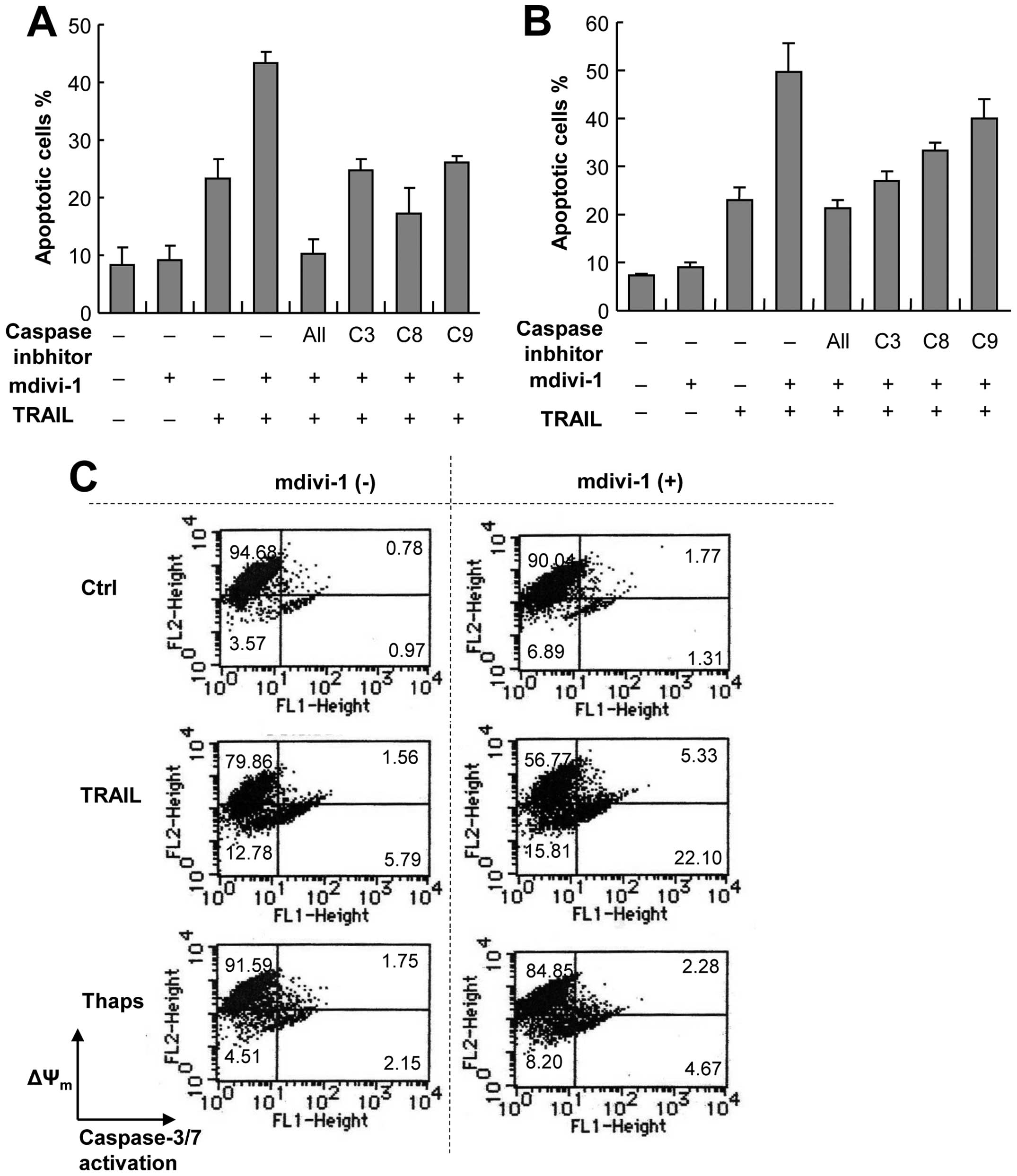
Potentiation of apoptosis by mdivi-1
occurs through a caspase-dependent pathway
Because of their easy handling, high responsivity to
mdivi-1, and the availability of normal counterparts, we used A375
and A549 cells as models for investigating the molecular mechanisms
underlying the potentiation effect of mdivi-1. To elucidate the
possible role of the caspase cascade, we examined the effects of an
array of caspase inhibitors on the potentiation mediated by
mdivi-1. The general caspase inhibitor z-VAD-FMK completely
abolished the potentiation of apoptosis in A375 cells, indicating
that the effect is caspase-dependent (Fig. 3A). The caspase-8-specific inhibitor
z-IETD-FMK, the caspase-9-specific inhibitor z-LEHD-FMK and the
caspase-3/7-specific inhibitor z-DEVD-FMK all abolished the
potentiation of apoptosis almost completely, suggesting the
involvement of both the extrinsic and intrinsic death pathways in
the potentiation (Fig. 3A).
Similar results were obtained with A549 cells except that the
caspase-8 and caspase-9 inhibitors were less effective than the
caspase-3 inhibitor (Fig. 3B).
These results show that the potentiation of apoptosis occurs
through a caspase-dependent pathway involving caspase-3 activation.
To provide insight into the mechanisms underlying the potentiation,
we examined the effects of TRAIL and mdivi-1 on ΔΨm and
caspase-3 activation, because loss of ΔΨm impairs
mitochondrial integrity disruption, resulting in the release of
pro-apoptotic factors and caspase-3 activation. Simultaneous
measurements using a ΔΨm-specific dye and a
caspase-3-specific substrate revealed that TRAIL induced
substantial ΔΨm dissipation and caspase-3 activation in
A375 cells in a dose-dependent manner within 24 h (Fig. 3C). Although mdivi-1 up to 50 μM
alone caused only modest loss of ΔΨm and caspase-3
activation, it markedly potentiated the effects of TRAIL (Fig. 3C–E). In contrast, mdivi-1 had
little effects on ΔΨm or caspase-3 activation induced by
thapsigargin (Fig. 3C).
Immunoblotting provided additional evidence for the potentiation of
TRAIL-induced caspase-3 activation by Mdivi-1. Mdivi-1 or TRAIL
alone induced minimal cleavage of procaspase-3 (p32), while the
combination of TRAIL and mdivi-1 caused a robust increase in levels
of cleaved (active) caspase-3 (p17/p12). The cleaved caspase-3 was
first detected at 8 h after stimulation and levels continued to
increase for another 4 h. Thereafter levels decreased probably due
to additional processing (Fig.
3F). These results show that the potentiation of apoptosis by
mdivi-1 occurs through a caspase-dependent pathway including the
intrinsic pathway.
Potentiation of apoptosis by mdivi-1 is
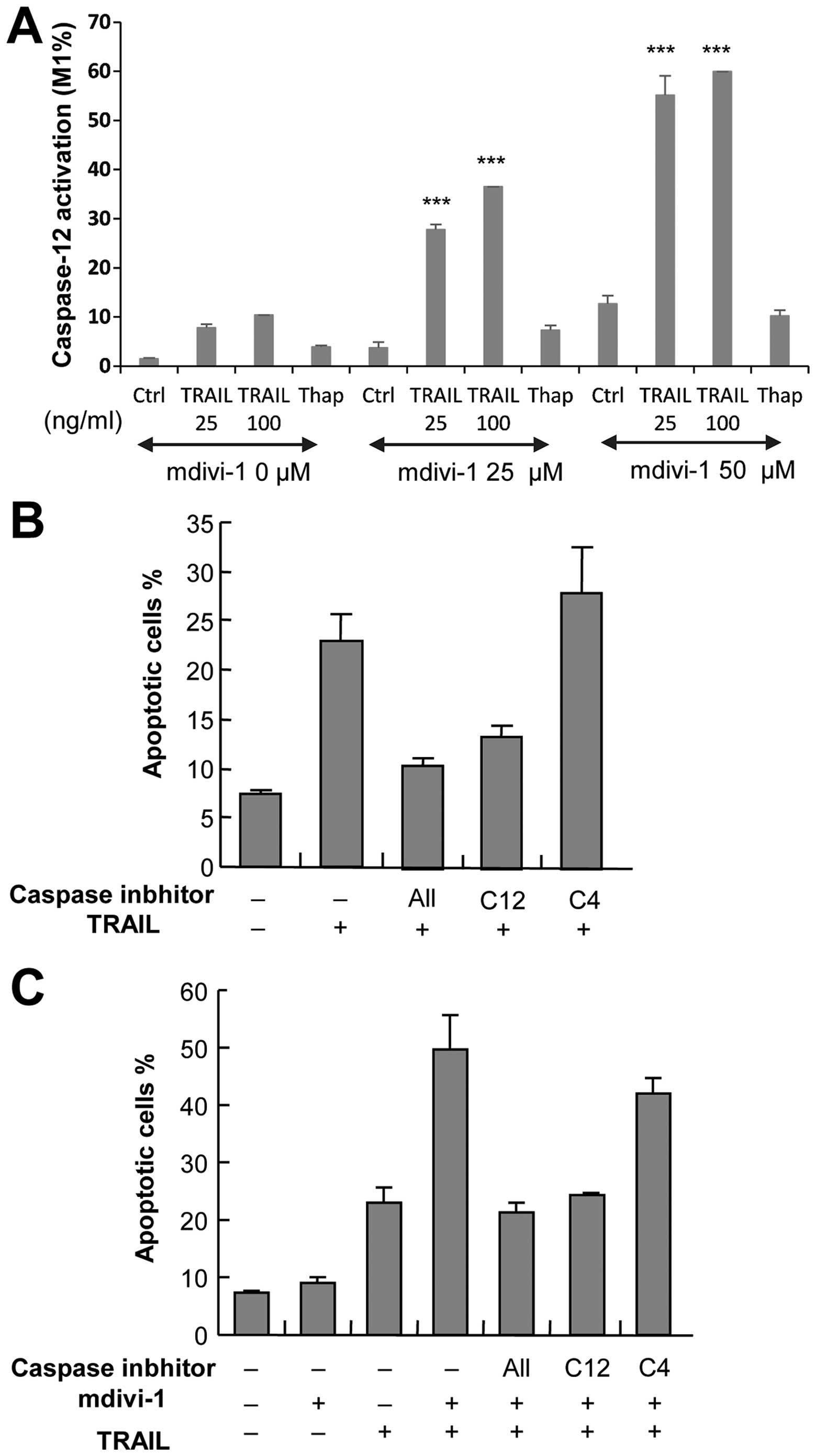
mediated by caspase-12 activation
To explore the role of ER death pathway in the
potentiation of apoptosis by mdivi-1, we examined the ability of
mdivi-1 to modulate activation of caspase-12, an ER-associated
caspase, which occurs independently of the extrinsic and intrinsic
death pathways. Measurements of caspase-12 activity using a
specific substrate revealed that basal caspase-12 activation was
low (1.46±0.19%, n=3), and 100 ng/ml TRAIL and 50 μM mdivi-1
individually induced caspase-12 activation to a similar extent
(10.4 and 15.4%, respectively). However, synergistic effects were
observed when these two agents were used together (59.9%) (Fig. 4A). Next, we examined the effect of
z-ATAD-FMK, a caspase-12-specific inhibitor, on apoptosis.
TRAIL-induced apoptosis was strongly (maximum of 60%) inhibited by
z-ATAD-FMK, while z-LEVD-FMK, an inhibitor of another ER-associated
enzyme, caspase-4, was ineffective (Fig. 4B). Moreover, the potentiation of
TRAIL-induced apoptosis by mdivi-1 was completely abolished by
treatment with z-ATAD-FMK, while z-LEVD-FMK exhibited only modest
(<10%) inhibition (Fig. 4C).
Taken together, these results suggest that potentiation of
apoptosis by mdivi-1 is mediated by caspase-12 activation.
Potentiation of apoptosis by mdivi-1 is
preceded by depolarization and increased mitochondrial oxidative
stress and mass
Persistent cell membrane depolarization is an early
and prerequisite event in apoptosis and caspase-3 activation in
human malignant tumor cells induced by diverse pro-apoptotic
stimuli including TRAIL (24,28–31).
To elucidate the possible role of depolarization in the
TRAIL-sensitizing effect of mdivi-1, we examined the ability of
mdivi-1 to induce depolarization using the anionic dye bis-oxonol
in a flow cytometer. Mdivi-1 alone up to 50 μM caused only modest
depolarization. However, mdivi-1 (≥25 μM) potentiated TRAIL-induced
depolarization as effectively as 5 μg/ml antimycin A (Fig. 5A). Since increase in mitochondrial
ROS (mROS) levels is a major cause of mitochondrial integrity
disruption and the intrinsic pathway, we measured the effect of
mdivi-1 on mROS levels using MitoSOX, a fluoroprobe that targets
mitochondria and serves as a selective probe for superoxide in
these organelles (32,33). As shown in Fig. 5B, 25 and 100 ng/ml TRAIL increased
MitoSOX fluorescence by 1.5- and 3-fold, respectively, at 4 h.
Mdivi-1 alone also robustly increased MitoSOX fluorescence
(3-fold). When TRAIL and mdivi-1 were used together, their effects
were additive (Fig. 5B). We also
assessed oxidation of cardiolipin, a phospholipid that is
associated with cytochrome c in the outer surface of the
inner mitochondrial membrane. Because the fluorescent dye NAO binds
to non-oxidized cardiolipin, but not to oxidized cardiolipin,
measurement of NAO fluorescence can be used to monitor oxidation of
cardiolipin in mitochondria (34).
Fig. 5C shows a representative
histogram. TRAIL increased oxidation of cardiolipin in a
dose-dependent manner, as shown by the decrease in NAO fluorescence
(an increase in the M1 population). Although mdivi-1 alone did not
affect the oxidation of cardiolipin, it markedly potentiated the
effect of TRAIL (Fig. 5C). In
contrast, thapsigargin alone had a minimal effect on the oxidation
of cardiolipin, and mdivi-1 only slightly altered the effect of the
compound. A considerable increase in NAO fluorescence (an increase
in the M2 population) was also observed in cells treated with TRAIL
alone or TRAIL+mdivi-1, suggesting an increase in mitochondrial
mass. However, unlike the MitoSOX signals and cardiolipin
oxidation, the mitochondrial mass changes were not TRAIL
dose-dependent. In contrast, thapsigargin had a minimal effect on
mitochondrial mass. Collectively, these results show that the
potentiation of TRAIL-induced apoptosis by mdivi-1 is preceded by
depolarization and increased mitochondrial oxidative stress and
mass, all of which are potentiated by the compound.
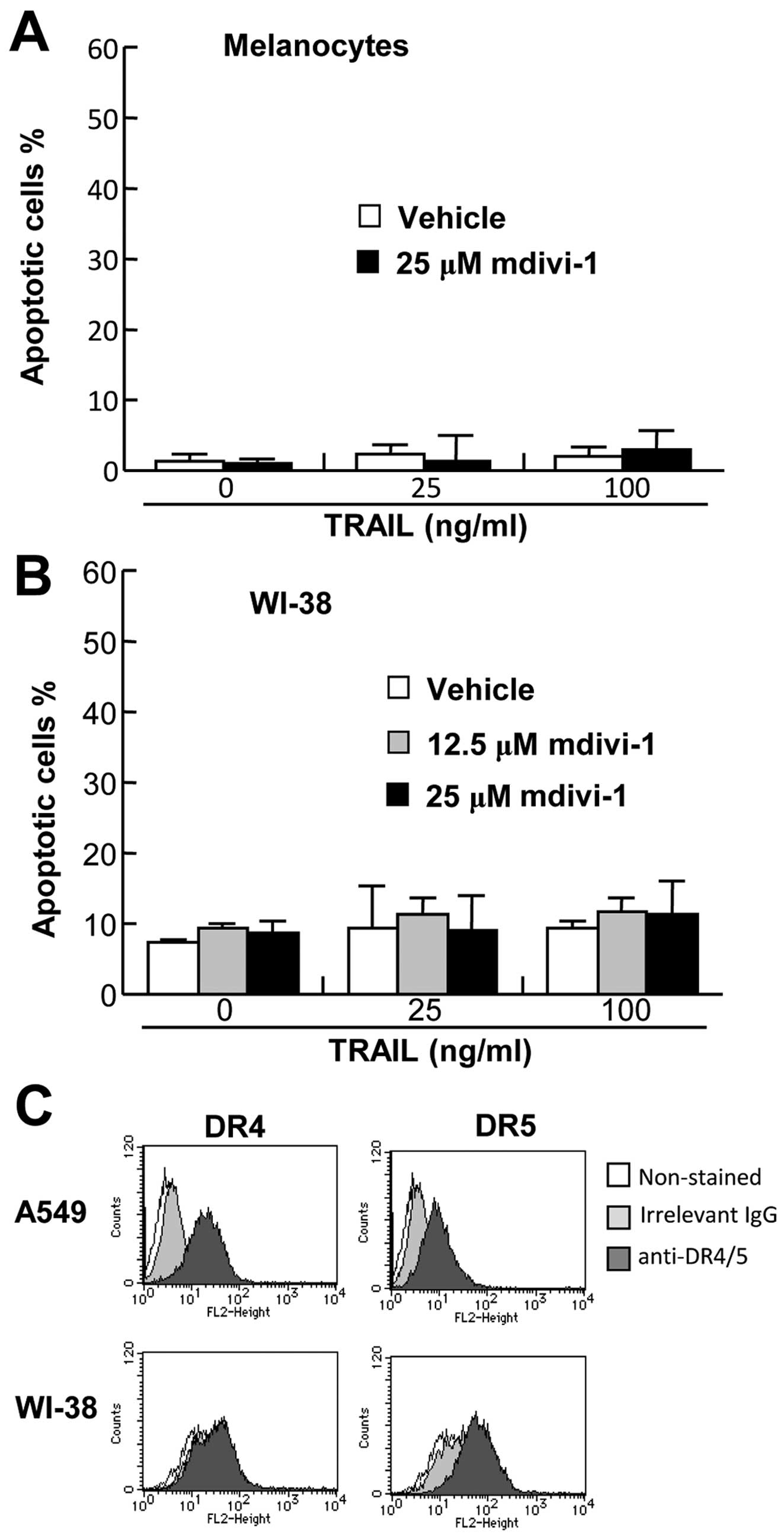
Effects of mdivi-1 are
tumor-selective
To examine the effect of mdivi-1 on cell survival,
primary melanocytes were treated with TRAIL and/or mdivi-1, and
analyzed for Annexin V and PI double staining by flow cytometry. As
shown in Fig. 6A, TRAIL and
mdivi-1 alone or in combination induced minimal apoptosis in the
cells despite robust cell surface expression of DR4 and DR5
(25). We also examined the effect
of TRAIL and/or mdivi-1 on the survival of WI-38 fibroblast-like
lung cells. Again, TRAIL and mdivi-1 alone or in combination
induced minimal apoptosis in these cells (Fig. 6B) despite substantial cell surface
expression of DR5 (Fig. 6C).
Collectively, these findings show that the effects of mdivi-1 are
tumor-selective.
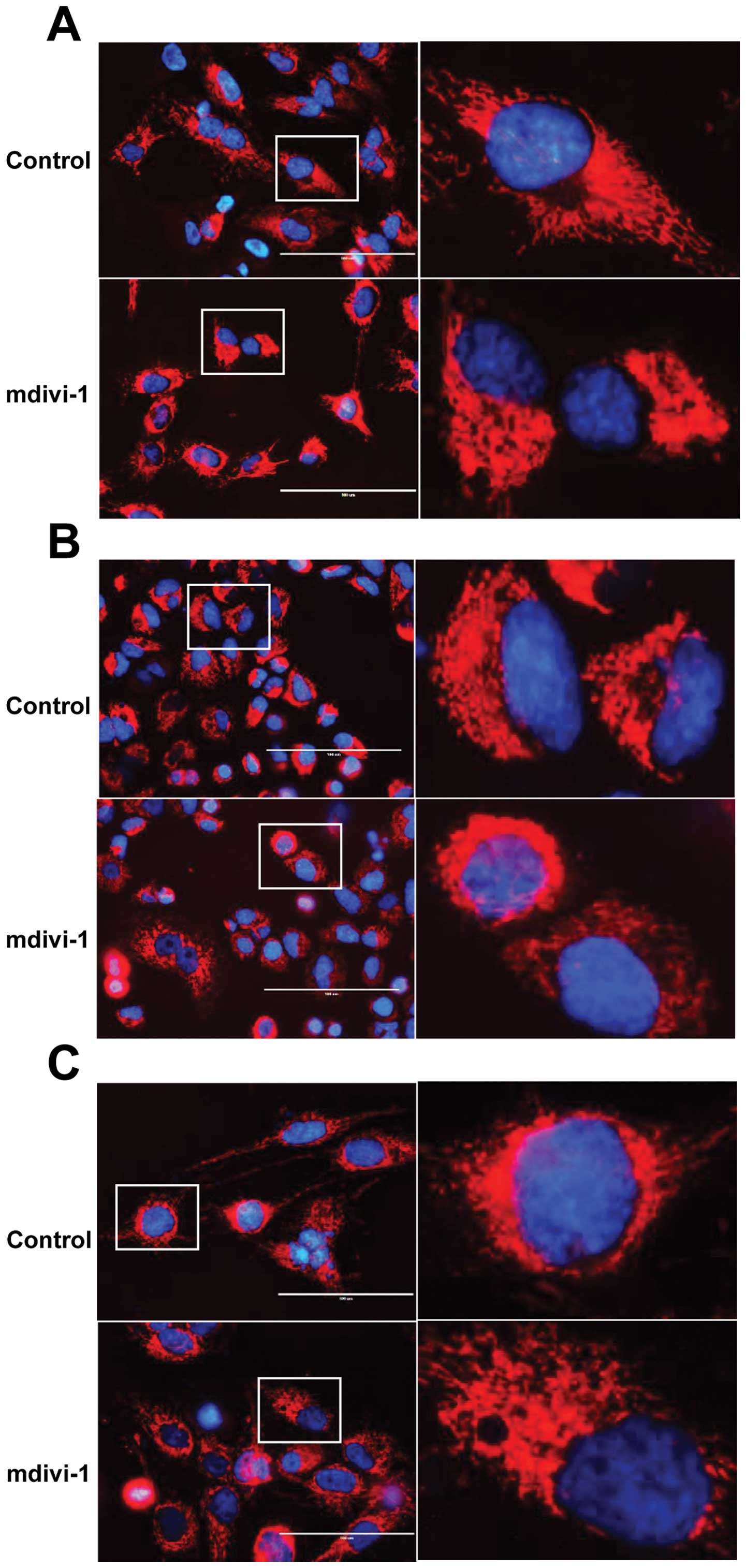
Mdivi-1 exerts its TRAIL-sensitizing
effects through the modulation of mitochondrial morphology
To evaluate the role of mitochondrial morphology
dynamics in the TRAIL-sensitizing effect of mdivi-1, we examined
its effect on mitochondrial morphology. Cells were loaded with the
mitochondria-targeting dye MitoTracker Red and examined under a
fluorescence microscope. A reticular network radiating from the
nucleus was clearly observed in untreated A375 cells (Fig. 7A). As expected, marked
mitochondrial morphological changes were observed at 24 h after the
addition of mdivi-1. Robust mitochondrial hyperfusion with a bias
towards one of the poles of the nucleus was observed (Fig. 7A). Mdivi-1 caused similar
mitochondrial hyperfusion in A549 and MG63 cells (Fig. 7B and C). To examine whether these
effects of mdivi-1 are mediated by an ability to modulate
mitochondrial fission, we analyzed the impact of Drp1 gene
silencing on the mitochondrial morphology, because Drp1 GTPase
activity is essential for mitochondrial fission (14,15).
Treatment with Drp1 siRNA for 72 h resulted in a marked decrease in
Drp1 protein expression compared with control cells treated with a
control siRNA (Fig. 7D, left
panel). In the Drp1 knockdown cells, prominent mitochondrial
hyperfusion was observed, as in mdivi-1-treated cells (Fig. 7E). Moreover, Drp1 knockdown by
itself did not increase basal apoptosis, while A375 and A549 Drp1
knockdown cells were more susceptible than control cells to
TRAIL-induced apoptosis, and this sensitization became more
apparent as TRAIL concentrations increased (Fig. 7F and G). The effects of Drp1
knockdown were specific for TRAIL, because it had minimal effects
on cellular sensitivity to thapsigargin in either cell type
(Fig. 7F and G). These results
show that mdivi-1 sensitizes cancer cells to TRAIL by modulating
mitochondrial morphology.
Discussion
In this study we demonstrate for the first time that
mdivi-1 kills and sensitizes different human cancer cell types to
TRAIL-induced apoptosis. Several lines of evidence indicate that
these effects are mediated by mitochondrial morphology modulation:
i) After mdivi-1 treatment, marked mitochondrial morphological
changes, including mitochondrial hyperfusion with a bias towards a
pole of the nucleus, were detected; ii) similar mitochondrial
hyperfusion was observed in Drp1 knockdown cells; and iii) Drp1
knockdown also sensitized different cancer cell types to
TRAIL-induced apoptosis. Collectively, the present findings
indicate that inhibition of mitochondrial fission potentiates
apoptosis, suggesting that mitochondrial fission inhibits cancer
cell apoptosis. Our findings are similar to previous studies
showing that etoposide and arsenic trioxide induce mitochondrial
aggregation, an event upstream of cytochrome c release
during apoptosis in glioblastoma cells (35,36)
and suggest a universal role for mitochondrial network changes
during cancer cell apoptosis.
Healthy mitochondria are required for cell health,
and numerous diseases including cancer are associated with aberrant
mitochondrial network dynamics (37). Consequently, drugs that modulate
mitochondrial fission and fusion may restore proper dynamics and
have therapeutic potential (37).
However, the role of mitochondrial fission in cancer cell apoptosis
remains elusive, because controversial results have been reported
with regard to its role. Various apoptosis inducers cause extensive
fragmentation of mitochondria in cancer cells concomitant with
mitochondrial outer membrane permeabilization and cytochrome
c release (19,21). Moreover, inhibition of
mitochondrial fission by downregulation of Drp1 or Fis1, a
counterpart of Drp1, has been shown to delay cytochrome c
release, suggesting that mitochondrial fission is important for
apoptosis (16,17,20).
In contrast, other studies have revealed that Drp1-dependent
mitochondrial fragmentation is not a prerequisite for apoptosis and
that they are separate events, because mitochondrial fission was
neither essential for cytochrome c release nor obligatory
for apoptosis (19,22). In addition, several lines of
evidence suggest that mitochondrial fission is protective against
Ca2+-induced apoptosis (14). Human lung cancer cell lines exhibit
an imbalance of Drp1/Mfn-2 expression compared with normal human
lung epithelial and vascular cells and a state of mitochondrial
fragmentation. Lung tumor tissue samples from patients exhibit
similar increase in mitochondrial fragmentation compared with
normal lung tissue. Consistent with a protective role of
mitochondrial fission, inhibition of mitochondrial fragmentation by
Drp1 knockdown or mdivi-1 or promotion of mitochondrial fusion by
Mfn2 overexpression increases spontaneous apoptosis in lung cancer
cells and regress tumor growth in vivo (38). Our data expand these observations
and suggest that mitochondrial fission protects different human
cancer cell types including melanoma, lung cancer and osteosarcoma
cells from apoptosis. Importantly, mdivi-1 had minimal cytotoxicity
and TRAIL-sensitizing effect in normal melanocytes and fibroblasts.
This indicates that mdivi-1 may be selectively cytotoxic toward
tumor cells. Although the mechanisms underlying this selectivity
remain to be elucidated, it is plausible that disruption of
mitochondrial morphology dynamics preferentially causes
mitochondrial dysfunction in cancer cells.
We found that mdivi-1 potentiates multiple
pro-apoptotic events, including membrane depolarization, loss of
ΔΨm, caspase-3 and caspase-12 activation, mROS
generation and cardiolipin oxidation. These observations are in
accordance with our previous studies showing that diverse compounds
commonly potentiate these events and TRAIL-induced apoptosis in
human malignant tumor cells. These compounds include high
K+, ATP-sensitive K+ channel inhibitors such
as glibenclamide and U37883A (24,28),
diallyltrisulfide, a major garlic organosulfur compound (25), the cell-permeable oxidant,
H2O2 (39)
and mitochondrial inhibitors such as antimycin A and FCCP (26). A loss of ΔΨm is the main
cause of mitochondrial outer membrane permeabilization,
mitochondrial integrity disruption and pro-apoptotic protein
release. Mitochondrial outer membrane permeabilization allows
release of several pro-apoptotic proteins including cytochrome
c and apoptosis-inducing factor-1, thereby promoting the
activation of the caspase-9-caspase-3 cascade and processing of key
regulatory and structural proteins. Although the precise mechanisms
of mitochondrial outer membrane permeabilization are poorly
understood, it is widely accepted that an increase in
permeabilization of the inner mitochondrial membrane is involved
and that mitochondrial permeability transition pores (mPTPs) are
thought to play pivotal roles in this process (40,41).
mPTPs are putative high-conductance non-specific channels in the
inner mitochondrial membrane, which are composed of proteins that
link the inner and outer mitochondrial membrane. Several
mitochondrial proteins localized in these membranes such as
voltage-dependent anion channels and adenine nucleotide translocase
are thought to constitute the mPTP. It is thought that concurrent
opening of multiple mPTPs causes excess mitochondrial swelling, the
physical disorganization of the outer mitochondrial membrane,
mitochondrial pro-apoptotic protein release and apoptosis (42). In this regard, it is noteworthy
that the potentiation of apoptosis by mdivi-1 was associated with
increased mitochondrial mass, a hallmark of mitochondrial swelling.
Because adenine nucleotide translocase has three cysteine residues
whose oxidation is critical for mPTP opening/closing, mPTPs are
particularly vulnerable to ROS (40,41,43).
Consequently, mitochondrial permeability transition can be
triggered by excess mROS generation and/or disruption of the
mitochondrial redox homeostasis (44–47).
In addition, mROS can control cytochrome c release through
the oxidation of the mitochondrial phospholipid cardiolipin
(46). The enhancement of membrane
depolarization may be also important in the potentiation of
apoptosis, because persistent depolarization is an early and
prerequisite event in TRAIL-induced apoptosis in cancer cells
(24,28). Importantly, depolarization and mROS
mutually control one another. Depolarization potentiates
TRAIL-induced mROS generation, while scavenging of mROS by
antioxidants reduces depolarization, and mROS generation by
mitochondrial metabolic dysfunction potentiates the depolarization
(28). In addition, scavenging of
mROS inhibits pro-apoptotic events in the intrinsic and ER death
pathways, including ΔΨm dissipation, caspase-3 and
caspase-12 activation, suggesting that mROS plays a central role in
the intrinsic and ER death pathways (48). Collectively, our data suggest that
mitochondrial oxidative stress play a key role in the
TRAIL-sensitizing effect of mdivi-1.
In conclusion, the present study shows that cancer
cells are more vulnerable than normal cells to disruption of
mitochondrial network dynamics and that this higher susceptibility
can be exploited in cancer treatment, in particular in combination
therapy with TRAIL.
Acknowledgements
The authors thank Dr M. Murai and Dr T. Inoue for
technical assistance. This study was supported in part by a
Grant-in-Aid from the Ministry of Education, Culture, Sports,
Science and Technology (KAKENHI 23591631; to Y.S.-K.) and
Grants-in-Aid from Nihon University (to Y.S.-K.).
References
|
1
|
Almasan A and Ashkenazi A: Apo2L/TRAIL:
apoptosis signaling, biology, and potential for cancer therapy.
Cytokine Growth Factor Rev. 14:337–348. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johnstone RW, Frew AJ and Smyth MJ: The
TRAIL apoptotic pathway in cancer onset, progression and therapy.
Nat Rev Cancer. 8:782–798. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang S: The promise of cancer therapeutics
targeting the TNF-related apoptosis-inducing ligand and TRAIL
receptor pathway. Oncogene. 27:6207–6215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gonzalvez F and Ashkenazi A: New insights
into apoptosis signaling by Apo2L/TRAIL. Oncogene. 29:4752–4765.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
LeBlanc HN and Ashkenazi A: Apo2L/TRAIL
and its death and decoy. Cell Death Differ. 10:66–75. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kischkel FC, Lawrence DA, Chuntharapai A,
Schow P, Kim KJ and Ashkenazi A: Apo2L/TRAIL-dependent recruitment
of endogenous FADD and caspase-8 to death receptors 4 and 5.
Immunity. 12:612–620. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dyer MJ, MacFarlane M and Cohen GM:
Barriers to effective TRAIL-targeted therapy of malignancy. J Clin
Oncol. 25:4505–4506. 2007. View Article : Google Scholar
|
|
8
|
Dimberg LY, Anderson CK, Camidge R,
Behbakht K, Thorburn A and Ford HL: On the TRAIL to successful
cancer therapy? Predicting and counteracting resistance against
TRAIL-based therapeutics. Oncogene. 32:1341–1350. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Landes T and Martinou JC: Mitochondrial
outer membrane permeabilization during apoptosis: the role of
mitochondrial fission. Biochim Biophys Acta. 1813:540–545. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Elgass K, Pakay J, Ryan MT and Palmer CS:
Recent advances into the understanding of mitochondrial fission.
Biochim Biophys Acta. 1833:150–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hoppins S, Lackner L and Nunnari J: The
machines that divide and fuse mitochondria. Annu Rev Biochem.
76:751–780. 2007. View Article : Google Scholar
|
|
12
|
Twig G and Shirihai OS: The interplay
between mitochondrial dynamics and mitophagy. Antioxid Redox
Signal. 14:1939–1951. 2011. View Article : Google Scholar
|
|
13
|
Rouzier C, Bannwarth S, Chaussenot A, et
al: The MFN2 gene is responsible for mitochondrial DNA instability
and optic atrophy ‘plus’ phenotype. Brain. 135:23–34.
2012.PubMed/NCBI
|
|
14
|
Chen H, Chomyn A and Chan DC: Disruption
of fusion results in mitochondrial heterogeneity and dysfunction. J
Biol Chem. 280:26185–26192. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chang CR and Blackstone C: Dynamic
regulation of mitochondrial fission through modification of the
dynamin-related protein Drp1. Ann NY Acad Sci. 1201:34–39. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Frank S, Gaume B, Bergmann-Leitner ES, et
al: The role of dynamin-related protein 1, a mediator of
mitochondrial fission, in apoptosis. Dev Cell. 1:515–525. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee YJ, Jeong SY, Karbowski M, Smith CL
and Youle RJ: Roles of the mammalian mitochondrial fission and
fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell.
15:5001–5011. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Arnoult D, Grodet A, Lee YJ, Estaquier J
and Blackstone C: Release of OPA1 during apoptosis participates in
the rapid and complete release of cytochrome c and subsequent
mitochondrial fragmentation. J Biol Chem. 280:35742–35750. 2005.
View Article : Google Scholar
|
|
19
|
Alirol E, James D, Huber D, Marchetto A,
Vergani L and Martinou JC: The mitochondrial fission protein hFis1
requires the endoplasmic reticulum gateway to induce apoptosis. Mol
Biol Cell. 17:4593–4605. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Estaquier J and Arnoult D: Inhibiting
Drp1-mediated mitochondrial fission selectively prevents the
release of cytochrome c during apoptosis. Cell Death Differ.
14:1086–1094. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Suen DF, Norris KL and Youle RJ:
Mitochondrial dynamics and apoptosis. Genes Dev. 22:1577–1590.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sheridan C, Delivani P, Cullen SP and
Martin SJ: Bax- or Bak-induced mitochondrial fission can be
uncoupled from cytochrome c release. Mol Cell. 31:570–585. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cassidy-Stone A, Chipuk JE, Ingerman E, et
al: Chemical inhibition of the mitochondrial division dynamin
reveals its role in Bax/Bak-dependent mitochondrial outer membrane
permeabilization. Dev Cell. 14:193–204. 2008. View Article : Google Scholar
|
|
24
|
Suzuki Y, Inoue T, Murai M,
Suzuki-Karasaki M, Ochiai T and Ra C: Depolarization potentiates
TRAIL-induced apoptosis in human melanoma cells: Role for
ATP-sensitive K+ channels and endoplasmic reticulum
stress. Int J Oncol. 41:465–475. 2012.PubMed/NCBI
|
|
25
|
Murai M, Inoue T, Suzuki-Karasaki M,
Ochiai T, Ra C, Nishida S, et al: Diallyl trisulfide sensitizes
human melanoma cells to TRAIL-induced cell death by promoting
endoplasmic reticulum-mediated apoptosis. Int J Oncol.
41:2029–2037. 2012.
|
|
26
|
Inoue T and Suzuki-Karasaki Y:
Mitochondrial superoxide mediates mitochondrial and endoplasmic
reticulum dysfunctions in TRAIL-induced apoptosis in Jurkat cells.
Free Radic Biol Med. 61:273–284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang CC, Chen LH, Gillespie S, et al:
Tunicamycin sensitizes human melanoma cells to tumor necrosis
factor-related apoptosis-inducing ligand-induced apoptosis by
up-regulation of TRAIL-R2 via the unfolded protein response. Cancer
Res. 67:5880–5888. 2007. View Article : Google Scholar
|
|
28
|
Bortner CD, Gomez-Angelats M and Cidlowski
JA: Plasma membrane depolarization without repolarization is an
early molecular event in anti-Fas-induced apoptosis. J Biol Chem.
276:4304–4314. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nolte F, Friedrich O, Rojewski M, Fink RH,
Schrezenmeier H and Körper S: Depolarisation of the plasma membrane
in the arsenic trioxide (As2O3)-and
anti-CD95-induced apoptosis in myeloid cells. FEBS Lett. 578:85–89.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yin W, Li X, Feng S, et al: Plasma
membrane depolarization and Na,K-ATPase impairment induced by
mitochondrial toxins augment leukemia cell apoptosis via a novel
mitochondrial amplification mechanism. Biochem Pharmacol.
78:191–202. 2009. View Article : Google Scholar
|
|
31
|
Suzuki-Karasaki M, Ochiai T and
Suzuki-Karasaki Y: Crosstalk between mitochondrial ROS and
depolarization in the potentiation of TRAIL-induced apoptosis in
human tumor cells. Int J Oncol. 44:616–628. 2014.PubMed/NCBI
|
|
32
|
Robinson KM, Janes MS, Pehar M, et al:
Selective fluorescent imaging of superoxide in vivo using
ethidium-based probes. Proc Natl Acad Sci USA. 103:15038–15043.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mukhopadhyay P, Rajesh M, Kashiwaya Y,
Haskó G and Pacher P: Simple quantitative detection of
mitochondrial superoxide production in live cells. Biochem Biophys
Res Commun. 358:203–208. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Petit JM, Maftah A, Ratinaud MH and Julien
R: 10-N-nonyl acridine orange interacts with cardiolipin and allows
the quantification of this phospholipid in isolated mitochondria.
Eur J Biochem. 209:267–273. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Haga N, Fujita N and Tsuruo T:
Mitochondrial aggregation precedes cytochrome c release from
mitochondria during apoptosis. Oncogene. 22:5579–5585. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Haga N, Fujita N and Tsuruo T: Involvement
of mitochondrial aggregation in arsenic trioxide
(As2O3)-induced apoptosis in human
glioblastoma cells. Cancer Sci. 96:825–833. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lackner LL and Nunnari J: Small molecule
inhibitors of mitochondrial division: tools that translate basic
biological research into medicine. Chem Biol. 17:578–583. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rehman J, Zhang HJ, Toth PT, et al:
Inhibition of mitochondrial fission prevents cell cycle progression
in lung cancer. FASEB J. 26:2175–2186. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tochigi M, Inoue T, Suzuki-Karasaki M,
Ochiai T, Ra C and Suzuki-Karasaki Y: Hydrogen peroxide induces
cell death in human TRAIL-resistant melanoma through intracellular
superoxide generation. Int J Oncol. 42:863–872. 2013.PubMed/NCBI
|
|
40
|
Halestrap AP and Brennerb C: The adenine
nucleotide translocase: A central component of the mitochondrial
permeability transition pore and key player in cell death. Curr Med
Chem. 10:1507–1525. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lemasters JJ, Theruvath TP, Zhong Z and
Nieminen AL: Mitochondrial calcium and the permeability transition
in cell death. Biochim Biophys Acta. 1787:1395–1401. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hail N Jr: Mitochondria: A novel target
for the chemoprevention of cancer. Apoptosis. 10:687–705. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhivotovsky B, Galluzzi L, Kepp O and
Kroemer G: Adenine nucleotide translocase: a component of the
phylogenetically conserved cell death machinery. Cell Death Differ.
16:1419–1425. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Skulachev VP: Why are mitochondria
involved in apoptosis? Permeability transition pores and apoptosis
as selective mechanisms to eliminate superoxide-producing
mitochondria and cell. FEBS Lett. 397:7–10. 1996. View Article : Google Scholar
|
|
45
|
Orrenius S: Reactive oxygen species in
mitochondria-mediated cell death. Drug Metab Rev. 39:443–455. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ott M, Gogvadze V, Orrenius S and
Zhivotovsky B: Mitochondria, oxidative stress and cell death.
Apoptosis. 12:913–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ralph SJ, Rodríguez-Enríquez S, Neuzil J
and Moreno-Sánchez R: Bioenergetic pathways in tumor mitochondria
as targets for cancer therapy and the importance of the ROS-induced
apoptotic trigger. Mol Aspects Med. 31:29–59. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Suzuki-Karasaki Y, Suzuki-Karasaki M,
Uchida M and Ochiai T: Depolarization controls TRAIL sensitization
and tumor-selective killing of cancer cells: crosstalk with ROS.
Front Oncol. (In press).
|