Introduction
Although osteosarcomas have been treated with
chemotherapy for >30 years, patients with recurrent or
metastatic osteosarcomas still have very poor prognosis (1–3).
Finding new strategies to treat recurrent or metastatic
osteosarcoma remains an important but unmet clinical need.
Myocardin has been reported to be a key regulator of
cardiac and smooth muscle differentiation and its expression is
restricted to smooth and cardiac muscle lineages (4). Myocardin drives transcription through
the interaction with the MADS box-containing transcription factor,
serum response factor (SRF), which binds to a consensus DNA
sequence, CC[A/T]6GG (CArG box) (5,6). In
addition, myocardin functions as an effective inducer of growth
arrest and differentiation of some tumor and is frequently
repressed during human malignant transformation (7). Although the importance of myocardin
in cardiac and smooth muscle has firmly established, its role in
osteosarcomas is poorly understood, let alone why it cannot be
expressed in osteosarcoma.
Recently, deregulation of expression of several
miRNAs has been identified between osteosarcoma samples and normal
human osteoblasts as well as several important studies have focused
on the impact of microRNAs (miRNA, miR) on tumorigenesis and
progression of osteosarcoma (8–13).
The expression of miR-135b is upregulated in osteosarcoma (8) and miR-135b functions as an oncogene
in other malignant tumors suggesting that it might be a target for
malignant tumor therapy (14–18),
but its roles still keep emerging in osteosarcoma.
Our results demonstrated that myocardin expression
is specifically attenuated in osteosarcoma MG63 cells. We next
performed an analysis of potential microRNA target sites employing
3 commonly used prediction algorithms, miRanda, TargetScan and
miRDB. All 3 algorithms predicted that miR-135a and miR-135b can
target 3′UTR of myocardin. However, results of microarray assay and
real-time PCR showed that only miR-135b was significantly
upregulated in osteosarcoma tissues. Thus, we reasoned that due to
the overexpression of miR-135b, myocardin was suppressed or
degraded by miR-135b in osteosarcoma. Following experiments showed
that miR-135b degraded myocardin mRNA through targeting its 3′UTR
in osteosarcoma MG63 cells. Moreover, miR-135b promoted
proliferation, invasion and migration of MG63 cells. Finally, we
found that myocardin has the opposite effects of miR-135b, which
suppressed proliferation, migration and invasion in MG63 cells.
Thus, it is probably that miR-135b promotes proliferation, invasion
and migration of osteosarcoma cells by degrading myocardin.
Materials and methods
Cell culture and expression plasmids
MG63 human osteosarcoma cells were maintained in
minimum essential medium (Life Technologies, Inc., Carlsbad, CA,
USA) containing 10% fetal bovine serum, 1% non-essential amino
acids, and 1% penicillin/streptomycin. A7r5 cells were maintained
as described previously (19). All
expression plasmids were purchased from Tiangene (Tianjin, China).
Briefly, the pcDNA3.1-myocardin contains 3′UTR of myocardin
(1214nt) and a cDNA encoding amino acids 1-935 of mouse myocardin,
named myocardin-WT. Site-directed mutagenesis of the miR-135b
target-site in the myocardin-3′UTR was carried out using Quik
change-mutagenesis kit (Stratagene, Heidelberg, Germany),
myocardin-WT as a template. For transfection experiments, the cells
were cultured in serum-free medium without antibiotics at 60%
confluence for 24 h, and then transfected with transfection reagent
(Lipofectamine 2000, Invitrogen, Carlsbad, CA, USA) according to
the manufacturer’s instructions. After incubation for 6 h, the
medium was removed and replaced with normal culture medium for 48
h.
miRNA precursors, anti-miRNA
oligonucleotides
The locked nucleic acid (LNA)-modified
oligonucleotide inhibitors (anti-miR-135b) used for miRNA
knock-down and scramble were purchased from Exiqon. The miR-135b
miRNA precursor (pre-miR-135b) and a control precursor (scramble)
were purchased from Ambion, Inc.
Reverse transcription-polymerase chain
reaction
Total RNA was isolated from cells using TRIzol
reagent (Invitrogen). cDNA was synthesized from 1 μg of total RNA
in a 20-μl reverse transcription (RT) system followed by PCR
amplification in a 50-μl PCR system performed using an RT-PCR kit
(Promega, Madison, WI, USA). Housekeeping gene
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as RNA
loading control. The PCR primer sequences are as follows:
myocardin: 5′-GGACTGCTCTGGCAACCCAGTGC′;
5′-CATCTGCTGACTCCGGGTCATTTGC-3′. The PCR was conducted according to
the manufacturer’s instructions and the PCR products were analyzed
by agarose gel electrophoresis. Gels were photographed and
densities of the bands were determined with a computerized image
analysis system (Alpha Innotech, San Leandro, CA, USA). The area of
each band was calculated as the integrated density value (IDV).
Mean values were calculated from three separate experiments. The
IDV ratio of myocardin to GAPDH was calculated for each sample.
Cell counting assay
Forty-eight hours after transfection, MG63 cells
were seeded in 96-well plates in triplicate at a density of
5×103 cells/well in 100 μl of RPMI-1640 medium
containing 10% FBS and antibiotics. Cell proliferation was
evaluated using Cell Counting Kit-8 (Dojindo Lab, Kumamoto),
according to the manufacturer’s instructions and the absorbance
value for each well was measured at 450 nm using a microplate
reader (Spectra Max 180, Molecular Devices).
MTT assay
The effect on the cell proliferation was assessed by
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium (MTT; Sigma,
St. Louis, MO, USA) assay was performed as described before
(20). Absorbance was directly
proportional to the number of survival cells.
BrdU proliferation analysis
Cell proliferation was assessed by using a
colorimetric BrdU proliferation kit according to the manufacturer’s
instructions (Roche, Indianapolis, IN, USA). The cells transfected
with myocardin or empty vector or pre-miR-135b or anti-miR-135b
were labeled with BrdU for 3–4 h. The genomic DNA was fixed and
denatured, and then incubated with peroxidase-conjugated anti-BrdU
antibody for 90 min. A substrate for the conjugated peroxidase was
then added and the reaction product was quantified by measuring the
absorbance. The results were then normalized by the number of total
viable cells.
Migration and invasion assay
For Transwell migration assays,
2.5×104–5.3×104 cells were plated in the top
chamber with the non-coated membrane (24-well insert; pore size, 8
mm; BD Biosciences). For invasion assays, 1.25×105 cells
were plated in the top chamber with Matrigel-coated membrane
(24-well insert; pore size, 8 mm; BD Biosciences). In both assays,
cells were plated in medium without serum or growth factors, and
medium supplemented with serum was used as a chemoattractant in the
lower chamber. The cells were incubated for 24 h and cells that did
not migrate or invade through the pores were removed by a cotton
swab. Cells on the lower surface of the membrane were stained with
the Diff-Quick Staining Set (Dade) and counted.
Western blot analysis
Western blot analysis was performed as described
before (21). Antibodies used are
listed in Table I.
 | Table IAntibody used in the western blot
assay. |
Table I
Antibody used in the western blot
assay.
| Primary antibody | Secondary
antibody |
|---|
| Anti-CDK1 (1:500;
Abcam) | Anti-rabbit secondary
antibodies (Li-COR) |
| Anti-CDK2 (1:500;
Abcam) |
| Anti-c-myc (1:200;
Abcam) |
| Anti-PCNA (1:500;
Abcam) |
| Anti-Ki67 (1:500;
Abcam) |
| Anti-p53 (1:500;
Abcam) |
| Anti-p27 (1:500;
Abcam) |
| Anti-myocardin
(1:200; Abcam) |
| Anti-β-actin (1:500;
Abcam) |
miRNA microarray
Total RNA from cultured cells, with efficient
recovery of small RNAs, was isolated using the mirVana miRNA
Isolation kit (Ambion, Austin, TX, USA). cRNA for each sample was
synthesized by using 3′ IVT Express kit (Affymetrix) according to
the manufacturer’s protocols. The purified cRNA was fragmented by
incubation in fragmentation buffer (provided in the 3′IVT express
kit) at 95°C for 35 min and chilled on ice. The fragmented labeled
cRNA was applied to MicroRNA2.0 array (Affymetrix) and hybridized
in Genechip hybridization oven 640 (Affymetrix) at 45°C for 18 h.
After washing and staining in Genechip fluidics station 450
(Affymetrix), the arrays were scanned by using Genechip scanner
3000 (Affymetrix). The gene expressions levels of samples were
normalized and compared by using Partek GS 6.5 (Partek).
Average-linkage hierarchical clustering of the data was applied by
using the Cluster and the results were displayed by using
TreeView.
Real-time PCR
Total RNA from cultured cells, with efficient
recovery of small RNAs, was isolated using the mirVana miRNA
Isolation kit (Ambion). Detection of the mature form of miRNAs was
performed using the mirVana qRT-PCR miRNA Detection kit and qRT-PCR
primer sets, according to the manufacturer’s instructions (Ambion).
The U6 small nuclear RNA was used as an internal control.
Gene microarray
Preparation of RNA from cells and analysis of
Affymetrix gene chip microarray data were as previously described
(22). Total RNA was prepared
using TRIzol reagent (Invitrogen) according to the manufacturer’s
instructions. RNA was further purified using RNeasy columns
(Qiagen, Chatsworth, CA, USA) and treatment with RNase-free DNase I
(Qiagen). Total RNA was used to generate cRNA, which was labeled
with biotin as recommended by Affymetrix. cRNA was then hybridized
to Affymetrix Hu95A GeneChips, which contain ~12,000 human
oligonucleotide probe sets. After washing, the chips were scanned
and analyzed using Microarray Suite 5.0 (Affymetrix). Average
intensities for each Gene Chip were globally scaled to a target
intensity of 150. Further analysis was performed using Gene Spring
software version 5.0.1 to obtain expression level information, fold
change, and P-values for each gene relative to control.
Luciferase reporter assay
The 3′ untranslated region (3′UTR) of human
myocardin mRNA was cloned in between the Not1 and Xba1 sites
of pRL-TK (Promega) using PCR-generated fragment. Site-directed
mutagenesis of the miR-135b target-site in the myocardin-3′UTR was
carried out using Quik change-mutagenesis kit (Stratagene), with
myocardin-WT-luc as a template. For reporter assays, MG63 was
transiently transfected with WT or mutant reporter plasmid and
microRNA using Lipofectamine 2000 (Invitrogen). Reporter assays
were performed 36 h post-transfection using the
Dual-luciferaseassay-system (Promega), normalized for transfection
efficiency by cotransfected Renilla-luciferase.
Northern blot analysis
Northern blot analysis of miRNAs, were performed as
described previously (23). Probes
were labeled with [γ-32P]-ATP complementary to miR-135b
and U6 snRNA.
Results
Myocardin expression is specifically
attenuated in osteosarcoma MG63 cells
To identify the level of myocardin expression, we
performed RT-PCR and western blotting in A7r5 cells (as a positive
control) as well as MG63 cells. We could not detect any myocardin
mRNA (Fig. 1A) and protein
expression (Fig. 1B) in MG63
cells, although stable expression of myocardin could be detected in
A7r5 cells. Because myocardin functions as an effective inducer of
growth arrest and differentiation of some tumor and is frequently
repressed during human malignant transformation (7), we reasoned that the defect of
myocardin was associated with pathogenesis and progression of
osteosarcoma. To test this hypothesis and study biological effects
of myocardin, we transfected MG63 cells with myocardin-WT
expression plasmids. Although myocardin protein was significantly
increased by myocardin-WT plasmids in A7r5 cells (Fig. 1C), we did not detect any protein
expression of myocardin in MG63 cells (Fig. 1D).
Myocardin as a potential target of
miR-135b in osteosarcoma MG63 cells
Having demonstrated that myocardin expression is
specifically attenuated in MG63 cells and myocardin-WT plasmids
cannot be expressed in the cells, we investigated the mechanisms
inhibiting myocardin expression. MicroRNAs (miRNAs) are a new class
of small (~22 nucleotide) non-coding RNAs that negatively regulate
protein-coding gene expression by targeting mRNA degradation or
translation inhibition (24).
Thus, we considered that myocardin was degraded by miRNA.
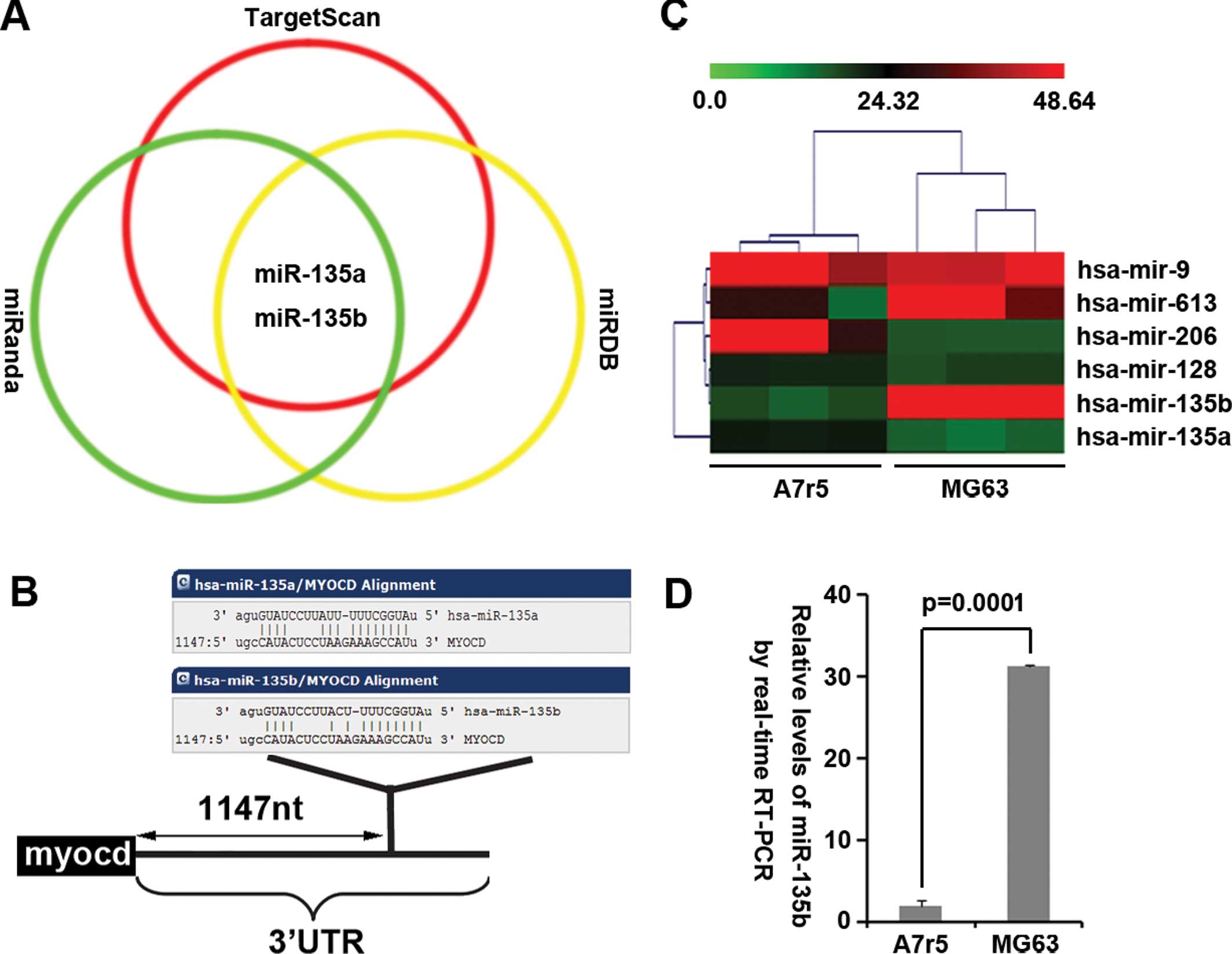
As a further confirmation, we used 3 common
prediction algorithms - miRanda (http://www.microrna.org/), TargetScan (http://www.targetscan.org) and miRDB (http://mirdb.org/miRDB/index.html) to analyze
3′UTR of myocardin. All 3 algorithms predicted that miR-135a and
miR-135b could target 3′UTR of myocardin (Fig. 2A) and the predicted target is shown
in Fig. 2B. We also performed
microRNA profiling and real-time PCR to detect and analyze the
difference of miRNA profiling in A7r5 cells (myocardin-positive
cells) and MG63 cells (myocardin-negative cells). The results of
the microarray (Fig. 2C) showed
that miR-135b expression was significantly upregulated in MG63
cells. To confirm the results of microarray, we performed real-time
PCR to analyze the level of miR-135b expression between A7r5 cells
and MG63 cells. Consistent with the results of microarray,
real-time PCR showed that miR-135b expression was significantly
upregulated in MG63 cells (Fig.
2D). All the results make us reason that it is probable that
downregulation of myocardin was associated with miR-135b
overexpression in MG63 cells.
Silencing miR-135b restores the
expression of myocardin and miR-135b inhibits myocardin expression
by targeting its 3′UTR in osteosarcoma MG63 cells
To examine whether myocardin was indeed regulated by
miR-135b, we transfected MG63 cells with anti-miR135b, in which
endogenous myocardin is undetectable by RT-PCR and western
blotting. Our results showed that anti-miR-135b effectively
inhibited miR-135b expression in MG63 cells (Fig. 3A). We next performed RT-PCR and
western blotting to detect myocardin expression in MG63 cells
transfected with anti-miR-135b. The results showed that myocardin
mRNA (Fig. 3B) and myocardin
protein (Fig. 3C) were
significantly upregulated in MG63 cells transfected with
anti-miR-135b. To further demonstrate the direct regulation of
myocardin by miR-135b, we constructed luciferase reporters with the
targeting sequences of wild-type (myocardin-WT-luc) and mutated
myocardin 3′UTRs (myocardin-MUT-luc) (Fig. 3D). Both the wild-type and mutant
reporters were introduced into MG63 cells (miR-135b high
expression) and A7r5 cells (miR-135b low expression). The
luciferase activities of myocardin-WT-luc but not myocardin-MUT-luc
were significantly suppressed in MG63 cells but not in A7r5 cells
(Fig. 3E).
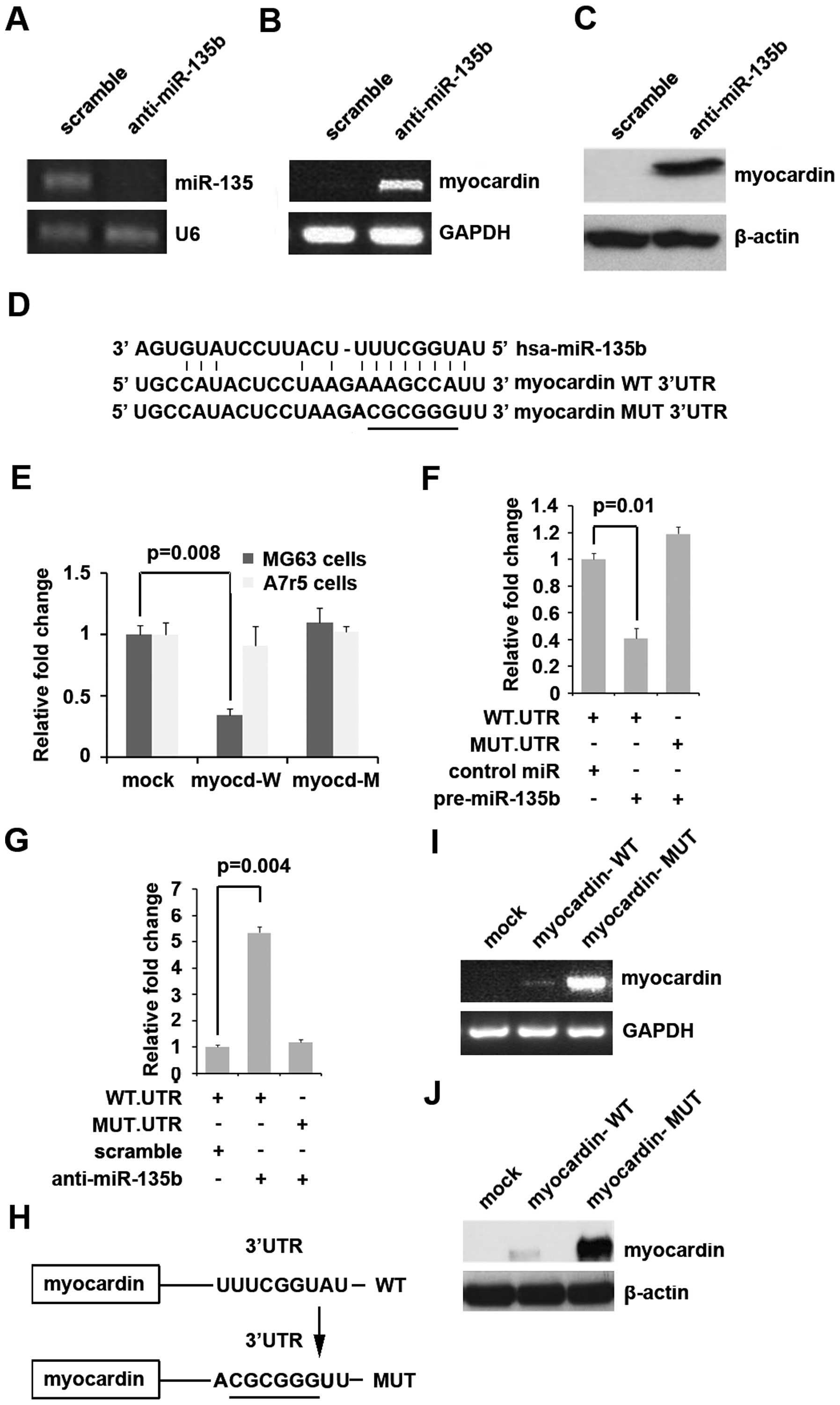 | Figure 3(A) Real-time PCR for miR-135b in MG63
cells. U6 was a loading control. n=3. (B) RT-PCR for myocardin in
MG63 cells. GAPDH was a loading control. n=3. (C) Western blotting
for myocardin in MG63 cells. β-actin was a loading control. n=3.
(D) Diagram of myocardin-3′UTR-containing reporter constructs. MUT,
contains 6-base-mutation at the miR-135b-target region, abolishing
its binding. (E) Reporter assay, WT-UTR but not MUT-UTR reporter
activity is reduced only in MG63 cells. n=3. (F) Reporter assay,
with cotransfection of 500 ng WT- or MUT-reporter and 50 nM
control-miR (scramble), or pre-miR-135b as indicated. n=3. (G)
Reporter assay, with cotransfection of 500 ng WT- or MUT-reporter
and 50 nM scramble, or anti-miR-135b as indicated. n=3. (H) Diagram
of myocardin expression plasmids, containing the predicted target.
MUT, contains 6-base-mutation at the miR-135b-target region,
abolishing its binding. (I) RT-PCR for myocardin in MG63 cells.
GAPDH was a loading control. n=3. (J) Western blotting for
myocardin in MG63 cells. β-actin was a loading control. n=3. |
To detect whether miR-135b targets 3′UTR of
myocardin, luciferase assay was performed. Our results showed that
miR-135b inhibited myocardin-WT-luc plasmids, but not
myocardin-MUT-luc plasmids (Fig.
3F). To further confirm that miR-135b can target the sites of
3′UTR of myocardin as predicated, we transfected MG63 cells with
anti-miR-135b. The results demonstrated that silencing miR-135b was
able to increase luciferase activity of 3′UTR of myocardin-WT-luc
in MG63 cells, but not myocardin-MUT-luc (Fig. 3G). All the results indicated that
miR-135b directly targeted the sites of 3′UTR of myocardin as
predicted by the bioinformatics methods.
In order to further demonstrate that miR-135b
inhibits myocardin expression through targeting its predicted sites
of 3′UTR, we purchased myocardin expression plasmids containing
predicted sites of 3′UTR (myocardin-WT) and mutated the 6 bases
predicted in its 3′UTR (myocardin-MUT) (Fig. 3H). Both the wild-type and mutant
expression plasmids were introduced into MG63 cells
(myocardin-negative) and then RT-PCT and western blotting were
performed to detect the expression of myocardin. The results showed
that only myocardin-MUT significantly increased myocardin mRNA
(Fig. 3I) and its protein
expression (Fig. 3J) in MG63
cells, indicating that miR-135b indeed inhibited myocardin
expression by targeting its 3′UTR.
miR-135b are highly expressed in
osteosarcoma tissues and it promotes MG63 cells proliferation,
migration and invasion
Having demonstrated that silencing miR-135b restores
the expression of myocardin and miR-135b inhibits myocardin
expression by targeting its 3′UTR in MG63 cells. We considered that
miR-135b played an important role in MG63 cells. Thus, in an
attempt to identify the level of miR-135b expression in
osteosarcoma and normal tissues, we performed miRNA profiling in
osteosarcoma tissues. RNAs isolated from 6 pairs of osteosarcoma
tissue and matched adjacent normal tissue samples were hybridized
to a custom miRNA microarray platform. After three hybridizations,
quantification, and normalization, we found that miR-135b, was
elevated in the primary tumors compared with normal tissues
(Fig. 4A).
To further assess the expression of miR-135b in
osteosarcoma, northern blot analysis was conducted in 6 pairs of
osteosarcoma tissue and matched adjacent normal tissue samples. The
expression of miR-135b was consistently higher in osteosarcoma
tissues than in normal tissues (Fig.
4B). Consistent with the miRNA microarray data and northern
blot analysis, real-time PCR analysis also revealed the expression
of miR-135b in the primary tumors compared with normal tissues was
elevated (Fig. 4C).
To determine whether miR-135b overexpression would
affect cell proliferation, we transfected MG63 cells with
pre-miR-135b, and then real-time PCR was performed to detect
miR-135b expression in the cells. The results showed that miR-135b
expression was effectively increased by pre-miR-135b in MG63 cells
(Fig. 4D). MTT assays were
performed and the results showed that pre-miR-135b could promote
proliferation of MG63 cells (Fig.
4E). Consistent with the results of MTT assay, transfection of
MG63 cells with pre-miR-135b increased BrdU incorporation in the
cells (Fig. 4F). We had showed
that anti-miR-135b could significantly decrease the expression of
miR-135b in MG63 cells (Fig. 3A).
To further confirm the relationship of miR-135b and proliferation,
we performed MTT assay and BrdU incorporation assay and the results
showed that silencing miR-135b led to a >2.5-fold reduction in
the proliferation properties (Fig.
4G) and DNA synthesis of these cells (Fig. 4H).
To determine whether miR-135b would increase the
basal levels of cell migration or invasion, we overexpressed
miR-135b in MG63 cells. In MG63 cells, ectopic expression of
miR-135b resulted in a four- to five-fold increase in cell
migration (Fig. 5A) and invasion
(Fig. 5A). These results indicated
that overexpression of miR-135b is sufficient to promote both
migration and invasion in vitro. Consistent with the results
of miR-135b overexpression, silencing miR-135b led to ~4-fold
reduction in the migration and invasion of these cells, as gauged
by Transwell migration assay (Fig.
5B) and Matrigel invasion assay (Fig. 5B).
Myocardin suppresses proliferation,
migration and invasion in MG63 cells
Having demonstrated the miR-135b can promote
proliferation, migration and invasion of MG63 cells and that
knockdown of miR-135b can restore myocardin expression, we reasoned
that myocardin had the opposite effects of miR-135b in MG63 cells.
To determine whether myocardin expression in the MG63 cells would
also affect cell proliferation, migration and invasion, we
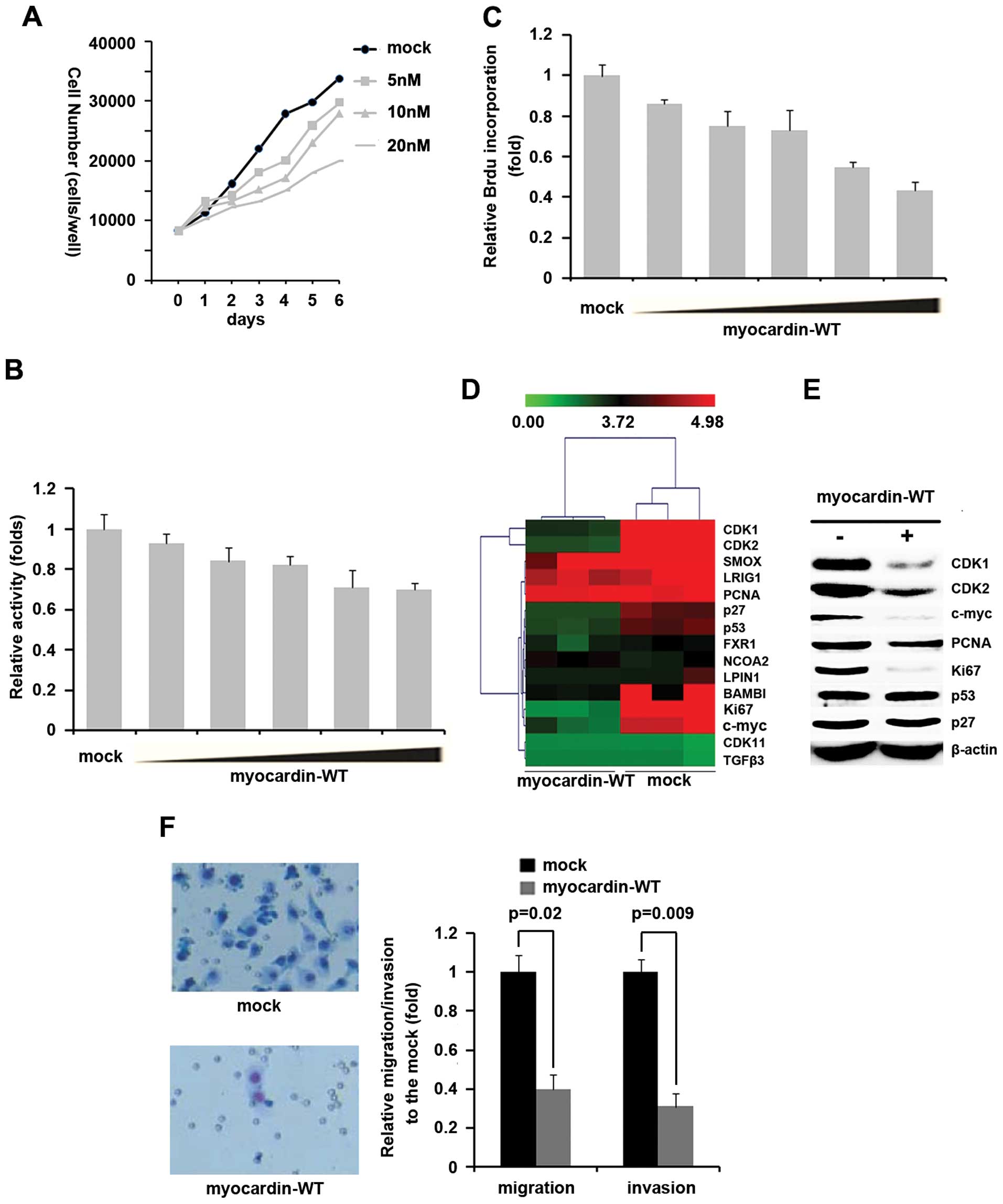
performed cell counting assay in MG63 cells transfected with
myocardin-MUT. We observed a dose-dependent suppression of MG63
cell proliferation (Fig. 6A).
Consistent with cell counting assay, MTT assay showed that
myocardin suppressed cell proliferation in a dose-dependent manner
(Fig. 6B). To further confirm that
myocardin can inhibit MG63 cell proliferation, BrdU incorporation
assay was performed. The results showed that myocardin inhibited
BrdU incorporation in a dose-dependent manner in MG63 cells
(Fig. 6C). We next performed gene
profiling to search for proliferation-associated genes, and found
that CDK1, CDK2, c-myc and Ki67 were significantly downregulated
and p27 and p53 were also downregulated in myocardin-MUT
transfected groups (Fig. 6D). To
further confirm the regulation of CDK1, CDK2, p27, P53, c-myc and
Ki67 by myocardin, we performed western blotting to analyze CDK1,
CDK2, p27, p53, c-myc, PCNA and Ki67 protein expression in MG63
cells transfected with myocardin-MUT. Our results showed that
myocardin-MUT significantly inhibited CDK1, CDK2, c-myc and Ki67
protein expression in MG63 cells (Fig.
6E).
We next identified whether restoration of myocardin
affected migration and invasion of MG63 cells. Transwell migration
assay and Matrigel invasion assay were performed and the results
showed that myocardin-MUT significantly inhibited migration and
invasion (Fig. 6F).
Discussion
Deregulated expression of miR-135b in
osteosarcoma
miR-135b levels are elevated in a variety of cancers
including breast, non-small cell lung cancer (NSCLC), prostate, and
colon as well as its upregulation is far more robust in highly
invasive lines compared to the less invasive in NSCLC (25–27).
Overexpression of miR-135b conferred an increased tumorigenic
ability to the relatively benign CL1-0 cells, resulting in
>4-fold greater tumor burden in xenograft mouse models. In
vivo, stable expression of a miR-135b antagonist decreased the
number of metastatic tumor nodules in mice injected with highly
invasive CL1-5-F4 cells shown to have high levels of miR-135b
(15). Clinically, high levels of
miR-135b in lung cancer specimens significantly correlated with
decreased overall survival (15).
Although the expression of miR-135b is upregulated in osteosarcoma
and miR-135b functions as an oncogene in other malignant tumors
suggesting that it might be a target for malignant tumor therapy,
its roles still keep emerging in osteosarcoma. We showed that
miR-135b inhibited proliferation, migration and invasion and
silencing its expression restored myocardin expression in MG63
cells. Due to the overexpression of miR-135b, myocardin mRNA was
degraded in MG63 cells. Next we will study the roles of miR-135b
in vivo.
Myocardin as a tumor suppressor in
osteosarcoma
Myocardin has been reported to be a key regulator of
cardiac and smooth muscle differentiation and its expression is
restricted to smooth and cardiac muscle lineages (4). Myocardin drives transcription through
the interaction with the MADS box-containing transcription factor,
serum response factor (SRF), which binds to a consensus DNA
sequence, CC[A/T]6GG (CArG box) (5,6). In
addition, myocardin functions as an effective inducer of growth
arrest and differentiation of some tumors and is frequently
repressed during human malignant transformation (7). Although the importance of myocardin
in cardiac and smooth muscle has been firmly established, its role
in osteosarcoma is still keep emerging. In this study, we found
that myocardin, as a suppressor gene, inhibited proliferation,
migration and invasion of MG63 cells. To further confirm the roles
of myocardin, its roles in vivo need to be studied.
Reference
|
1
|
Bielack SS, Marina N, Ferrari S, Helman
LJ, Smeland S, Whelan JS, et al: Osteosarcoma: the same old drugs
or more? J Clin Oncol. 26:3102–3103. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chou AJ, Geller DS and Gorlick R: Therapy
for osteosarcoma: where do we go from here? Paediatr Drugs.
10:315–327. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
O’Day K and Gorlick R: Novel therapeutic
agents for osteosarcoma. Expert Rev Anticancer Ther. 9:511–523.
2009.
|
|
4
|
Wang D, Chang PS, Wang Z, Sutherland L,
Richardson JA, et al: Activation of cardiac gene expression by
myocardin, a transcriptional cofactor for serum response factor.
Cell. 105:851–862. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yoshida T, Sinha S, Dandré F, Wamhoff BR,
Hoofnagle MH, et al: Myocardin is a key regulator of CArG-dependent
transcription of multiple smooth muscle marker genes. Circ Res.
92:856–864. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shore P and Sharrocks AD: The MADS-box
family of transcription factors. Eur J Biochem. 229:1–13. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pipes GC, Creemers EE and Olson EN: The
myocardin family of transcriptional coactivators: versatile
regulators of cell growth, migration, and myogenesis. Genes Dev.
20:1545–1556. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lulla RR, Costa FF, Bischof JM, Chou PM,
de Bonaldo FM, Vanin EF and Soares MB: Identification of
Differentially expressed microRNAs in osteosarcoma. Sarcoma.
2011:7326902011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jones KB, Salah Z, Del Mare S, Galasso M,
Gaudio E, Nuovo GJ, Lovat F, LeBlanc K, Palatini J, Randall RL,
Volinia S, Stein GS, Croce CM, Lian JB and Aqeilan RI: miRNA
signatures associate with pathogenesis and progression of
osteosarcoma. Cancer Res. 72:1865–1877. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang H, Cai X, Wang Y, Tang H, Tong D and
Ji F: microRNA-143, down-regulated in osteosarcoma, promotes
apoptosis and suppresses tumorigenicity by targeting Bcl-2. Oncol
Rep. 24:1363–1369. 2010.PubMed/NCBI
|
|
11
|
Duan Z, Choy E, Harmon D, Liu X, Susa M,
Mankin H and Hornicek F: MicroRNA-199a-3p is downregulated in human
osteosarcoma and regulates cell proliferation and migration. Mol
Cancer Ther. 10:1337–1345. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Osaki M, Takeshita F, Sugimoto Y, Kosaka
N, Yamamoto Y, Yoshioka Y, Kobayashi E, Yamada T, Kawai A, Inoue T,
Ito H, Oshimura M and Ochiya T: MicroRNA-143 regulates human
osteosarcoma metastasis by regulating matrix metalloprotease-13
expression. Mol Ther. 19:1123–1130. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ziyan W, Shuhua Y, Xiufang W and Xiaoyun
L: MicroRNA-21 is involved in osteosarcoma cell invasion and
migration. Med Oncol. 28:1469–1474. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Arigoni M, Barutello G, Riccardo F, Ercole
E, Cantarella D, Orso F, Conti L, Lanzardo S, Taverna D, Merighi I,
Calogero RA, Cavallo F and Quaglino E: miR-135b coordinates
progression of ErbB2-driven mammary carcinomas through suppression
of MID1 and MTCH2. Am J Pathol. 182:2058–2070. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lin CW, Chang YL, Chang YC, Lin JC, Chen
CC, Pan SH, Wu CT, Chen HY, Yang SC, Hong TM and Yang PC:
MicroRNA-135b promotes lung cancer metastasis by regulating
multiple targets in the Hippo pathway and LZTS1. Nat Commun.
4:18772013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang L, Sun ZJ, Bian Y and Kulkarni AB:
MicroRNA-135b acts as a tumor promoter by targeting the
hypoxia-inducible factor pathway in genetically defined mouse model
of head and neck squamous cell carcinoma. Cancer Lett. 331:230–238.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matsuyama H, Suzuki HI, Nishimori H,
Noguchi M, Yao T, Komatsu N, Mano H, Sugimoto K and Miyazono K:
miR-135b mediates NPM-ALK-driven oncogenicity and renders
IL-17-producing immunophenotype to anaplastic large cell lymphoma.
Blood. 118:6881–6892. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Khatri R and Subramanian S: MicroRNA-135b
and its circuitry networks as potential therapeutic targets in
colon cancer. Front Oncol. 3:2682013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Holycross BJ, Blank RS, Thompson MM, Peach
MJ and Owens GK: Platelet-derived growth factor-BB-induced
suppression of smooth muscle cell differentiation. Circ Res.
71:1525–1532. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luo XG, Zou JN, Wang SZ, Zhang TC and Xi
T: Novobiocin decreases SMYD3 expression and inhibits the migration
of MDA-MB-231 human breast cancer cells. IUBMB Life. 62:194–199.
2010.PubMed/NCBI
|
|
21
|
Liao XH, Wang N, Liu QX, Qin T, Cao B, et
al: Myocardin-related transcription factor-A induces cardiomyocyte
hypertrophy. IUBMB Life. 63:54–61. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Frasor J, Danes JM, Komm B, Chang KC,
Lyttle CR and Katzenellenbogen BS: Profiling of estrogen up- and
downregulated gene expression in human breast cancer cells:
insights into gene networks and pathways underlying estrogenic
control of proliferation and cell phenotype. Endocrinology.
144:4562–4574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu J, Ryan DG, Getsios S,
Oliveira-Fernandes M, Fatima A and Lavker RM: MicroRNA-184
antagonizes microRNA-205 to maintain SHIP2 levels in epithelia.
Proc Natl Acad Sci USA. 105:19300–19305. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
Downing JR, Jacks T, Horvitz HR and Golub TR: MicroRNA expression
profiles classify human cancers. Nature. 435:834–838. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bandres E, Cubedo E, Agirre X, Malumbres
R, Zarate R, Ramirez N, et al: Identification by real-time PCR of
13 mature microRNAs differentially expressed in colorectal cancer
and non-tumoral tissues. Mol Cancer. 5:292006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lowery AJ, Miller N, Devaney A, Mcneill
RE, Davoren PA, Lemetre C, et al: MicroRNA signatures predict
oestrogen receptor, progesterone receptor and HER2/neu receptor
status in breast cancer. Breast Cancer Res. 11:R272009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tong AW, Fulgham P, Jay C, Chen P, Khalil
I, Liu S, et al: MicroRNA profile analysis of human prostate
cancers. Cancer Gene Ther. 16:206–216. 2013.
|