Introduction
Prostate cancer is currently the most common and
frequently occurring malignant disease throughout the world. It is
estimated that prostate cancer is the second leading cause of death
among men in the USA (1,2). A recent study reported that prostate
cancer patients have a higher risk of death due to causes other
than prostate cancer itself (3).
Despite the serious implications of prostate cancer, little is
known about the molecular mechanism underlying this disease. To
date, only a few risk factors have been clearly established for
prostate cancer including age, a genetic history, and a complex
interplay between environmental factors such as lifestyle and diet
(4,5). Chemotherapy remains one of the major
options for effective treatment of hormone refractory prostate
cancer (HRPC). The ultimate goal of successful chemotherapy is
simultaneous suppression of survival signaling circuits of cancer
cells with minimal toxicity to normal cells.
Viriditoxin (VDT) has been isolated from
Paecilomyces variotii fungus (derived from the jellyfish
Nemopilema nomurai) and offers a new approach for
controlling antibiotic-resistant bacterial infection (6–8).
Bacterial cell division is modulated by a group of proteins called
divisomes. The filamenting temperature-sensitive mutant Z (FtsZ) is
one of these proteins and plays a key role in its own cell
division. VDT inhibits FtsZ by affecting cell morphology,
macromolecular synthesis and DNA damage responses; thus, more
recently, FtsZ has been considered as a novel therapeutic target of
antibiotics (8). FtsZ is also a
close structural homologue of eukaryotic tubulin, which forms
microtubules during cell division and plays a critical role in the
cytokinesis of cells (9).
Therefore, we hypothesized that VDT may also inhibit eukaryotic
cell division by blocking tubulin. Evidence shows that, after
testing several compounds using SRB assays, the NSC159628 (VDT)
compound causes cell cycle arrest at the G2/M phase after 12 h of
treatment. In addition, when cells are further treated for more
than 24 h, NSC159628 induces apoptosis in HeLa cells (10).
Microtubules are major dynamic structural components
of cytoskeleton formation during cell development, maintenance of
cell shape, cell division, intracellular transport and cell
movement (11). Microtubule
binding drugs are used as anticancer agents for the treatment of a
variety of cancers. Different types of microtubule stabilizing
agents including taxol, docetaxel and paclitaxel have been shown to
have potent anticancer activity against various types of human
cancers (12,13). Now, it is widely believed that,
like taxol, docetaxel binds to β-tubulin, stabilizes spindle
microtubules, and impairs mitosis by retarding cell cycle
progression in the G2/M phase (14). Moreover, evidence has revealed that
p53, a tumor suppressor gene, plays an important role in apoptosis
in prostate and colorectal cancer cells after treatment with
microtubule-targeting agents (15). In spite of the clear relationship
between mitotic mechanisms and apoptosis, tumor suppression by p53
has been generally accepted as a predominant mechanism of cell
death in response to chemotherapy drugs targeting microtubules
(16–18). Mitotic catastrophe can also be
triggered by microtubule instability leading to an abnormal mitotic
checkpoint (19). Mitotic
catastrophe is a mechanism of cell death characterized by the
occurrence of aberrant mitosis and is induced by improper
chromosomal segregation and cell division with characteristic
features of polynucleated cells (20). It results from premature or
inadequate entry of cells into mitosis and represents an
intermediate stage between prolonged mitotic arrest and the
induction of cell death (19).
Stilbene 5c, a stilbene derivative, is a potentially potent
antitumor agent that acts by binding to tubulin. Stilbene 5c was
shown to simultaneously trigger multiple mechanistic pathways
leading to cell death including the promotion of apoptosis,
autophagic cell death and mitotic catastrophe (21). Microtubules play an important role
in autophagy through their association with autophagosomes. A
previous study showed that microtubule disruption induced by
nocodazole or vinblastine dramatically inhibits autophagy-mediated
protein degradation (22) or
delays the transport of proteolytic enzymes to the lysosome
(23). In contrast, microtubules
facilitate autophagosome formation (24) and microtubule depolymerizing
agents, such as naphthazarin, induce autophagy in A549 lung cancer
cells (25). Also, evidence has
shown that different microtubular interfering agents enhance the
fusion of autophagosomes by acetylation of microtubules (26). However, the exact role of
microtubules in autophagic cell death is still unclear. Thus, we
investigated the antitumor effect of VDT against prostate cancer
cells. According to our data, VDT effectively inhibited
proliferation of prostate cancer cells through the induction of
cell cycle arrest at the G2/M phase and autophagic cell death.
Materials and methods
Chemicals and reagents
The novel compound VDT was kindly provided by
Professor Jee H. Jung, Laboratory of Marine Natural Products,
College of Pharmacy, Pusan National University, South Korea
(Fig. 1). Reference compound
docetaxel (DOC, cat. no. 01885) was purchased from Sigma-Aldrich
(St. Louis, MO, USA). Medium (RPMI-1640, cat. no. 11875),
antibiotics (Antibiotic-Antimycotic, cat. no. 15240), HEPES (cat.
no. 15630-080), Dulbecco’s phosphate-buffered saline (DPBS, cat.
no. 21600-010), trypsin-EDTA (cat. no. 15400) and fetal bovine
serum (FBS, cat. no. 16000) were purchased from Gibco Invitrogen
Corporation (Carlsbad, CA, USA). Primary antibodies against cyclin
A (sc-751), cyclin B1 (sc-245), cyclin E (sc-481), cyclin-dependent
kinases 2 (Cdk2, sc-6248), Cdk4 (sc-260), Cdc2 (sc-954),
poly-ADP-ribose polymerase (PARP, sc-7150), Bax (sc-7480), Bcl-2
(sc-7382), cytochrome c (sc-7159), p53(sc-126) and p21
(sc-6246) as well as horseradish peroxidase-conjugated secondary
antibodies (sc-2004, sc-2005) were from Santa Cruz Biotechnology
(Santa Cruz, CA, USA). Primary antibodies against LC3 (#3868), Atg5
(#8540), Atg7 (#2631), Beclin 1 (#3495), cleaved caspase-3 (#9664),
cleaved caspase-7 (#8438) and cleaved caspase-9 (#7237) were from
Cell Signaling Technology (Danvers, MA, USA). The Annexin V-FITC
apoptosis detection kit I (cat. no. 556547) was purchased from BD
Biosciences (San Diego, CA, USA). All other chemicals were
purchased from Sigma-Aldrich. VDT and DOC were dissolved in
dimethyl sulfoxide (DMSO, D2650, Sigma-Aldrich) and stored at −20°C
until use. These agents were diluted to appropriate concentrations
with culture medium containing 1% FBS for all experiments. The
final concentration of DMSO was less than 0.1% (vol/vol).
Cell lines and culture media
Human prostate cancer cell lines LNCaP (ATCC
CRL-1740), PC3 (ATCC CRL-1435), and DU145 (ATCC HTB81) were
obtained from the American Type Culture Collection (Manassas, VA,
USA). The cells were grown in RPMI-1640 containing 10%
heat-inactivated FBS, 1.25 mM HEPES and 100 U/ml
penicillin/streptomycin. Cells were maintained as a monolayer at
37°C in a humidified atmosphere containing 5% CO2, and
culture medium was replaced every 2 days. After 48 h of incubation,
culture medium was replaced with treatment medium containing the
desired concentration of drug.
Cell viability assay
Cell viability was determined using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT,
M5655, Sigma-Aldrich). The cultures were seeded in 96-well plates
at a density of 2×103 cells per well. After 48 h of
incubation, cells were treated with various concentrations of VDT
or DOC and incubated for 24 and 48 h. After treatment, 15 μl of MTT
reagent (5 mg/ml) was added and the cells were incubated in the
dark for 4 h at 37°C. Then, the supernatant was aspirated and
formazan crystals were dissolved in 100 μl of DMSO at 37°C for 15
min with gentle agitation. The absorbance was measured at 540 nm
using the VERSA Max Microplate Reader (Molecular Devices Corp.,
Sunnyvale, CA, USA). Data were analyzed from three independent
experiments and normalized to the absorbance of wells containing
media only (0%) or untreated cells (100%). IC50 values
were calculated from sigmoidal dose response curves using SigmaPlot
10.0 software (Systat Software, Inc., Point Richmond, CA, USA).
Western blot analysis
Cells were treated with VDT (0.1, 0.5 and 1 μM) or
DOC (0.5 μM) for 48 h and then harvested by trypsinization and
washed twice with cold PBS. For protein isolation, cells were
suspended in PRO-PREP™ solution (cat. no. 17081, Intron, Seongnam,
Korea) and placed on ice for 30 min. The suspension was collected
after centrifugation at 12,000 × g for 15 min at 4°C. To isolate
cytosolic and nuclear proteins, cells were suspended in 50 μl of
lysis buffer I containing 10 mM HEPES, pH 7.9, 1.5 mM
MgCl2 (cat. no. M8266, Sigma-Aldrich), 10 mM KCl (cat.
no. P9333, Sigma-Aldrich), 0.5 mM DTT (cat. no. 43815,
Sigma-Aldrich), and 0.5 mM PMSF (cat. no. 93482, Sigma-Aldrich),
and placed on ice for 20 min. Supernatant was removed after
centrifugation at 12,000 × g for 10 min, and the pellet was
resuspended in 30 μl of lysis buffer II containing 10 mM HEPES, pH
7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.5 mM PMSF,
and 0.5% NP-40 (cat. no. 74385, Sigma-Aldrich), and placed on ice
for 20 min. Cells were lysed by gently vortexing, and nuclei were
separated from the cytosol by centrifugation at 12,000 × g for 10
min. Nuclei were resuspended in 40 μl of buffer III containing 5 mM
HEPES, pH 7.9, 300 mM NaCl (cat. no. S3014, Sigma-Aldrich), 1.5 mM
MgCl2, 0.2 mM EDTA (cat. no. E9884), 0.5 mM DTT, 0.5 mM
PMSF, and 26% glycerol (cat. no. G2025, Sigma-Aldrich) and placed
on ice with shaking for 30 min. Nuclear extracts were obtained by
centrifugation at 12,000 × g for 30 min and stored at −70°C.
Protein concentration was measured with protein assay reagents
(cat. no. 500-0002, Bio-Rad, Hercules, CA, USA) according to the
manufacturer’s instructions. Equivalent amounts of proteins were
resolved on a 6–15% SDS-PAGE gradient, transferred to
polyvinylidene difluoride membrane (PVDF, cat. no. IPVH00010,
Millipore, Billerica, MA, USA), and probed sequentially with the
primary antibodies. Proteins were visualized with horseradish
peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz
Biotechnology) using the ECL-plus kit (cat. no. RPN2132, GE
Healthcare, Pittsburgh, PA, USA) for detection.
Cell cycle analysis
Cells were treated with VDT (0.1, 0.5 and 1 μM) or
DOC (0.5 μM) for 48 h. The total sample of cells, both in
suspension and adhered, was harvested and washed with PBS
containing 1% bovine serum albumin (BSA, cat. no. A4503,
Sigma-Aldrich) before fixing in 95% ice-cold ethanol containing
0.5% Tween-20 (cat. no. P9416, Sigma-Aldrich) for at least 1 h at
−20°C. The cells (1×106) were washed in 1% BSA, stained
with cold propidium iodide (PI, cat. no. P4864, Sigma-Aldrich)
staining solution including 100 μg/ml ribonuclease A (RNase, cat.
no. R6513, Sigma-Aldrich), and incubated in the dark for 30 min at
room temperature. DNA content was analyzed by flow cytometry (BD
Accuri™ C6, Becton-Dickinson, San Jose, CA, USA).
DAPI staining
Morphological changes of the nuclear chromatin of
apoptotic cells were identified by staining with the DNA binding
dye 4′,6-diamidino-2-phenylindole (DAPI, cat. no. D8417,
Sigma-Aldrich). Cells were grown in 6-well plates at a density of
1×105 cells per well followed by the desired treatment.
After 48 h of incubation, the cells were washed with cold PBS,
fixed with methanol for 30 min, rewashed and then stained with 200
μl of DAPI solution (1 μg/ml) at 37°C for 30 min. After removing
the staining solution, apoptotic cells were visualized using a
fluorescence microscope (Carl Zeiss Axiovert 200, Oberkochen,
Germany).
Annexin V-FITC binding assay
An Annexin V-FITC binding assay was performed
according to the manufacturer’s instruction using the Annexin
V-FITC apoptosis detection kit I (BD Biosciences, San Diego, CA,
USA). The cells were treated with VDT or DOC for 48 h. The total
number of cells was collected by trypsinization and washed twice
with cold PBS. The pellet was resuspended in 100 μl of 1X binding
buffer at a density of 1×105 cells per ml and incubated
with 5 μl of FITC-conjugated Annexin V and 5 μl of PI for 15 min at
room temperature in the dark. Binding buffer (1X, 400 μl) was added
to each sample tube, and immediately the samples were analyzed by
FACS (Becton-Dickinson) and quantified using Cell Quest
software.
Acridine orange staining
Cells were grown in cover glass bottom dishes at a
density of 1×105 cells per dish, cultured for 24 h, and
then incubated with the indicated drug in RPMI containing 1% FBS
for 48 h. Following incubation, the media was removed and the cells
were stained with 1 μg/ml of acridine orange (cat. no. A8097,
Sigma-Aldrich) at 37°C for 15 min. After removing the staining
solution, PBS was added to the dish and the cells were examined
using a fluorescence microscope at ×600 magnification (Olympus
FV10i, Tokyo, Japan).
Mitotic catastrophe assay
Cells were grown in cover glass bottom dishes at a
density of 1×105 followed by treatment with VDT or DOC
at the desired concentration. After 48 h of incubation, the cells
were washed with cold PBS, fixed with 4% paraformaldehyde (cat. no.
158127, Sigma-Aldrich) and isopropanol (cat. no. I9516,
Sigma-Aldrich) for 15 min, rewashed, and then stained with DAPI
solution (1 μg/ml) at 37°C for 15 min. After removing the staining
solution, the cells were visualized and images were taken using a
fluorescence microscope at ×400 magnification (Olympus FV10i). Five
separate experiments were visualized and evaluated. Cells were
assigned to three groups as follows: normal nuclei; abnormal
multinucleated (MN) cells characteristic of mitotic catastrophe;
and apoptotic nuclei. Small round evenly stained nuclei were
considered as normal nuclei and apoptotic was defined as the
presence of condensed fragmented chromatin. Multinucleated enlarged
nuclei, complete aberrant nuclei, catastrophic clusters and nuclei
undergoing multinucleation were considered as multinucleated
mitotic catastrophe nuclei (27).
Statistical analysis
All the data shown represent the mean ± SEM of
triplicate experiments performed in a parallel manner unless
otherwise indicated. Statistical significance was determined using
the paired Student’s t-test. A p<0.05 or p<0.01 was
considered statistically significant.
Results
Viriditoxin inhibits the proliferation of
prostate cancer cells
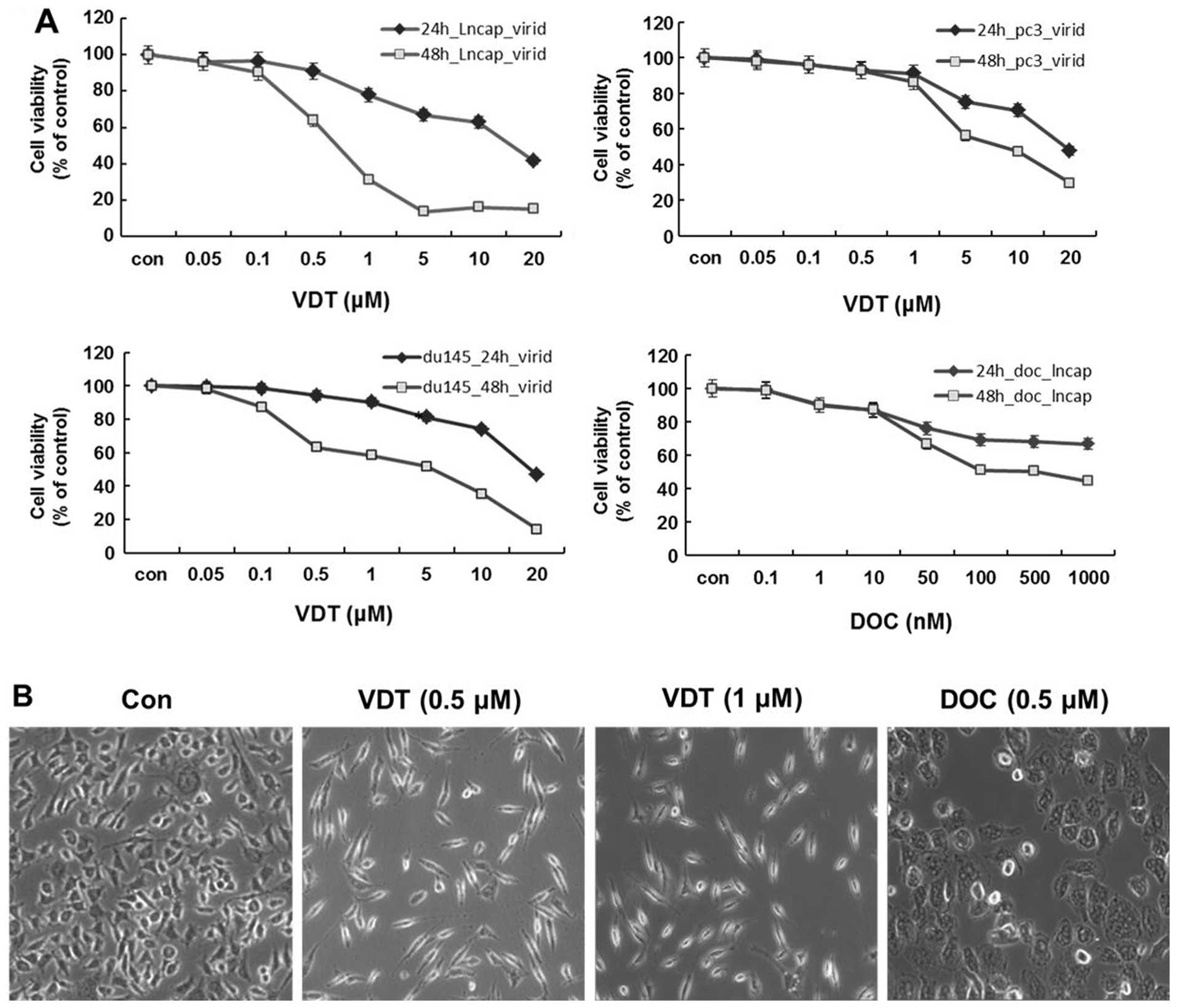
To determine the cytotoxicity of VDT on human
prostate cancer cells, LNCaP, DU145 and PC3 cells were treated with
various concentrations of VDT for 24 and 48 h. As shown in Fig. 2A, VDT significantly inhibited the
growth of the three human prostate cancer cell lines in a
concentration- and time-dependent manner. After 48 h of treatment,
the IC50 values of VDT against LNCaP, DU145 and PC3
cells were 0.63, 5.36 and 7.6 μM, respectively (Table I). VDT appeared to be more potent
in LNCaP cells compared to DU145 and PC3 cells. VDT treatment also
induced marked morphological changes, including cytoplasmic
shrinkage and cellular flattening following 48 h of treatment
(Fig. 2B).
 | Table IThe IC50 values of
viriditoxin on prostate cancer cell lines. |
Table I
The IC50 values of
viriditoxin on prostate cancer cell lines.
| IC50
values (μM) |
|---|
|
|
|---|
| Time | LNCaP | DU145 | PC3 |
|---|
| 24 h | 14.84 | 18.47 | 18.72 |
| 48 h | 0.63 | 5.36 | 7.60 |
VDT induces G2/M phase arrest and affects
cell cycle regulatory proteins
To examine the effect of VDT on cell cycle
progression, cells were treated with the indicated concentration of
VDT (0.1, 0.5 and 1 μM) or DOC (0.5 μM) for 48 h and then flow
cytometric analysis was performed. VDT significantly increased the
percentage of cells in the G2/M phase and concomitantly decreased
cells in the S phase in LNCaP cells (Fig. 3). LNCaP cells treated with 0.5 μM
DOC completely arrested the G2/M phase resulting in 63.1% of cells
in this phase (Fig. 3A). VDT also
increased the proportion of cells in the sub-G1 phase, which is an
apoptosis indicator in LNCaP cells (Fig. 3C). To explore the mechanism of VDT
on regulation of the cell cycle, we examined the expression levels
of cell cycle-related proteins by western blot analysis. Cell cycle
progression is tightly regulated by cyclins and cyclin-dependent
kinases (CDKs). Generally, the cyclin D1-CDK4 complex regulates the
cell cycle at the G1 phase while the cyclin E-CDK2 complex
initiates the G2/M transition. The CDK inhibitors p21Cip1 and
p27Kip1 regulate cell cycle progression from the G0/G1 phase to the
S phase; induction of p21Cip1 and p27Kip1 may lead to blockade of
the G1/S transition (28–29). As shown in Fig. 3, VDT decreased expression levels of
cyclin A, cyclin B1 and Cdc2, whereas expression of cyclin E1, Cdk2
was increased. In particular, VDT significantly increased
expression of p27, p53 and p21 in a concentration-dependent manner
in LNCaP cells (Fig. 3).
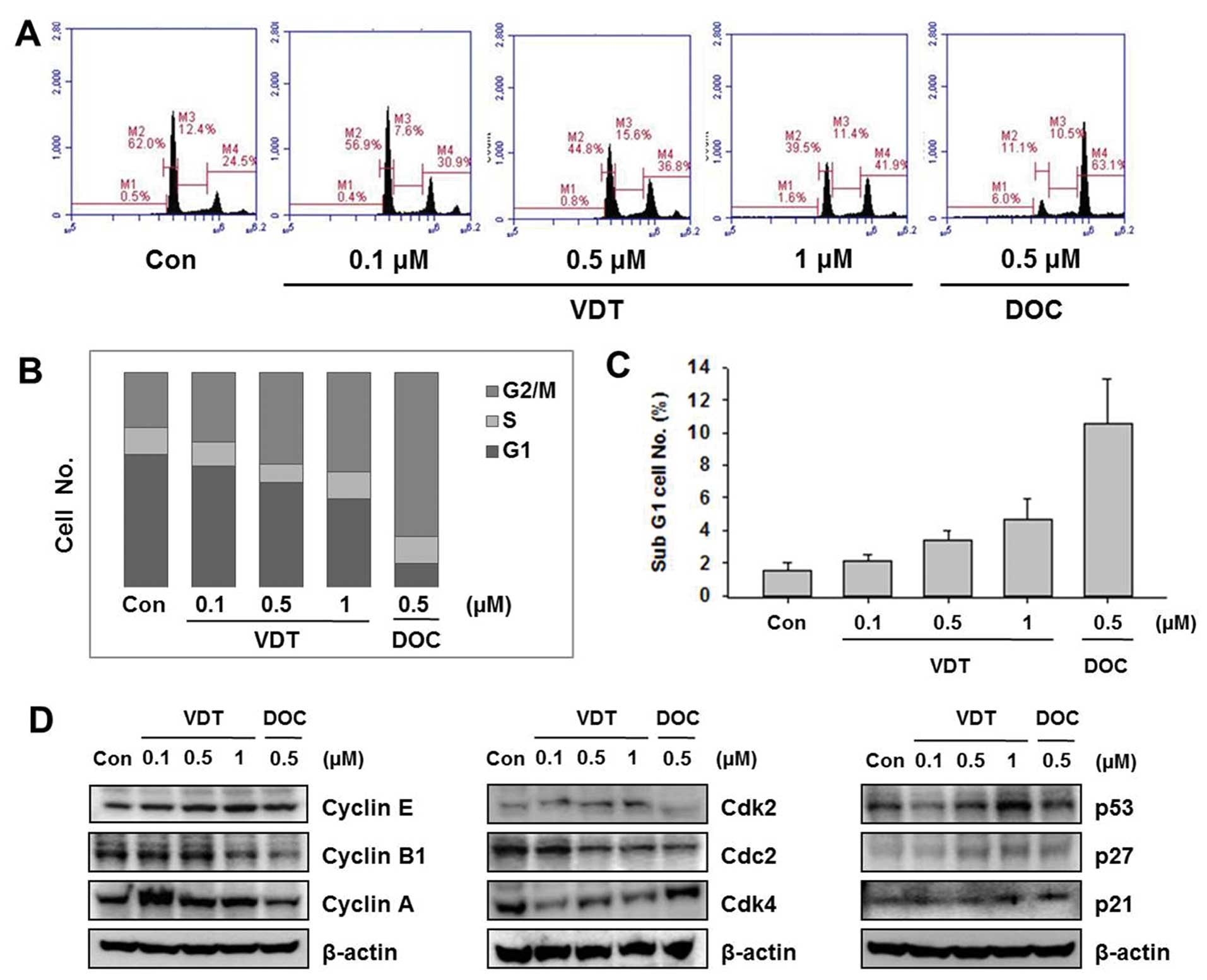 | Figure 3Effect of viriditoxin on cell cycle
distribution and expression of cell cycle regulatory protein in
LNCaP cells. Cells were treated for 48 h with the indicated
concentrations of viriditoxin (VDT) or docetaxel (DOC), stained
with propidium iodide (PI) and then analyzed by flow cytometry to
determine the distribution of cells in each phase of the cell
cycle. (A) Quantification of cell cycle distribution by flow
cytometry. (B) Percentage of LNCaP cells following drug treatment.
(C) Percentage of cells in the sub-G1 phase. (D) Effect of VDT and
DOC on expression of cell cycle regulatory proteins. Cells were
treated with the indicated concentrations of VDT for 48 h,
harvested, and then western blot analysis was performed using the
following antibodies: cyclin E, cyclin B1, cyclin A, Cdk2, Cdk4,
Cdc2, p53, p21, p27 and actin as an internal loading control. All
data are representative of three independent experiments. |
VDT slightly induces apoptosis in LNCaP
cells
Annexin VFITC-conjugated staining and western blot
analysis were performed to evaluate induction of apoptosis in LNCaP
cells after VDT treatment. Although a marked increase in
concentration-dependent cell death was observed in the cytotoxicity
assay, only a slight change in apoptotic cell death was detected at
the highest concentration of VDT (Fig.
4A). DAPI staining was used to confirm the effect of VDT on
apoptosis. Similar to observations of the sub-G1 phase of the cell
cycle, VDT induce increase amount number of apoptotic nuclei
(condensed or fragmented chromatin) compared to the untreated
control which showed enhanced fluorescence with DAPI staining
(Fig. 4B). Western blot analysis
also showed that high concentrations of VDT increased cleaved PARP,
Bax and cytochrome c, cleaved caspase-3 expression levels
and decreased Bcl-2 expression significantly in LNCaP cells
(Fig. 4C).
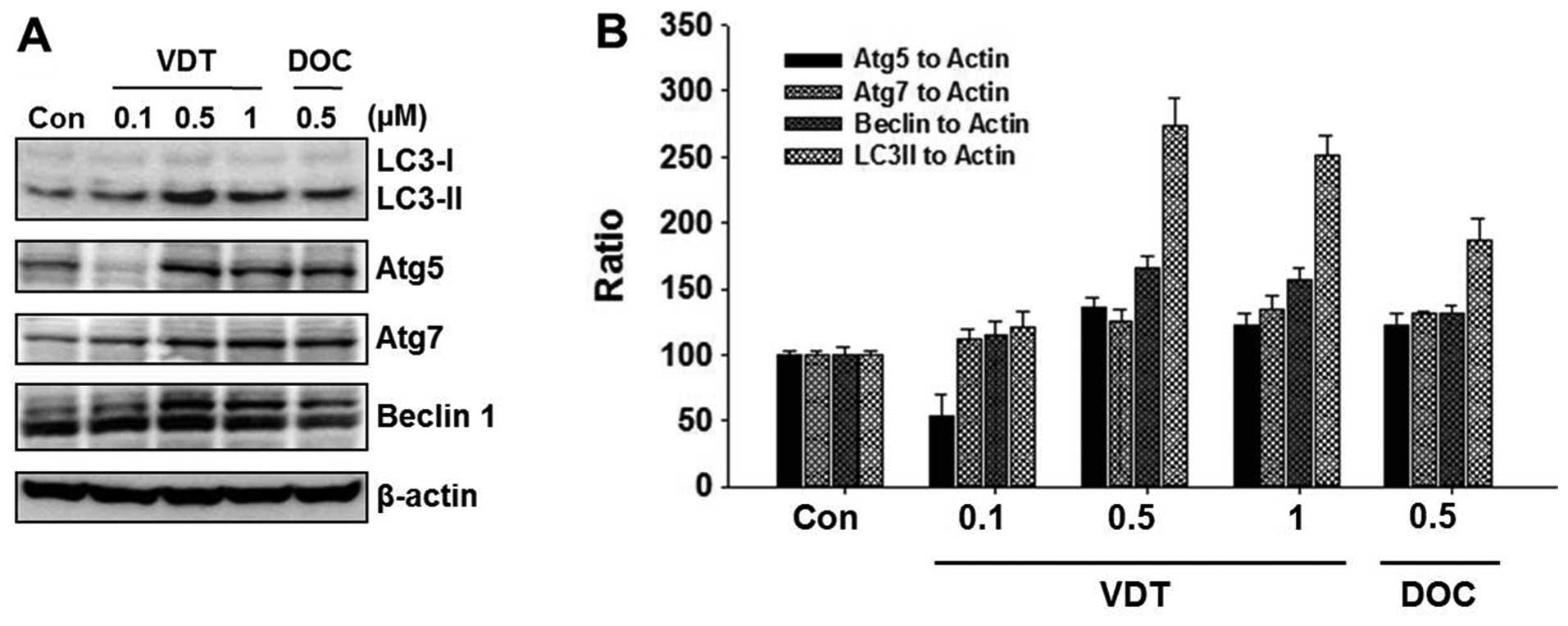
VDT induces autophagic cell death in
LNCaP cells
To elucidate the cell death mechanism, we
investigated autophagic cell death by western blot analysis and
acridine orange staining. VDT significantly increased autophagic
cell death in LNCaP cells. Conversion of the soluble form of LC3-I
to the autophagic vesicle-associated form LC3-II is considered a
specific marker of autophagosome production. As shown in Fig. 5A, VDT significantly increased the
level of LC3-II, whereas unconjugated LC3-I levels were slightly
decreased. In addition, beclin-1, Atg5 and Atg7 were required to
initiate the formation of autophagosomes. Similar to LC3-II, the
expression of beclin-1 was increased by VDT treatment (Fig. 5A). The ratio of conversion of
LC3-II, beclin-1, ATG5 and ATG7 to LC3-I and β-actin was calculated
by densitometry (Fig. 5B).
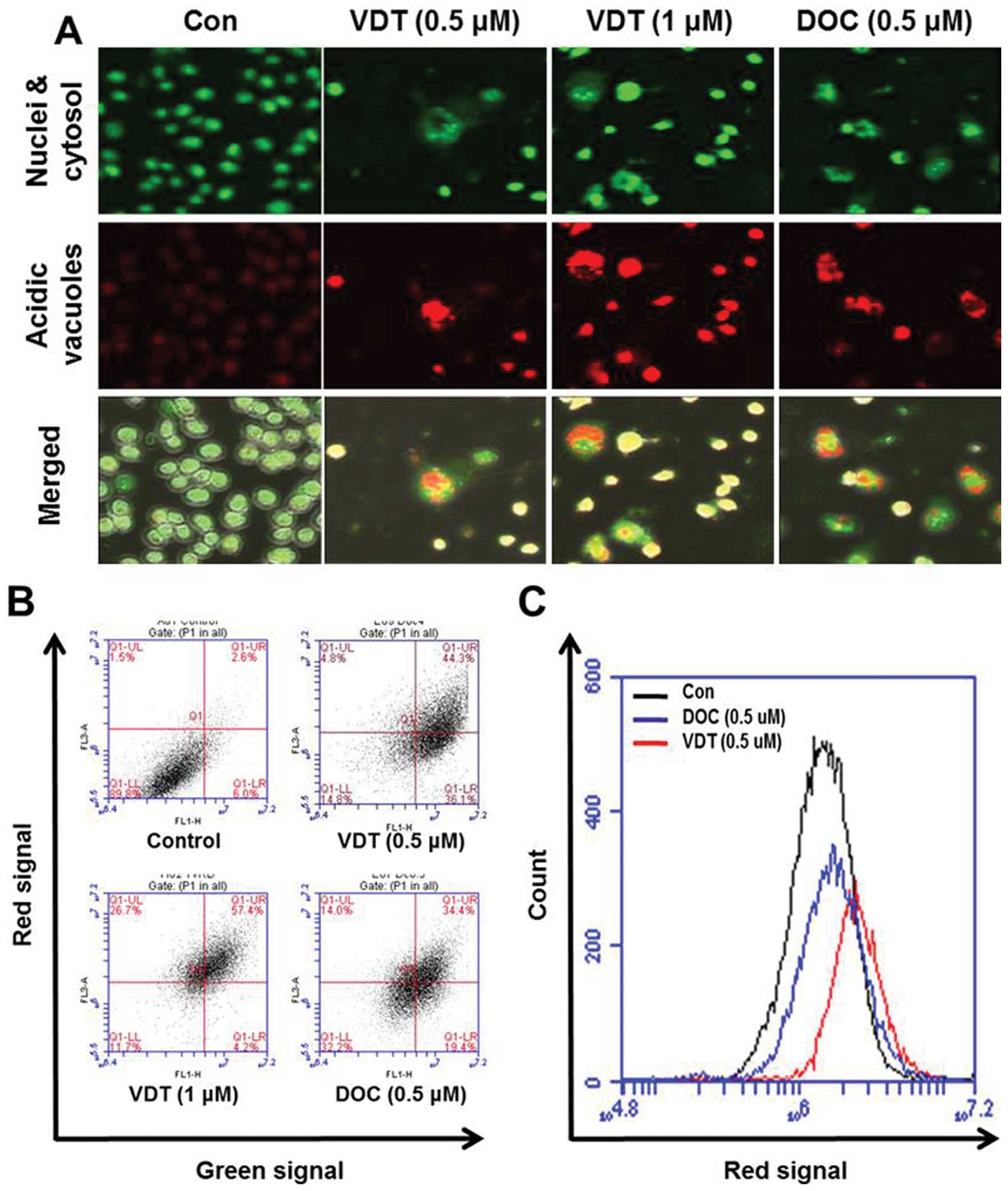
Next, induction of autophagy was confirmed by
acridine orange staining, which is commonly used to study
autophagy. Acridine orange is a lysotropic dye that accumulates in
acidic organelles in a pH-dependent manner. At neutral pH, acridine
orange is a hydrophobic green fluorescent molecule. However, within
acidic vesicles, acridine orange becomes protonated and trapped
within the organelle and forms aggregates that emit bright red
fluorescence (30). As shown in
Fig. 6A, control cells primarily
displayed green fluorescence with minimal red fluorescence,
indicating a lack of acidic vesicular organelles (AVOs). However,
drug-treated cells showed a fold-increase in red fluorescent AVOs
at 48 h post-treatment compared to the controls (Fig. 6A). Flow cytometric analysis after
acridine orange staining also showed an increase in red
fluorescence intensity after drug treatment indicating enhancement
of AVOs (Fig. 6B). Histogram
profiles in Fig. 6C show the mean
fluorescence intensity of control and drug-treated cells.
VDT induces mitotic catastrophe in LNCaP
cells
Analysis of nuclear morphology by fluorescence
microscopy showed that VDT significantly induced mitotic
catastrophe in LNCaP cells. Mitotic catastrophe was clearly
characterized by the appearance of enlarged multinucleated cells or
aberrant nuclei clusters (Fig.
7A). Approximately 45 to 60% of cells showed different
abnormalities of their nuclei compared to untreated control cells
after treatment with VDT (Fig.
7B).
Discussion
Tubulin inhibitors used as drugs for cancer
chemotherapy directly interfere with the tubulin system in multiple
solid tumors. Recently, the development of tubulin inhibitors has
been considerable interest because their function and biological
properties interfere with microtubule formation by affecting
polymerization or depolymerization of tubulin (31–33).
The disruption of microtubule formation as a primary target for
cancer chemotherapy can lead to cell cycle arrest in the M phase.
The majority of compounds blocking microtubule formation are
natural products that are remarkably diverse and have expanded
chemical structures. A number of microtubule targeting chemotherapy
drugs (paclitaxel, DOC or colchicine) are cytotoxic by stabilizing
or destabilizing microtubules. These drugs bind to distinct sites
on the microtubule or to the tubulin dimer and affect microtubule
dynamics by blocking the G2/M phase, thus prolonging the
pro/metaphase to anaphase transition time and inducing cell death
(34–37).
VDT is a novel compound isolated from
Paecilomyces variotii fungus (38) that inhibits FtsZ by affecting cell
morphology, macromolecular synthesis, and responses to DNA damage;
thus VDT has become a novel therapeutic target of antibiotics
(8,39). However, there is no data on the
antitumor activity of VDT. To clarify the molecular targets of VDT,
we first examined its cytotoxicity and the effect on cell cycle
progression in prostate cancer cells. VDT showed different
sensitivity in the three types of prostate cancer cell lines
evaluated in this study. LNCaP cells were very sensitive, DU145
cells were moderately sensitive, and PC3 cells showed low
sensitivity. LNCaP cells were mostly inhibited in a
concentration-dependent manner. Previous studies have indicated
that chemotherapeutic agents activating p53 can induce both
apoptosis and autophagic cell death in different cancer cells
(40,41). We predicted that VDT could induce
apoptotic cell death in LNCaP cells via p53-dependent induction of
p21. To confirm that VDT induces apoptotic cell death, the Annexin
V-FITC assay was performed. In this assay, a small amount of
apoptosis was observed. VDT exerted a broad spectrum of effects on
prostate cancer cells including arrest of the G2/M phase, p21
upregulation, and induction of apoptosis. A significant increase in
cytochrome c release in the cytoplasm, cleavage of PARP, and
caspase activation were observed in LNCaP cells after VDT
treatment. In the present study, VDT induced significant increases
in the population of cells in the G2/M phase. The G2/M transition
of the mitotic cycle is controlled by the cyclin A/B-CDC2/1 complex
and arrested by p21 (42,43). We found that VDT increased the
expression of p21, p27 and Cdc 2 with similar changes in p53
expression and decreased cyclin B1/cyclin A levels, which indicated
arrest of the G2/M phase in LNCaP cells. Several reports also
demonstrated that the principal pro-apoptotic transcription factor,
p53, can stimulate autophagy (44,45).
Autophagy is mainly a catabolic mechanism by which cellular
components are degraded through the action of lysosomes (46–49).
However, there is currently a debate over the role of autophagy in
ensuring continuous growth of cancer cells. Despite this lack of
clarity, it is believed that autophagy plays an important role in
cancer, both in protecting against cancer as well as potentially
contributing to the growth of cancer (50).
Although most of the microtubule targeting drugs
inhibits cancer cells through the apoptosis pathway, some can
inhibit apoptotic cell death in wild-type p53 prostate and
colorectal cancer cells (15).
Microtubule disrupting drugs can also trigger cell death via the
mitotic catastrophe pathway (51).
The DNA damage response and cell cycle checkpoints of cancer cells
make them more susceptible to mitotic catastrophe (52). Our data indicated that VDT markedly
increased cytotoxicity through apoptosis and mitotic catastrophe in
LNCaP cells. Previous studies also showed that various
tubulin-binding drugs, such as vinblastine, naphthazarin or
2-methoxyestradiol, contribute to anticancer activity by
stimulating autophagic cell death in different cancer cell lines
(24,25,53–55).
Specifically, vinblastine has been shown to increase formation of
autophagosomes as is indicated by increased levels of autophagic
cell death (56,57). In the present study, we evaluated
VDT as a microtubule targeting agent and clearly showed that it
induced autophagic cell death. To confirm the mechanism underlying
the autophagic cell death pathway, we investigated whether any
functional links between microtubules and autophagic cell death
exist. Similarly, other studies have also tried to gain
insights/knowledge on the mechanisms underlying the association
between microtubule targeting drugs and autophagy. LC31A/1B is a
microtubule-associated protein (MAP) (58). Moreover, both LC31 and lapidated
LC3II are found in subcellular fractions containing fragments of
microtubules, suggesting an interaction between LC3 and
microtubules (59). Therefore, we
predict that tubulin- or microtubulin-targeting drugs may impact
the initiation and termination of autophagy by releasing LC3 into
the cytoplasm. A previous study showed a similar association
between LC3 and autophagy, concluding that LC3 must be released
from microtubules to participate in the initiation of autophagy
(60). However, several previous
studies using different microtubule-associated agents including
taxol or nocodazole denied the participation of microtubules in
autophagosome formation in different cancer cell lines (22,24,61).
In contrast, disassembling microtubules treated with high doses of
nocodazole was shown to prevent autophagosome formation, thus
highlighting the function of microtubules in autophagy (23). In addition, another study showed
that vinca alkaloid enhances autophagosome formation (24). Recently, microtubules have been
suggested as global and local integrators of autophagic responses
in the formation and motility of autophagosomes rather than in
their fusion with lysosomes (62).
In conclusion, our data showed that VDT
significantly inhibited proliferation of prostate cancer cells and
induced autophagic cell death in LNCaP cells via disturbance of
tubulin formation. Further studies are necessary to better
understand the effect of autophagy inhibition on autophagosome
production and lysosomal degradation. However, the results of the
present study highlight autophagic cell death following VDT
treatment in LNCaP cells and also predict a functional relationship
between microtubules and autophagy in the context of anticancer
chemotherapy. Furthermore, we also showed that VDT treatment
induced slight apoptosis and cell death via the mitotic catastrophe
pathway. Despite the promise of VDT treatment demonstrated here,
further investigation is necessary to elucidate the relationship
between autophagy and microtubules after VDT treatment in LNCaP
cells.
Acknowledgements
This study was supported in part by the Korea
Research Foundation (KRF-2013-041-E00392) grant funded by the
Korean Government.
References
|
1
|
Jemal A, Bray F, Center MM, Ferly J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
6:69–90. 2011. View Article : Google Scholar
|
|
2
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 6:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Riihimäki M, Thomsen H, Brandt A,
Sundquist J and Hemminki K: What do prostate cancer patients die
of? Oncologist. 16:175–181. 2011.PubMed/NCBI
|
|
4
|
Hsing AW and Chokkalingam AP: Prostate
cancer epidemiology. Front Biosci. 11:1388–1413. 2006. View Article : Google Scholar
|
|
5
|
Parent ME and Siemiatycki J: Occupation
and prostate cancer. Epidemiol Rev. 23:138–143. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lock RL and Harry EJ: Cell-division
inhibitors: new insights for future antibiotics. Nat Rev Drug
Discov. 7:324–338. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Erickson HP, Anderson DE and Osawa M: FtsZ
in bacterial cytokinesis: cytoskeleton and force generator all in
one. Microbiol Mol Biol Rev. 74:504–528. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang J, Galgoci A, Kodali S, Herath KB,
Jayasuriya H, Dorso K, Vicente F, Gonzalez A, Cully D, Bramhill D
and Singh S: Discovery of a small molecule that inhibits cell
division by blocking FtsZ, a novel therapeutic target of
antibiotics. J Biol Chem. 278:44424–44428. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Löwe J and Amos LA: Crystal structure of
the bacterial cell-division protein FtsZ. Nature. 391:203–206.
1998.
|
|
10
|
Chung KS, Yim NH, Lee SH, Choi SJ, Hur KS,
Hoe KL, Kim DU, Goehle S, Kim HB, Song KB, Yoo HS, Bae KH, Simon J
and Won M: Identification of small molecules inducing apoptosis by
cell-based assay using fission yeast deletion mutants. Invest New
Drugs. 26:299–307. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Heggeness MH, Simon M and Singer SJ:
Association of mitochondria with microtubules in cultured cells.
Proc Natl Acad Sci. 75:3863–3866. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Diaz JF and Andreu JM: Assembly of
purified GDP-tubulin into microtubules induced by taxol and
taxotere: reversibility, ligand stoichiometry, and competition.
Biochemistry. 32:2747–2755. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lavelle F, Bissery MC, Combeau C, Riou JF,
Vrignaud P and André S: Preclinical evaluation of docetaxel
(Taxotere). Semin Oncol. 22:3–16. 1995.
|
|
14
|
Stein CA: Mechanisms of action of taxanes
in prostate cancer. Semin Oncol. 26:3–7. 1999.PubMed/NCBI
|
|
15
|
Kim JY, Chung JY, Lee SG, Kim YJ, Park JE,
Yun J, Park YC, Kim BG, Yoo YH and Kim JM: p53 interferes with
microtubule-stabilizing agent-induced apoptosis in prostate and
colorectal cancer cells. Int J Mol Med. 31:1388–1394.
2013.PubMed/NCBI
|
|
16
|
Wolter KG, Hsu YT, Smith CL, Nechushtan A,
Xi XG and Youle RJ: Movement of Bax from the cytosol to
mitochondria during apoptosis. J Cell Biol. 139:1281–1292. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Saunders DE, Lawrence WD, Christensen C,
Wappler NL, Ruan H and Deppe G: Paclitaxel-induced apoptosis in
MCF-7 breast-cancer cells. Int J Cancer. 70:214–220. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin HL, Liu TY, Chau GY, Lui WY and Chi
CW: Comparison of 2-methoxyestradiol-induced, docetaxel-induced,
and paclitaxel-induced apoptosis in hepatoma cells and its
correlation with reactive oxygen species. Cancer. 89:983–994. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vakifahmetoglu H, Olsson M and Zhivotovsky
B: Death through a tragedy: mitotic catastrophe. Cell Death Differ.
15:1153–1162. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roninson IB, Broude EV and Chang BD: If
not apoptosis, then what? Treatment-induced senescence and mitotic
catastrophe in tumor cells. Drug Resist Updat. 4:303–313. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alotaibi MR, Asnake B, Di X, Beckman MJ,
Durrant D, Simoni D, Baruchello R, Lee RM, Schwartz EL and Gewirtz
DA: Stilbene 5c, a microtubule poison with vascular disrupting
properties that induces multiple modes of growth arrest and cell
death. Biochem Pharmacol. 86:1688–1698. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aplin A, Jasionowski T, Tuttle DL, Lenk SE
and Dunn WA Jr: Cytoskeletal elements are required for the
formation and maturation of autophagic vacuoles. J Cell Physiol.
152:458–466. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fass E, Shvets E, Degani I, Hirschberg K
and Elazar Z: Microtubules support production of starvation-induced
autophagosomes but not their targeting and fusion with lysosomes. J
Biol Chem. 281:36303–36316. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kochl R, Hu XW, Chan EY and Tooze SA:
Microtubules facilitate autophagosome formation and fusion of
autophagosomes with endosomes. Traffic. 7:129–145. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Acharya BR, Bhattacharyya S, Choudhury D
and Chakrabarti G: The microtubule depolymerizing agent
naphthazarin induces both apoptosis and autophagy in A549 lung
cancer cells. Apoptosis. 16:924–939. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie R, Nguyen S, McKeehan WL and Liu L:
Acetylated microtubules are required for fusion of autophagosomes
with lysosomes. BMC Cell Biol. 11:89–100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mansila S, Bataller M and Portugal J:
Mitotic catastrophe as a consequence of chemotherapy. Anticancer
Agents Med Chem. 6:589–602. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Harper JW, Adami GR, Wei N, Keyomarsi K
and Elledge SJ: The p21 Cdk-interacting protein Cip1 is a potent
inhibitor of G1 cyclin-dependent kinases. Cell. 75:805–816. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Grimmler M, Wang Y, Mund T, Ciliensek Z,
Keidel EM, Waddell MB, Jäkel H, Kullmann M, Kriwacki RW and Hengst
L: Cdk-inhibitory activity and stability of p27Kip1 are directly
regulated by oncogenic tyrosine kinases. Cell. 128:269–280. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Millot C, Millot JM, Morjani H, Desplaces
A and Manfait M: Characterization of acidic vesicles in
multidrug-resistant and sensitive cancer cells by acridine orange
staining and confocal micro spectrofluorometry. J Histochem
Cytochem. 45:1255–1264. 1997. View Article : Google Scholar
|
|
31
|
Perez EA: Microtubule inhibitors:
differentiating tubulin-inhibiting agents based on mechanisms of
action, clinical activity, and resistance. Mol Cancer Ther.
8:2086–2095. 2009. View Article : Google Scholar
|
|
32
|
Lu Y, Chen J, Xiao M, Li W and Miller DD:
An overview of tubulin inhibitors that interact with the colchicine
binding site. Pharm Res. 29:2943–2971. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Longuet M, Serduc R and Riva C:
Implication of bax in apoptosis depends on microtubule network
mobility. Int J Oncol. 25:309–317. 2004.PubMed/NCBI
|
|
34
|
Botta M, Forli S, Magnani M and Manetti F:
Molecular modeling approaches to study the binding mode on tubulin
of microtubule destabilizing and stabilizing agents. Top Curr Chem.
286:279–328. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Georgiadis MS, Russell EK, Gazdar AF and
Johnson BE: Paclitaxel cytotoxicity against human lung cancer cell
lines increases with prolonged exposure durations. Clin Cancer Res.
3:449–454. 1997.PubMed/NCBI
|
|
36
|
Checchi PM, Nettles JH, Zhou J, Snyder JP
and Joshi HC: Microtubule-interacting drugs for cancer treatment.
Trends Pharmacol Sci. 24:361–365. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hernandez-Vargas H, Palacios J and
Moreno-Bueno G: Molecular profiling of docetaxel cytotoxicity in
breast cancer cells: uncoupling of aberrant mitosis and apoptosis.
Oncogene. 26:2902–2913. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Silva MRO, Kawai K, Hosoe T, Takaki GMC,
Gusmão NB and Fukushima K: Viriditoxin, an antibacterial substance
produced by mangrove endophytic fungus Paecilomyces
variotii. Microbial Pathogens and Strategies for Combating
Them: Science, Technology and Education. Méndez-Vilas A: Formatex
Research Center; Badajoz: pp. 1406–1411. 2013
|
|
39
|
Park YS, Grove CI, González-López M,
Urgaonkar S, Fettinger JC and Shaw JT: Synthesis of
(−)-viriditoxin: a 6,6′-binaphthopyran-2-one that targets the
bacterial cell division protein FtsZ. Angew Chem Int Ed Engl.
50:3730–3733. 2011.
|
|
40
|
Feng Z, Zhang H, Levine AJ and Jin S: The
coordinate regulation of the p53 and mTOR pathways in cells. Proc
Natl Acad Sci. 102:8204–8209. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Brady CA and Attardi LD: p53 at a glance.
J Cell Sci. 123:2527–2532. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Eastman A: Cell cycle checkpoints and
their impact on anticancer therapeutic strategies. J Cell Biochem.
91:223–231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Payne SR, Zhang S, Tsuchiya K, Moser R,
Gurley KE, Longton G, deBoer J and Kemp CJ: p27kip1 deficiency
impairs G2/M arrest in response to DNA damage, leading to an
increase in genetic instability. Mol Cell Biol. 28:258–268. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Maiuri MC, Galluzzi L, Morselli E, Kepp O,
Malik SA and Kroemer G: Autophagy regulation by p53. Curr Opin Cell
Biol. 22:181–185. 2010. View Article : Google Scholar
|
|
45
|
Tasdemir E, Maiuri MC, Galluzzi L, Vitale
I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C,
Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R,
Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G,
Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F and
Kroemer G: Regulation of autophagy by cytoplasmic p53. Nat Cell
Biol. 10:676–687. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lin NY, Beyer C, Giessl A, Kireva T,
Scholtysek C, Uderhardt S, Munoz LE, Dees C, Distler A, Wirtz S,
Krönke G, Spencer B, Distler O, Schett G and Distler JH: Autophagy
regulates TNFα-mediated joint destruction in experimental
arthritis. Ann Rheum Dis. 72:761–768. 2013.
|
|
47
|
Klionsky DJ: The molecular machinery of
autophagy: unanswered questions. J Cell Sci. 118:7–18. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Klionsky DJ: Autophagy: from phenomenology
to molecular understanding in less than a decade. Nat Rev Mol Cell
Biol. 8:931–937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mizushima N and Klionsky DJ: Protein
turnover via autophagy: implications for metabolism. Annu Rev Nutr.
27:19–40. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shimizu S, Yoshida T, Tsujioka M and
Arakawa S: Autophagic cell death and cancer. Int J Mol Sci.
15:3145–3153. 2014. View Article : Google Scholar
|
|
51
|
Nabha SM, Mohammad RM, Dandashi MH,
Coupaye-Gerard B, Aboukameel A, Pettit GR and Al-Katib AM:
Combretastatin-A4 prodrug induces mitotic catastrophe in chronic
lymphocytic leukemia cell line independent of caspase activation
and poly (adp-ribose) polymerase cleavage. Clin Cancer Res.
8:2735–2741. 2002.PubMed/NCBI
|
|
52
|
Burns TF, Fei P, Scata KA, Dicker DT and
El-Deiry WS: Silencing of the novel p53 target gene Snk/Plk2 leads
to mitotic catastrophe in paclitaxel (taxol)-exposed cells. Mol
Cell Biol. 23:5556–5571. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chen Y, McMillan-Ward E, Kong J, Israels
SJ and Gibson SB: Oxidative stress induces autophagic cell death
independent of apoptosis in transformed and cancer cells. Cell
Death Differ. 15:171–182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kamath K, Okouneva T, Larson G, Panda D,
Wilson L and Jordan MA: 2-Methoxyestradiol suppresses microtubule
dynamics and arrests mitosis without depolymerizing microtubules.
Mol Cancer Ther. 5:2225–2233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lorin S, Borges A, Ribeiro Dos Santos L,
Souquere S, Pierron G, Ryan KM, Codogno P and Djavaheri-Mergny M:
c-Jun NH2-terminal kinase activation is essential for
DRAM-dependent induction of autophagy and apoptosis in
2-methoxyestradiol-treated Ewing sarcoma cells. Cancer Res.
69:6924–6931. 2009. View Article : Google Scholar
|
|
56
|
Arstila AU, Nuuja IJ and Trump BF: Studies
on cellular autophagocytosis: vinblastine-induced autophagy in the
rat liver. Exp Cell Res. 87:249–252. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Marzella L, Sandberg PO and Glaumann H:
Autophagic degradation in rat liver after vinblastine treatment.
Exp Cell Res. 128:291–301. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tanida I, Ueno T and Kominami E: LC3 and
autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar
|
|
59
|
Geeraert C, Ratier A, Pfisterer SG, Perdiz
D, Cantaloube I, Rouault A, Pattingre S, Proikas-Cezanne T, Codogno
P and Pous C: Starvation-induced hyperacetylation of tubulin is
required for the stimulation of autophagy by nutrient deprivation.
J Biol Chem. 285:24184–24194. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shen S, Kepp O, Martins I, Vitale I,
Souquère S, Castedo M, Pierron G and Kroemer G: Defective autophagy
associated with LC3 puncta in epothilone-resistant cancer cells.
Cell Cycle. 9:377–383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Reunanen H, Marttinen M and Hirsimaki P:
Effects of griseofulvin and nocodazole on the accumulation of
autophagic vacuoles in Ehrlich ascites tumor cells. Exp Mol Pathol.
48:97–102. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Mackeh R, Perdiz D, Lorin S, Codogno P and
Pous C: Autophagy and microtubules - new story, old players. J Cell
Sci. 126:1071–1080. 2013. View Article : Google Scholar : PubMed/NCBI
|