Introduction
Acute myeloid leukemia (AML) is characterized by
differentiation arrest and inappropriate proliferation and survival
of immature myeloid progenitors. In the majority of patients with
AML who achieve a complete remission (CR), the leukemia will recur
within 3 years after diagnosis. The survival probability at 1 and 5
year is 70 and 46%, respectively, based on the favorable risk
evaluation (1). Acute
promyelocytic leukemia (APL) is a unique subtype of AML. APL
patients achieve CR with all-trans retinoic acid (ATRA) treatment.
However, the remission duration is short due to rapid development
of retinoid resistance in some APL patients (2). Deregulation of phosphatidylinositol
3-kinase (PI3K)/Akt pathway may contribute to tumorigenesis,
metastasis, and resistance to conventional chemotherapy. PI3K/Akt
signaling pathway is frequently activated in AML cells, and it has
been considered as a new target of therapeutic intervention for AML
patients (3–7). Thus, the interruption of PI3K/Akt
signaling pathway should be considered when designing anti-AML
therapeutic strategies.
Emodin is a natural component extracted from the
root and rhizome of Rheum palmatum L. It has multiple
biological activities including antimicrobial, antiviral,
anti-inflammatory, anti-ulcerogenic, immunosuppressive and
chemo-preventive activities (8–10).
The investigations of emodin-mediated anticancer effects are in
progress. For example, emodin, as a tyrosine kinase inhibitor,
suppressed growth of HER-2/neu− overexpressing breast
cancer cells in vivo (11).
Emodin and docosahexaenoic acid (DHA) potently promoted arsenic
trioxide (As2O3) and interferon-α
(IFN-α)-induced cell death in HTLV-I infected cells by generation
of reactive oxygen species and inhibition of Akt and AP-1 pathways
(12). It was also displayed that
emodin enhanced the activity of gemcitabine against pancreatic
cancer in mice by promoting the mitochondrial-dependent apoptotic
pathway (13). Previous studies in
our group have shown that emodin may induce apoptosis and reverse
multidrug resistance in HL-60 and HL-60/ADR cells (14–16),
but the molecular mechanisms have not been completely elucidated.
In the present study, we provided the first demonstration of the
potential roles and mechanisms of emodin alone or in combination
with ATRA in NB4 cells, ATRA-resistant MR2 cells and especially
those primary leukemic cells from newly diagnosed AML patients. We
showed that low dose of emodin could potentially enhance
ATRA-induced terminal differentiation in AML cells, especially, in
ATRA-resistant cells. Emodin could effectively inhibit
proliferation and induce apoptosis with a dose- and time-dependent
manner in AML cells as well as ATRA-resistant leukemic cells. The
critical mechanism underlying these effects is linked to the
inhibition of PI3K/Akt signaling pathway in AML cells. These
results suggest that emodin or emodin in combination with ATRA may
have benefits in AML therapy.
Materials and methods
Chemicals and reagents
Emodin (C15H10O5,
MW: 446.35, HPLC-determined purity >98%) was obtained from
Nanjing Qingze Medical Technology Co. (Nanjing, China). Emodin and
ATRA (Sigma, St. Louis, MO, USA) were reconstituted in 100%
dimethyl sulfoxide (DMSO) to 50,000 μM as a stock solution. The
stock solutions were maintained at −20°C and further diluted in
culture media before use.
Patients and cell lines
ATRA-resistant MR2 cell line and its parental NB4
cell line were obtained from Shanghai Institute of Hematology,
Shanghai Ruijing Hospital, China. Cell lines were cultured in
RPMI-1640 supplemented with 10% FBS, penicillin (100 IU/ml) and
streptomycin (100 μg/ml) based on the supplier’s instructions and
established procedures. The primary leukemia cells from peripheral
blood samples were obtained from 21 newly diagnosed AML patients.
Samples had ≥70% blasts in the initial isolation prior to
manipulation. Normal controls were recruited from 6 healthy donors.
All patient samples were referred to our hospital for
cytomorphological and cytogenetic diagnostics, and were diagnosed
as AML according to standard French-American-British (FAB) and WHO
criteria. Peripheral blood mononuclear cells (PBMCs) were isolated
by Ficoll-Paque density-gradient separation. Informed consent was
obtained from all patients and healthy donors in accordance with
the Declaration of Helsinki, and all manipulations of human
specimens were approved by the institutional review board in Fujian
Medical University Union Hospital.
Cell proliferation assay
NB4, MR2 and primary AML cells plated in 96-well
plates were treated in triplicates with emodin, ATRA alone or
emodin plus ATRA for 48 or 72 h at 37°C. Cells were then incubated
with 5 mg/ml MTT (Sigma) for 4 h. The supernatants were removed and
cells were pulsed with 100 μl DMSO. The optical density (OD) was
measured at 492/630 nm using a spectrophotometer (STAT FAX-2100).
The inhibitory rate on cell proliferation was calculated as
(1-ODtreated/ODcontrol) ×100%. The half
inhibitory concentration (IC50) values were obtained by
the Logit method.
Cell differentiation assay
NB4 and MR2 cells were seeded at a density of
5.0×104/ml in 6-well plates in the presence of either 10
μM emodin, 1.0 μM ATRA alone or 10 μM emodin in combination with
1.0 μM ATRA at 37°C for 96 h. Cells were harvested, washed twice
with phosphate-buffered saline (PBS). Cell differentiation effects
were determined by morphological examination, CD11b analysis and
NBT staining. Cell morphological features were observed by
microscopy after Wright-Giemsa staining. Cell surface
differentiation antigen CD11b (Biolegend, San Diego, CA, USA) was
measured according to the manufacture’s instruction. Data were
acquired by flow cytometry (Beckman Coulter FC500, Fullerton, CA,
USA). The nitroblue tetrazolium chloride (NBT) reduction assay was
performed as previously described (17,18).
Total of 200 cells on each slide were counted under light
microscope, and the percentages of NBT-positive cells were
calculated in each group.
Apoptosis assay
Briefly, 1.0×105/ml of NB4 and MR2 cells
in RPMI-1640 medium with 10% fetal bovine serum were plated in
6-well plates. After 12 h of incubation with emodin, cells were
harvested and washed with PBS, and then stained with Annexin
V-FITC/PI (Becton-Dickinson, NJ, USA) according to the
manufacturer’s instructions. The early apoptotic cells were
quantified by flow cytometry. To further analyze the cell cycle
distribution and the percentages of late apoptotic cells, NB4 and
MR2 cells were harvested after 48 h of treatment with emodin. Cell
pellets were incubated with the DNA PREP LPR solutions (Beckman
Coulter), followed by staining with propidium iodide on ice. Flow
cytometry analysis was performed to determine the fraction of cells
in G0/G1, S, and G2/M. The cells
undergoing apoptosis were obtained from the distinct
sub-G1 region of the DNA distribution histograms.
DNA fragmentation assay
NB4, MR2 and primary AML cells were exposed to
different concentrations of emodin for 48 h. Cells were harvested
by centrifugation. After washed with PBS, DNA fragmentation was
analyzed using the manufacturer’s procedures (Beyotime, Shanghai,
China). In brief, genomic DNA was extracted, then absorbed by a
miniprep spin column and eluted with buffer. Electrophoresis for
purified DNA was performed in a 1.5% agarose gel at 35 V for 3 h.
DNA was visualized by Goldview Nucleic Acid Stain on Gel Image
Analysis System (Peiqing, JS-380A, Shanghai, China).
Western blotting
NB4, MR2 and primary AML cells were exposed to
emodin at varying concentrations and time points. Total protein was
extracted and western blotting was performed as described
previously (18). Quantification
of the band densitometry was performed by Quantity One Version
4.6.2 Software (Bio-Rad, Hercules, CA, USA). Primary antibodies
against human caspase-9, caspase-3, poly(ADP-ribose) polymerase
(PARP), p-Akt (Ser473), Akt, p-mTOR (Ser2448), mTOR, 4E-BP1
(53H11), p-4E-BP1 (Thr70), p70S6K and p-p70S6K (Thr389) were
obtained from Cell Signaling Technology (Beverly, MA, USA). Bcl-2,
RARα and β-actin antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Rapamycin was obtained
from Calbiochem EMD Millipore Corp. (Billerica, MA, USA). LY294002
and PD98059 were obtained from Sigma-Aldrich Corp. (St. Louis, MO,
USA).
Statistical analysis
Results are presented as mean ± standard deviation,
and statistical comparisons of experimental groups were evaluated
by Student’s t-test. Statistical significance was defined as
P<0.05.
Results
Emodin inhibits cell proliferation in NB4
cells, MR2 cells and primary AML cells
MTT assay was used to examine the effects of emodin
on ATRA-resistant MR2 cells and the parental NB4 cells. Consistent
with the results of our previous study in HL-60 and HL-60/ADR
cells, NB4 and MR2 cells were also sensitive to emodin in a
dose-dependent manner (Fig. 1A).
Emodin inhibited MR2 cell proliferation with an average
IC50 value of 34.01±2.40 μM, which was similar to the
effect on the parental NB4 cells yielding an average
IC50 of 37.99±2.30 μM. To identify whether similar
effects were found in the primary AML cells, 21 samples from AML
patients were treated with increasing concentrations of emodin
(ranging from 10 to 80 μM) in vitro for 72 h. As shown in
Fig. 1B, the results demonstrated
that all of the primary AML cells isolated from 21 different
subtypes of AML patients including three AML cases unclassified,
although variable in degree, were sensitive to emodin with the
average IC50 value of 31.22±15.58 μM. While less
cytotoxicity was found when healthy PBMCs were exposed to the same
setting of emodin in vitro. More than 70% cells from the 6
healthy donors remained alive even though administered with emodin
as high as 80 μM (Fig. 1C).
Enhancements of the sensitivity of ATRA in NB4 and
MR2 cells were also observed by MTT assay. ATRA at 0.2 μM and
emodin at 10 μM alone only induced mild proliferation inhibition in
NB4 and MR2 cells. However, the combination of 0.2 μM ATRA and 10
μM emodin significantly raised inhibitory rates in NB4 cells and
MR2 cells (P<0.05), which were similar to those achieved by 1 μM
ATRA treatment in the two cell lines (P>0.05). When increased up
the concentration of ATRA to 1 μM in combination with 10 μM of
emodin, the average levels of growth inhibition were significantly
enhanced from 54.29±13.85 and 26.58±3.57% in 1 μM ATRA
mono-treatment group to 85.77±2.89 and 43.95±4.99% in the
combination group in NB4 and MR2 cells, respectively, (P<0.05)
(Fig. 1D).
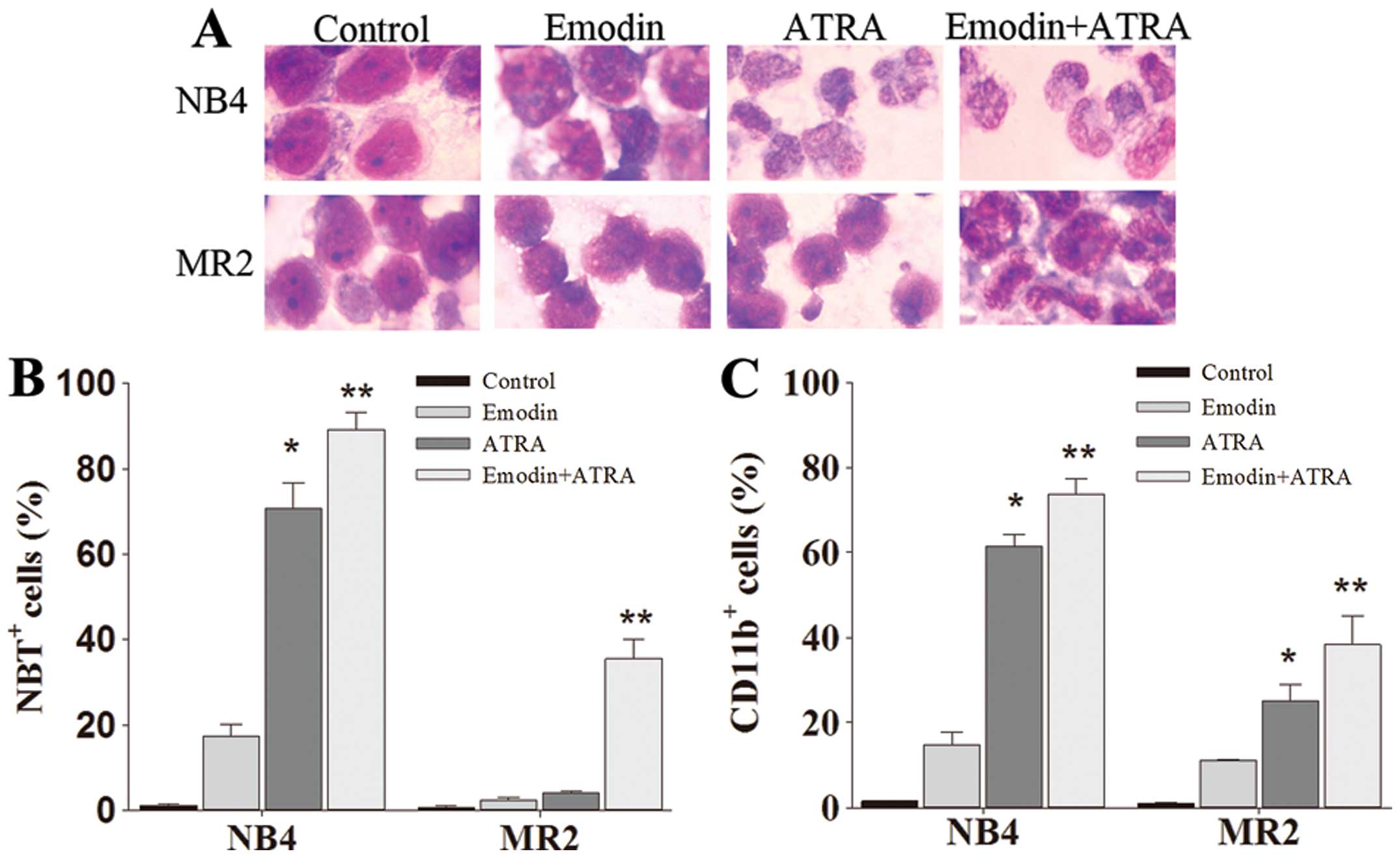
Emodin enhances differentiation induction
of ATRA in retinoid-responsive NB4 cells as well as in
retinoid-resistant MR2 cells
As shown in Fig.
1D, >90% NB4 and MR2 cells remained alive when treated with
emodin at a concentration of 10 μM. To identify whether this
non-toxic dose of emodin may enhance ATRA-sensitivity in NB4 cells,
especially in its ATRA-resistant subclone MR2 cells, we detected
the cellular differentiation effects of 10 μM emodin combined with
1.0 μM ATRA in the two cell lines. Compared with the untreated and
ATRA-treated cells, emodin only-treated cells presented slight
differentiation-inducing effects in either NB4 cells or MR2 cells.
As shown in Fig. 2A, the
combination of emodin and ATRA synergistically induced terminal
granulocytic differentiation manifested as smaller cell size, the
disappearance of nucleoli, reduced nuclei-cytoplasm ratio, and
condensed, distorted and stab form nuclei. Moreover, the
acceleration processes of NB4 and MR2 cell differentiation after
the combination treatment were further confirmed by CD11b
expression analysis and NBT reduction assay. The averaged levels of
CD11b-positive cells in co-treatment group were 73.67±3.76% in NB4
cells and 38.29±6.68% in MR2 cells, NBT-positive cells were
89.17±4.01 and 35.50±4.44%, respectively. CD11b and NBT-positive
cells in the combination treatment group were significantly higher
than those obtained from either emodin or ATRA alone group
(P<0.05) (Fig. 2B and C).
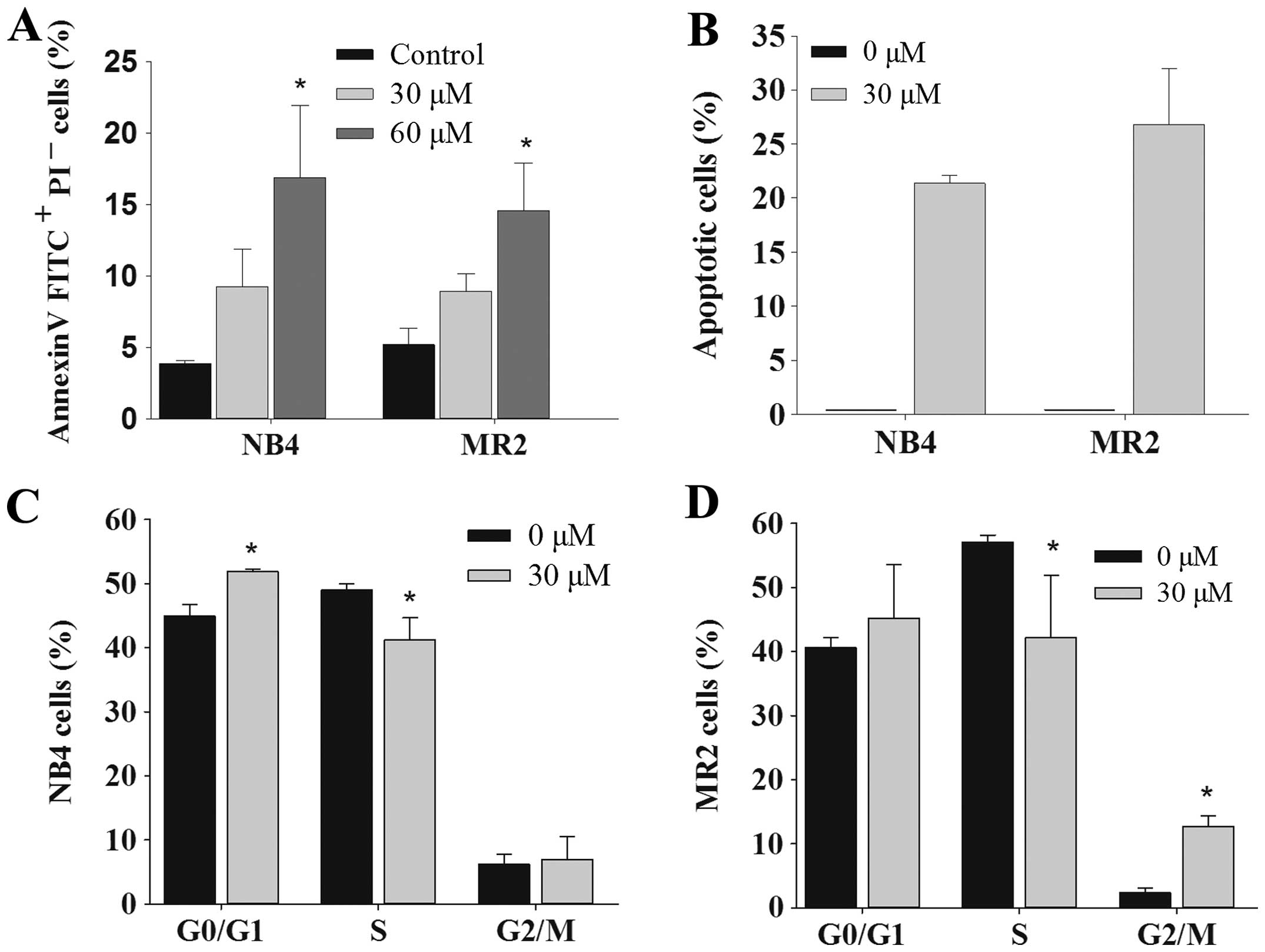
Emodin induces cell apoptosis in NB4
cells, MR2 cells and primary AML cells
Next, we assessed the role of emodin on the
induction of apoptosis in AML cells. The results showed that emodin
exerted apoptotic effects in NB4 and MR2 cells dose-dependently, as
assessed by Annexin V-FITC/PI staining assay (Fig. 3A). Quantitative evaluation of
apoptotic cells was further determined using cell cycle analysis.
When compared with the control group, the sub-G1
(apoptotic cell) population was clearly observable in NB4 and MR2
cells treated with 30 μM emodin. The ratio of apoptotic cells was
21.37±0.72 and 26.78±5.19% in NB4 and MR2 cells, respectively
(Fig. 3B). Results also revealed a
marked G0/G1 and G2/M phase arrest
in NB4 and MR2 cells in the presence of emodin. Furthermore, we
observed significantly decreased S phase cell population in the two
cell lines under the same experimental condition (Fig. 3C and D). DNA fragmentation assay
was applied to assess the effect of emodin on the induction of
apoptosis in AML cells. Consistently, we found that both of NB4 and
MR2 cells succumbed to emodin-induced the formation of DNA
fragments at 48-h treatment (Fig.
3E). Moreover, primary AML cells were also sensitive to emodin
stimuli in vitro. As shown in Fig. 3F, the apoptotic DNA fragmentation
was present in each emodin-treated primary AML sample group, which
further confirmed that emodin exhibited apoptotic induction effects
in AML cells.
Emodin leads to activation of the
caspase-dependent pathway and the degradation of RARα protein in
NB4 and MR2 cells
The mechanisms underlying emodin-induced apoptosis
in AML cells were also analyzed. As shown in Fig. 4A, emodin treatment reduced the
expression levels of procaspase-3 and procaspase-9 in NB4 and MR2
cells in a concentration- and time-dependent manner. The cleavage
of caspase-3 was induced in both cell lines. Consistently, results
also showed that emodin induced cleavage of PARP, one of the
important caspase-3 substrates. PARP was activated by cleaved
116-kDa fragment to 85-kDa fragment. The activation modulated by
emodin was enhanced with the longer incubation time and higher dose
exposure.
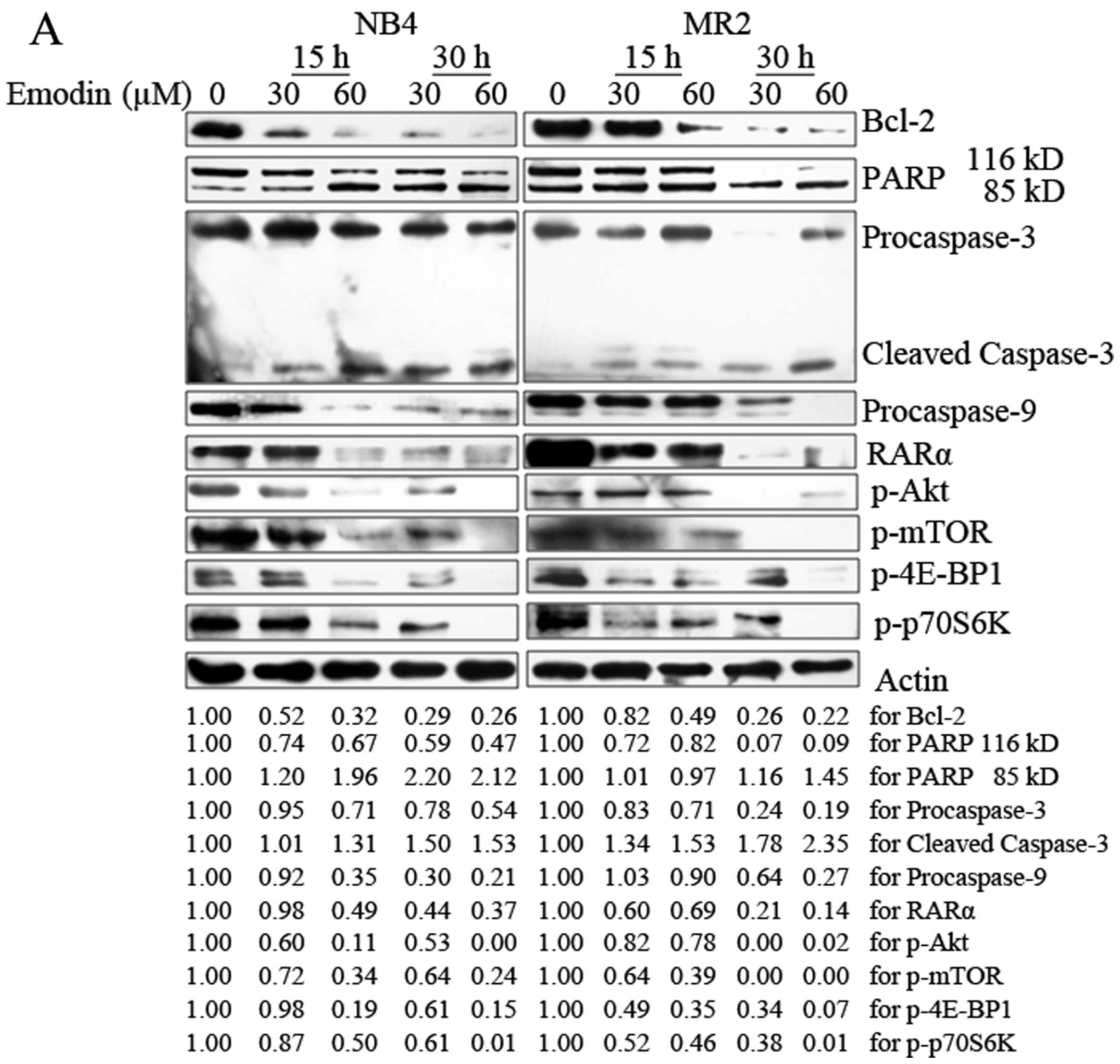 | Figure 4Emodin activates the
caspase-dependent apoptotic pathway and inhibits PI3K/Akt signaling
pathway in NB4 and MR2 cells. (A) NB4 and MR2 cells were treated
with emodin at indicated concentrations and time-points. Proteins
were extracted from harvested cells. Western blotting was performed
to visualize the expression of Bcl-2, 116 kDa PARP, 85 kDa PARP,
procaspase-3, cleaved caspase-3, procaspase-9, RARα,
phosphorylation of Akt (Ser473), mTOR (Ser2448), 4E-BP1 (Thr70) and
p-70S6K (Thr389) proteins along with β-actin as loading controls.
The experiments were repeated three times. The intensity of
different bands was quantified. Changes in protein expression
levels are represented as percentage change from untreated control
levels, which were set to 100% (1.00). The lower set of numbers
indicates the relative protein level. (B) NB4 and MR2 cells were
cultured with emodin (8 and 80 μM), LY294002 (40 μM), rapamycin
(1.0 μM) and PD98059 (20 μM) for 12 h. Protein extracts from
harvested cells were subjected to western blotting using
anti-p-Akt, anti-p-mTOR, anti-p-4E-BP1 and anti-p-p70S6K
antibodies. All experiments were repeated three times. |
Anti-apoptotic protein, Bcl-2, plays an important
role during caspase cascade activation and apoptosis induction.
PML/RARα oncoprotein was specifically expressed in APL cells, which
inhibits the granulocyte development at the promyelocytic stage of
differentiation. Therefore, we further examined the changes of
expression levels of Bcl-2 and RARα protein in NB4 and MR2 cells
following the treatment with emodin. Spot densitometry
quantification analysis revealed a decrease in Bcl-2 expression
from 52 to 26% in NB4 cells and from 82 to 22% in MR2 cells,
respectively, when administered with increasing doses of emodin at
different time points. Although RARα protein was not affected in
NB4 cells with the addition of 30 μM emodin for 15 h, decreased
RARα ranging from 49 to 37% were observed when longer incubation
time and higher dose were applied. Similarly, the presence of
emodin also resulted in a dose- and time-dependent degradation
(ranging from 69 to 14%) of RARα protein in MR2 cells (Fig. 4A).
Emodin inhibits activation of the
PI3K/Akt signaling pathway in AML cells
Constitutive PI3K/Akt activation is frequently
observed in AML samples and sustains leukemic cell growth (5–7,19,20).
Hence, we tested whether emodin may affect PI3K/Akt signaling
pathway in AML cells. The results demonstrated that emodin markedly
abrogated phosphorylation of Akt at Ser473 and of mTOR at Ser2448
in both NB4 and MR2 cells. Of note, emodin also inhibited mTOR
downstream targeted proteins p-4E-BP1 and p-p70S6K in the two cell
lines in a dose- and time-dependent manner (Fig. 4A). To further validate our
observation that emodin downregulates the activation of crucial
molecules in PI3K/Akt pathway, PI3K/Akt inhibitor (LY294002), mTOR
inhibitor (rapamycin) and MAPK inhibitor (PD 98059) were
administered parallelly with two different concentrations of emodin
in NB4 and MR2 cells. Compared with the untreated group and the
lower dose of emodin (8 μM) treatment group, the inhibitory effects
on the phosporylation of Akt, mTOR, 4E-BP1 and p70S6K induced by 80
μM emodin were as strong as those following by LY294002 and
rapamycin treatment. As expected, neither Akt nor mTOR or its
downstream target activity in PI3K/Akt pathway were suppressed by
PD 98059 in NB4 and MR2 cells (Fig.
4B).
To identify the potential clinical efficacy using
emodin as a new PI3K/Akt inhibitor, 21 primary AML samples were
included in this study. Eighteen out of the 21 AML samples
presented constitutive Akt activation. It is similar to previously
reported data that Akt phosphorylation at Ser473 can be detected in
50–80% of AML patients (5,21). Bcl-2 protein could be seen in all
of the 21 patient samples as well. The results show that emodin
suppressed Akt phosphorylation in a dose-dependent manner among
these 18 primary AML samples, which was consistent with the results
we observed in NB4 and MR2 cells. Similar downregulation effects on
Bcl-2 expression were found in all of the 21 detected samples. The
phosphorylation form of mTOR was detectable in 8 out of the 18 AML
samples with constitutive Akt activation. Interestingly, the
presence of increasing doses of emodin significantly abrogated mTOR
activation along with phosphorylated Akt inhibition in these 8
samples. The results from three representative AML samples (patient
nos. 8, 13 and 17) are shown in Fig.
5. These strongly indicate that the inhibition of PI3K/Akt
activation involved in the antileukemia activity is triggered by
emodin.
Discussion
AML is a clonal hematopoietic stem cell disorder
characterized by differentiation arrest, inappropriate
proliferation and survival of immature myeloid progenitors. The
prognosis of the disease remains poor, and the mortality rates in
AML patients are high despite considerable improvements in therapy.
The long-term follow-up studies showed a relapse rate exceeding 20%
in high-risk APL patients (22–24),
a unique subtype of AML, although the targeted therapy with ATRA
and As2O3 was introduced in the clinic. The
investigation of novel anti-leukemic agent might generate
considerable benefits for AML patients.
The antitumor activities of emodin have been
reported. For example, emodin in combination with clinically
achievable doses of DHA reduced arsenic concentrations by 100-fold
while still remaining highly toxic to tumor cells (12). Emodin may improve the expression of
globin genes in K562 cells and also induce K562 cells to erythroid
differentiation possibly via upregulating ALAS2 and c-KIT and
downregulating miR-221 and miR-222 (25). In this study, emodin exhibited
significant anti-leukemic effects in vitro. We provided the
first demonstration that nontoxic dose of emodin (<
IC10) inhibited growth and induced differentiation to a
low degree in NB4 and MR2 cells. However, the administration of low
concentration of emodin along with ATRA synergistically enhanced
ATRA-induced terminal differentiation in NB4 cells. Especially, the
emodin/ATRA combination also significantly increased CD11b-positive
cells, NBT-response and differentiation-related morphological
features in ATRA-resistant MR2 cells. More interestingly, when
increasing dose of emodin was administered individually, we found
that it presented proliferation inhibition dose-dependently in the
two cell lines as well as in primary leukemic cells from AML
patients. We further showed that administration with emodin caused
AML cells to undergo apoptosis as evidenced by increasing
proportion of Annexin V-FITC-positive cells, sub-G1 cell
cycle arrest and obvious DNA fragmentation.
The underlying mechanisms responsible for
emodin-induced apoptosis in AML cells were also explored in this
study. Muto et al showed that the activation of caspase-3
and -9 can be triggered by emodin in multiple myeloma cells
(26). AMAD, an emodin azide
methyl anthraquinone derivative, was reported to induce apoptosis
in the human breast cancer cell line MDA-MB-453 and the human lung
adenocarcinoma Calu-3 cells via a collapse of the mitochondrial
membrane potential and the activation of caspase cascades (27). As shown in this study, caspase-3,
caspase-9 and PARP precursors were decreased, while the cleavage of
caspase-3 and PARP were induced in AML cells in response to emodin.
Emodin triggered the activation of caspases in AML cells consistent
with the previous findings in multiple myeloma cells, breast cancer
cells and lung adenocarcinoma cells (26,27).
Bcl-2 is one of the important anti-apoptotic
proteins of the Bcl-2 family, which has been shown to contribute to
drug resistance in various human malignancies including hematologic
malignancies and solid tumors. Overexpression of Bcl-2 is
associated with poor prognosis (28–31).
We observed that emodin suppressed Bcl-2 expression in
ATRA-resistant MR2 cells as well as in its parental NB4 cells. Of
note, dose-dependent blockage of Bcl-2 expression was further
identified in 21 cases of primary AML cell samples when cultured in
the presence of emodin. An in vivo study in our group
recently demonstrated that consecutive treatment with emodin could
effectively downregulate Bcl-2 in AML in a xenograft model in nude
mice (data not shown). These indicate suppression of Bcl-2 may
contribute apoptosis execution in emodin-treated AML cells.
The promyelocytic leukemia (PML)/RARα fusion protein
formed as a result of the t(15;17) translocation was found in most
of APL patients. As retinoic acid receptors, APL blasts express
large amounts of PML/RARα, RARα and RXR isoforms. To activate
PML/RARα oncoprotein degradation appears to represent a critical
parameter for successful treatment of APL (32–35).
Broad networks of post-transcriptional suppressive pathways are
activated during ATRA-induced growth inhibition processes in APL
cells (36). Interestingly, we
found emodin significantly induced loss of RARα protein in the same
manner as Bcl-2 protein in NB4, especially in MR2 cells. It remains
to be further investigated how emodin exerts degradation effects on
RARα protein.
The constitutive activation of Akt pathway plays a
critical role in the progression of AML cases. In the present
study, we assessed whether the inhibition of PI3K/Akt pathway was
concurrently involved in emodin-mediated apoptosis in AML cells.
PI3K/Akt signaling pathway is comprised of a family of
intracellular protein kinases. Akt activation indirectly promotes
transcription of anti-apoptotic genes, while it directly
phosphorylates the downstream target, mTOR. Phosphorylated mTOR
promotes cell cycle transition from the G1 to S phase
via phosphorylation of the downstream targets p70S6K and 4E-BP1,
which favor translation of mRNAs for certain growth-promoting
proteins such as Cyclin D. We demonstrated that emodin is the
direct inhibitor of Akt activation in AML cells. Importantly, it
also inhibited mTOR leading to reduced phosphorylation of 4E-BP1
and p70S6K in AML cells. Accordingly, the downregulation of
anti-apoptotic protein Bcl-2, cell cycle arrest to G1
phase and reduction of S phase cell population were found in the
emodin-treated leukemic cells. Of note, emodin abrogated
phosphorylation of Akt at Ser473, which is the site responsible for
activating Akt in a feedback loop and has been implicated in
rapamycin failure after prolonged treatment (6,7,37,38).
Our results indicated emodin may prevent the feedback loop.
Moreover, the inhibition of PI3K/Akt pathway by emodin was further
confirmed by comparison with PI3K/Akt inhibitor LY294002 and mTOR
inhibitor rapamycin, as well as MAPK inhibitor PD98059. Thus, the
downregulation of PI3K/Akt pathway is an important molecular
mechanism underlying emodin-induced apoptosis in AML cells. Whether
there are other pathways, such as ERK1/2 pathway, in response to
emodin stimuli in AML cells will be further identified.
In conclusion, we have demonstrated that emodin
could enhance ATRA-induced differentiation in APL cells, even in
ATRA-resistant leukemic cells. We further showed that emodin
induced AML cell apoptosis dose- and time-dependently through
inhibition of the PI3K/Akt signaling pathway involving activation
of the caspase cascades. To this end, emodin may be considered as a
new promising anti-leukemic agent to overcome ATRA-resistance and
to improve clinical outcome of AML. Therefore, it would be worthy
of extensive experimental investigations in vivo.
Acknowledgements
This study was supported by National and Fujian
Provincial Key Clinical Specialty Discipline Construction Program,
China, National High Technology Research and Development Program of
China, 863 program (2012AA02A505), National Public Health Grand
Research Foundation (201202017), Fujian Provincial Key Laboratory
Foundation of Hematology (2009J1004), Fujian Provincial Natural
Science Foundation (2012J01353), Youth Foundation of Fujian
Provincial Health Bureau (2010-2-15).
References
|
1
|
Dohner H, Estey EH, Amadori S, Appelbaum
FR, Buchner T, Burnett AK, et al: Diagnosis and management of acute
myeloid leukemia in adults: recommendations from an international
expert panel, on behalf of the European LeukemiaNet. Blood.
115:453–474. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou GB, Zhang J, Wang ZY, Chen SJ and
Chen Z: Treatment of acute promyelocytic leukaemia with all-trans
retinoic acid and arsenic trioxide: a paradigm of synergistic
molecular targeting therapy. Philos Trans R Soc of Lond B Biol Sci.
362:959–971. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
West KA, Castillo SS and Dennis PA:
Activation of the PI3K/Akt pathway and chemotherapeutic resistance.
Drug Resist Updat. 5:234–248. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Franke TF, Hornik CP, Segev L, Shostak GA
and Sugimoto C: PI3K/Akt and apoptosis: size matters. Oncogene.
22:8983–8998. 2003. View Article : Google Scholar
|
|
5
|
Xu Q, Simpson SE, Scialla TJ, Bagg A and
Carroll M: Survival of acute myeloid leukemia cells requires PI3
kinase activation. Blood. 102:972–980. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tamburini J, Chapuis N, Bardet V, et al:
Mammalian target of rapamycin (mTOR) inhibition activates
phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like
growth factor-1 receptor signaling in acute myeloid leukemia:
rationale for therapeutic inhibition of both pathways. Blood.
111:379–382. 2008. View Article : Google Scholar
|
|
7
|
Zeng Z, Shi YX, Tsao T, et al: Targeting
of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in
AML cells under conditions mimicking the bone marrow
microenvironment. Blood. 120:2679–2689. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shrimali D, Shanmugam MK, Kumar AP, et al:
Targeted abrogation of diverse signal transduction cascades by
emodin for the treatment of inflammatory disorders and cancer.
Cancer Lett. 341:139–149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu L, Cai B, Zheng S, Liu X, Cai H and Li
H: Effect of emodin on endoplasmic reticulum stress in rats with
severe acute pancreatitis. Inflammation. 36:1020–1029. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
de Tamokou JD, Chouna JR, Fischer-Fodor E,
et al: Anticancer and antimicrobial activities of some
antioxidant-rich Cameroonian medicinal plants. PloS One.
8:e558802013.PubMed/NCBI
|
|
11
|
Zhang L, Lau YK, Xia W, Hortobagyi GN and
Hung MC: Tyrosine kinase inhibitor emodin suppresses growth of
HER-2/neu-overexpressing breast cancer cells in athymic mice and
sensitizes these cells to the inhibitory effect of paclitaxel. Clin
Cancer Res. 5:343–353. 1999.
|
|
12
|
Brown M, Bellon M and Nicot C: Emodin and
DHA potently increase arsenic trioxide interferon-alpha-induced
cell death of HTLV-I-transformed cells by generation of reactive
oxygen species and inhibition of Akt and AP-1. Blood.
109:1653–1659. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wei WT, Chen H, Ni ZL, et al: Antitumor
and apoptosis-promoting properties of emodin, an anthraquinone
derivative from Rheum officinale Baill, against pancreatic
cancer in mice via inhibition of Akt activation. Int J Oncol.
39:1381–1390. 2011.PubMed/NCBI
|
|
14
|
Huang LY, Hu JD, Chen XJ, Zhu LF and Hu
HL: Effects of emodin on the proliferation inhibition and apoptosis
induction in HL-60 cells and the involvement of c-myc gene.
Zhonghua Xue Ye Xue Za Zhi. 26:348–351. 2005.PubMed/NCBI
|
|
15
|
Chen YY, Zheng HY, Hu JD, et al:
Inhibitory effects of emodin on drug-resistant HL-60/ADR cell
proliferation and its induction of apoptosis. Zhongguo Shi Yan Xue
Ye Xue Za Zhi. 15:955–960. 2007.PubMed/NCBI
|
|
16
|
Chen YY, Li J, Hu JD, et al: Reversing
effects of emodin on multidrug resistance in resistant HL-60/ADR
cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 21:1413–1422.
2013.PubMed/NCBI
|
|
17
|
Lin M, Hu J, Liu T, Li J, Chen B and Chen
X: Knockdown of nucleophosmin by RNA interference reverses
multidrug resistance in resistant leukemic HL-60 cells.
Immunobiology. 218:1147–1154. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu J, Lin M, Liu T, Li J, Chen B and Chen
Y: DIGE-based proteomic analysis identifies nucleophosmin/B23 and
nucleolin C23 as over-expressed proteins in relapsed/refractory
acute leukemia. Leuk Res. 35:1087–1092. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tamburini J, Elie C, Bardet V, et al:
Constitutive phosphoinositide 3-kinase/Akt activation represents a
favorable prognostic factor in de novo acute myelogenous leukemia
patients. Blood. 110:1025–1028. 2007. View Article : Google Scholar
|
|
20
|
Martelli AM, Evangelisti C, Chiarini F and
McCubrey JA: The phosphatidylinositol 3-kinase/Akt/mTOR signaling
network as a therapeutic target in acute myelogenous leukemia
patients. Oncotarget. 1:89–103. 2010.PubMed/NCBI
|
|
21
|
Grandage VL, Gale RE, Linch DC and Khwaja
A: PI3-kinase/Akt is constitutively active in primary acute myeloid
leukaemia cells and regulates survival and chemoresistance via
NF-kappaB, Mapkinase and p53 pathways. Leukemia. 19:586–594.
2005.PubMed/NCBI
|
|
22
|
Niu C, Yan H, Yu T, et al: Studies on
treatment of acute promyelocytic leukemia with arsenic trioxide:
remission induction, follow-up, and molecular monitoring in 11
newly diagnosed and 47 relapsed acute promyelocytic leukemia
patients. Blood. 94:3315–3324. 1999.
|
|
23
|
Mathews V, George B, Chendamarai E, et al:
Single-agent arsenic trioxide in the treatment of newly diagnosed
acute promyelocytic leukemia: long-term follow-up data. J Clin
Oncol. 28:3866–3871. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghavamzadeh A, Alimoghaddam K, Rostami S,
et al: Phase II study of single-agent arsenic trioxide for the
front-line therapy of acute promyelocytic leukemia. J Clin Oncol.
29:2753–2757. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma YN, Chen MT, Wu ZK, et al: Emodin can
induce K562 cells to erythroid differentiation and improve the
expression of globin genes. Mol Cell Biochem. 382:127–136. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Muto A, Hori M, Sasaki Y, et al: Emodin
has a cytotoxic activity against human multiple myeloma as a
Janus-activated kinase 2 inhibitor. Mol Cancer Ther. 6:987–994.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yan Y, Su X, Liang Y, et al: Emodin azide
methyl anthraquinone derivative triggers mitochondrial-dependent
cell apoptosis involving in caspase-8-mediated Bid cleavage. Mol
Cancer Ther. 7:1688–1697. 2008. View Article : Google Scholar
|
|
28
|
Tauchi T, Sumi M, Nakajima A, Sashida G,
Shimamoto T and Ohyashiki K: BCL-2 antisense oligonucleotide
genasense is active against imatinib-resistant BCR-ABL-positive
cells. Clin Cancer Res. 9:4267–4273. 2003.PubMed/NCBI
|
|
29
|
Kim R, Emi M, Tanabe K and Toge T:
Preclinical evaluation of antisense bcl-2 as a chemosensitizer for
patients with gastric carcinoma. Cancer. 101:2177–2186. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Karnak D and Xu L: Chemosensitization of
prostate cancer by modulating Bcl-2 family proteins. Current Drug
Targets. 11:699–707. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Parrondo R, de Las Pozas A, Reiner T and
Perez-Stable C: ABT-737, a small molecule Bcl-2/Bcl-xL antagonist,
increases antimitotic-mediated apoptosis in human prostate cancer
cells. PeerJ. 1:e1442013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nasr R, Guillemin MC, Ferhi O, et al:
Eradication of acute promyelocytic leukemia-initiating cells
through PML-RARA degradation. Nat Med. 14:1333–1342. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hu J, Liu YF, Wu CF, et al: Long-term
efficacy and safety of all-trans retinoic acid/arsenic
trioxide-based therapy in newly diagnosed acute promyelocytic
leukemia. Proc Natl Acad Sci USA. 106:3342–3347. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Isakson P, Bjoras M, Boe SO and Simonsen
A: Autophagy contributes to therapy-induced degradation of the
PML/RARA oncoprotein. Blood. 116:2324–2331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang XW, Yan XJ, Zhou ZR, et al: Arsenic
trioxide controls the fate of the PML-RARalpha oncoprotein by
directly binding PML. Science. 328:240–243. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Harris MN, Ozpolat B, Abdi F, et al:
Comparative proteomic analysis of all-trans-retinoic acid treatment
reveals systematic posttranscriptional control mechanisms in acute
promyelocytic leukemia. Blood. 104:1314–1323. 2004. View Article : Google Scholar
|
|
37
|
Bayascas JR and Alessi DR: Regulation of
Akt/PKB Ser473 phosphorylation. Mol Cell. 18:143–145. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|