Introduction
Well-differentiated papillary and follicular
carcinomas typically have indolent behavior and can be effectively
treated by surgery followed by radioiodine therapy (1). However, tumors that lose
differentiation during progressive disease or already have
developed as undifferentiated entities (i.e., anaplastic thyroid
tumors) have lost the ability to trap iodine. As a consequence,
these types of tumors do not respond to conventional radioiodine
treatment and, thus, have a much less favorable prognosis (2). Hence, alternative concepts are
urgently warranted to improve therapeutic undertakings. In recent
years, small molecule kinase inhibitors have been introduced in
therapeutic treatments of several types of cancers (3,4).
Especially in regard to thyroid cancers focus has been directed to
the Ras/ERK1/2 and PI3K/AKT pathways because both are activated in
a high percentage of thyroid tumors. In more than 80% of papillary
cancers, for example, genes of the MAPK pathway are affected
(5). Activation of the classical
MAPK pathway occurs mainly via genetic alterations of Ras, Raf or
Ret genes (6). In addition to the
activation of the MAPK pathway, RET/PTC or BRAF mutations can also
result in the activation of PI3K. In thyroid cancer, moreover, the
activation of Akt can also be a result of PI3K mutation and
amplification, RET/PTC rearrangements, reduction or loss of PTEN
expression (7).
Clinical studies with specific ERK1/2 or Akt
inhibitors as well as with multikinase inhibitors such as sorafenib
have already been undertaken with promising results. Sorafenib (BAY
43-9006, Nexavar) is a second-generation, orally active multikinase
inhibitor that was recently approved for the treatment of patients
with advanced renal cell carcinoma and patients with unresectable
hepatocellular carcinoma (8). Some
clinical studies have also demonstrated the effectiveness of
sorafenib in advanced thyroid cancer (9,10).
The action of sorafenib affects a broader range of signal
transducers, such as vascular endothelial growth factor receptors,
platelet-derived growth factor receptor-β, c-kit or RET (8). Sorafenib downregulates activated Akt
and ERK1/2 in neuroblastoma (11)
and lymphoma cells (12), but,
unexpectedly, can also initiate ERK1/2 (13,14)
or Akt activation (15) that leads
to increased tumor cell proliferation and migration. The use of the
MEK inhibitors PD0325901 or U0126 is restricted to academic
research, as these drugs produced undesirable side effects in
clinical trials (16).
Nevertheless, these compounds serve as valuable experimental
paradigms to analyze the consequences of MEK/ERK1/2 inactivation on
the cellular level.
In the present study we show that in thyroid
carcinoma cells downregulation of Ras/MAPK and PI3K/Akt pathway
activation predominantly suppresses cell migration and
proliferation. In the anaplastic cell line Cal-62, however,
inactivation of Ras/MAPK signaling positively affects the migration
rate. This increase can be suppressed by PI3K/Akt pathway-dependent
(MK-2006 treatment) and, most likely, -independent (sorafenib
treatment) mechanisms.
Materials and methods
Cell lines and culture conditions
Human papillary thyroid cancer cell line B-CPAP and
human follicular thyroid cancer cell line FTC-133 were from
Professor G. Brabant (Department of Endocrinology, Christie
Hospital, Manchester, UK). Human anaplastic thyroid cancer cell
line Cal-62 was obtained from Leibniz Institute DSMZ, German
Collection of Microorganisms and Cell Cultures (Braunschweig,
Germany). B-CPAP cells were cultivated in RPMI-1640 medium, Cal-62
cells in DMEM, and FTC-133 cells in DMEM/Ham’s F12. Media were
supplemented with 10% fetal calf serum (FCS), 1%
penicillin-streptomycin and L-glutamine.
Cell extraction and western blot
analysis
Cell solubilisation in the presence of protease and
phosphatase inhibitors and western blot analyses were carried out
as described (17).
Drug treatment
Stock solutions and working concentrations were as
follows: for MK-2206 (Selleck Chemicals), 10 mM and 1 μM, for
PD0325901 (Axon Medchem) 10 mM and 2 μM, for sorafenib (Enzo Life
Sciences) 10 mM and 5 μM, for U0126 (Promega) 10 mM and 20 μM, for
wortmannin (Santa Cruz) 1 mM and 100 nM. All stock solutions were
prepared in DMSO and stored at −20°C. In a typical experiment,
cells were cultivated for three days in the absence or presence of
the compound that was added once after 36 h or twice after 24 and
48 h.
Cell proliferation assay
To analyze cell proliferation, 500 cells in 100 μl
culture medium were seeded in 96-well plates (12 wells per
time-point and treatment). Drugs were added immediately after 24 as
well as after 48 h in 100 μl of culture medium. Addition of pure
DMSO served as a medium control. Cells were fixed in 4%
formaldehyde in PBS just before treatment (T0) or after 72 h of
treatment (T1). Cells from both time-points were then stained with
DAPI (1 μg/ml in PBS) in order to determine microscopically the
cell number in a defined area of each well.
Collective cell migration assay
Cell suspensions (5,000 cells/μl) in a total volume
of 3 μl were seeded on defined regions in Petri dishes and allowed
to adhere for two to three hours. After floating the dishes with
culture medium, the adherent and confluent cells occupied a
circular area. Diameters of areas (12 per dish) were determined
microscopically subsequently after floating (T0) and after a 72-h
culture period (T1). Compounds were added at T0 as well as 24 and
48 h later.
Cell toxicity assay
Cytotoxicity was analyzed with the LDH Cytotoxicity
Assay Kit from Roche. Briefly, 10,000 cells (in 100 μl culture
medium) were seeded per well in 96-well plates. Twenty-four hours
later, cells were exposed to different drug concentrations for
24–72 h. After treatment, LDH activity was determined in the cell
culture supernatants. In parallel, cells that had been treated
identically were lysed in order to determine total LDH activity, a
value that allows to quantify cell proliferation.
Results
ERK1/2 and Akt activation in human
thyroid cancer cells
Human papillary cell line B-CPAP, anaplastic cell
line CAL-62 and follicular cell line FTC-133 were maintained for 48
h in the presence or absence of serum, or for 48 h in the absence
and finally for 20 min in the presence of serum. Under serum-free
and serum-containing culture conditions, no considerable
differences in the expression levels of pERK1/2 or pAkt were
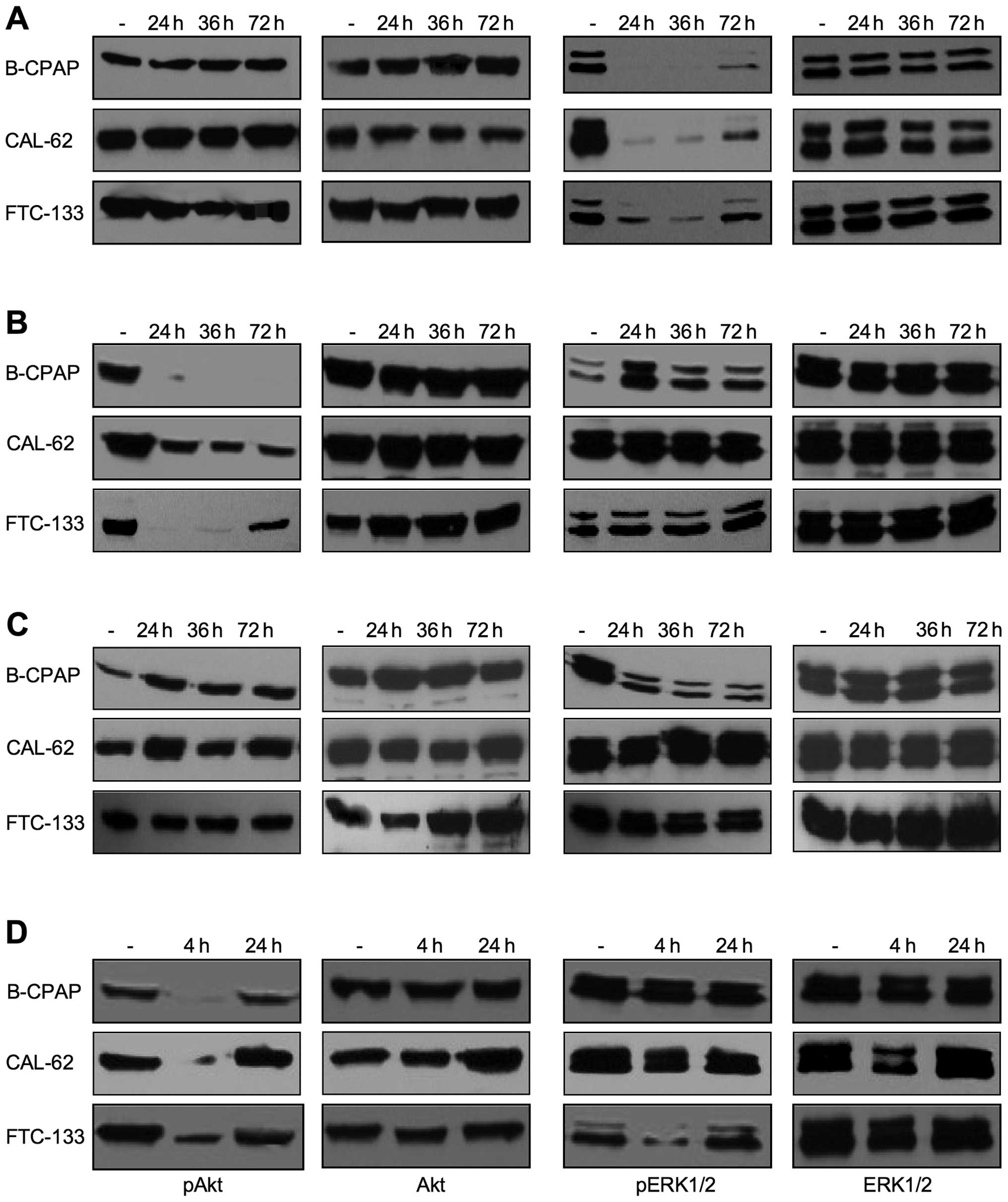
observed (Fig. 1). Stimulation of
quiescent cells with a serum pulse did not lead to an increased
pERK1/2 and pAkt activity, suggesting a constitutive activation of
Akt and ERK1/2 in thyroid cancer cells to an almost maximal
extent.
Pharmacological inhibition of
kinases
We first optimized the conditions to achieve maximal
inhibition rates after drug application. Drugs were applied for 72
h either without further medium change, with one medium change
after 36 h or with two medium changes after 24 and 48 h (Fig. 2). The most efficient inactivation
of ERK1/2 kinases was achieved when U0126 or PD0325901 were added
daily or at least once after 36 h (shown for U0126 in Fig. 2A). An almost complete inactivation
of Akt over the whole three-day incubation period was obtained with
a single addition of 1 μM MK-2206 in B-CPAP and with a twice
repeated addition in FTC-133 cells. In contrast, in Cal-62 only an
incomplete inhibition was detected, even when Mk-2206 was renewed
daily (Fig. 2B) or its
concentration was increased to 2 μM (not shown). Treatment with
multikinase inhibitor sorafenib did not affect the activation of
Akt and ERK1/2 in Cal-62 and FTC-133 cells, but strongly reduced
the pERK1/2 expression level in B-CPAP cells at a concentration of
5 μM (Fig. 2C). PI3K inhibitor
wortmannin efficiently inhibited pAkt expression, but due to the
instability of the drug, this effect was stable only for short time
periods after application (Fig.
2D, compare incubation times of 4 and 24 h). Such types of
experiments also allow to analyze the presence of crosstalk between
PI3K/Akt and the Ras/MAPK pathways. Indications for cross-talks of
the two pathways were mostly observed after a more frequent drug
application such as the upregulation of pERK1/2 upon MK-2206
application in B-CPAP cells or the downregulation of pERK1/2 upon
wortmannin treatment in FTC-133 cells (Fig. 2). Based on these results, in
three-day lasting functional assays, drugs were replaced every 24 h
and applied at a concentration of 1 μM for PD0325901, MK-2206 and
sorafenib and at 20 μM for U0126. Because of its low stability,
wortmannin was not included in long-lasting experiments.
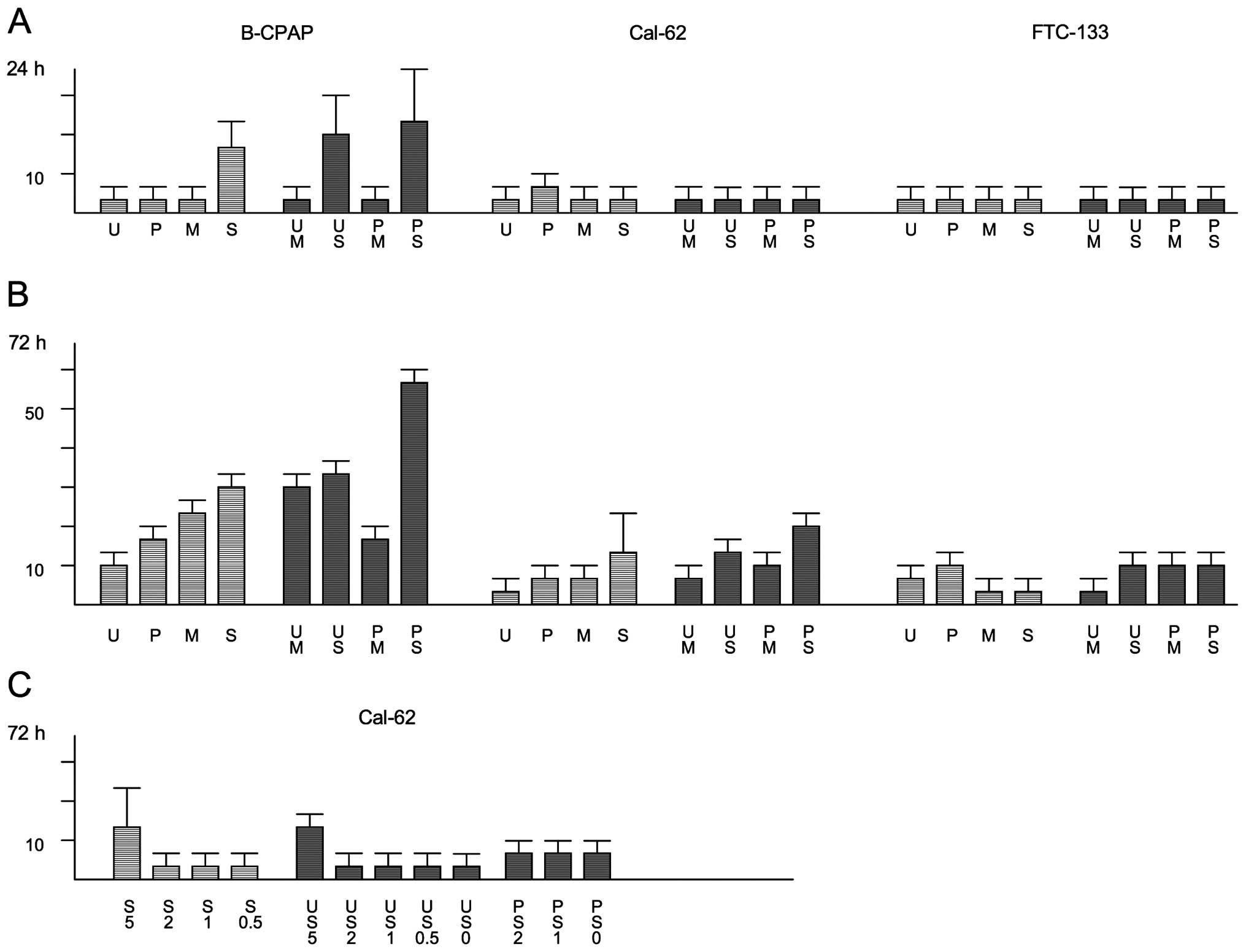
Impact of kinase inhibition on cell
proliferation
Low density thyroid carcinoma cells were allowed to
proliferate in 96-well plates for three days in the absence or
presence of compounds. Different sets of cells were stained with
DAPI before and after the 72-h incubation period and cell numbers
were determined microscopically (Fig.
3 and Table I). Perturbation
of the Ras/MAPK pathway led to a strong reduction in cell number,
whereby inhibition rates of 70–90% were observed for the different
cell lines after U0126 treatment (Fig.
3 and Table I). For PD0325901,
inhibition rates varied between 53% for FTC-113 cells and 84% for
B-CPAP cells (Table I). Also with
the multikinase inhibitor sorafenib proliferation of thyroid
carcinoma cells could be efficiently inhibited in the range of 70
to ~90% (Table I). Disruption of
the PI3K/Akt pathway with MK-2206 inhibited the proliferation rate
by ~65% in B-CPAP and by ~40% in Cal-62 and FTC-133 cells.
 | Table IImpact of kinase inhibition on the
proliferation rate of thyroid carcinoma cells. |
Table I
Impact of kinase inhibition on the
proliferation rate of thyroid carcinoma cells.
| Cell line | U0126 | PD0325901 | Sorafenib | MK-2206 |
|---|
| B-CPAP | 90±8c | 84±2c | 91±17c | 65±24b |
| Cal-62 | 81±11c | 74±27b | 68±6c | 42±14b |
| FTC-133 | 72±8c | 53±13b | 69±6c | 43±16b |
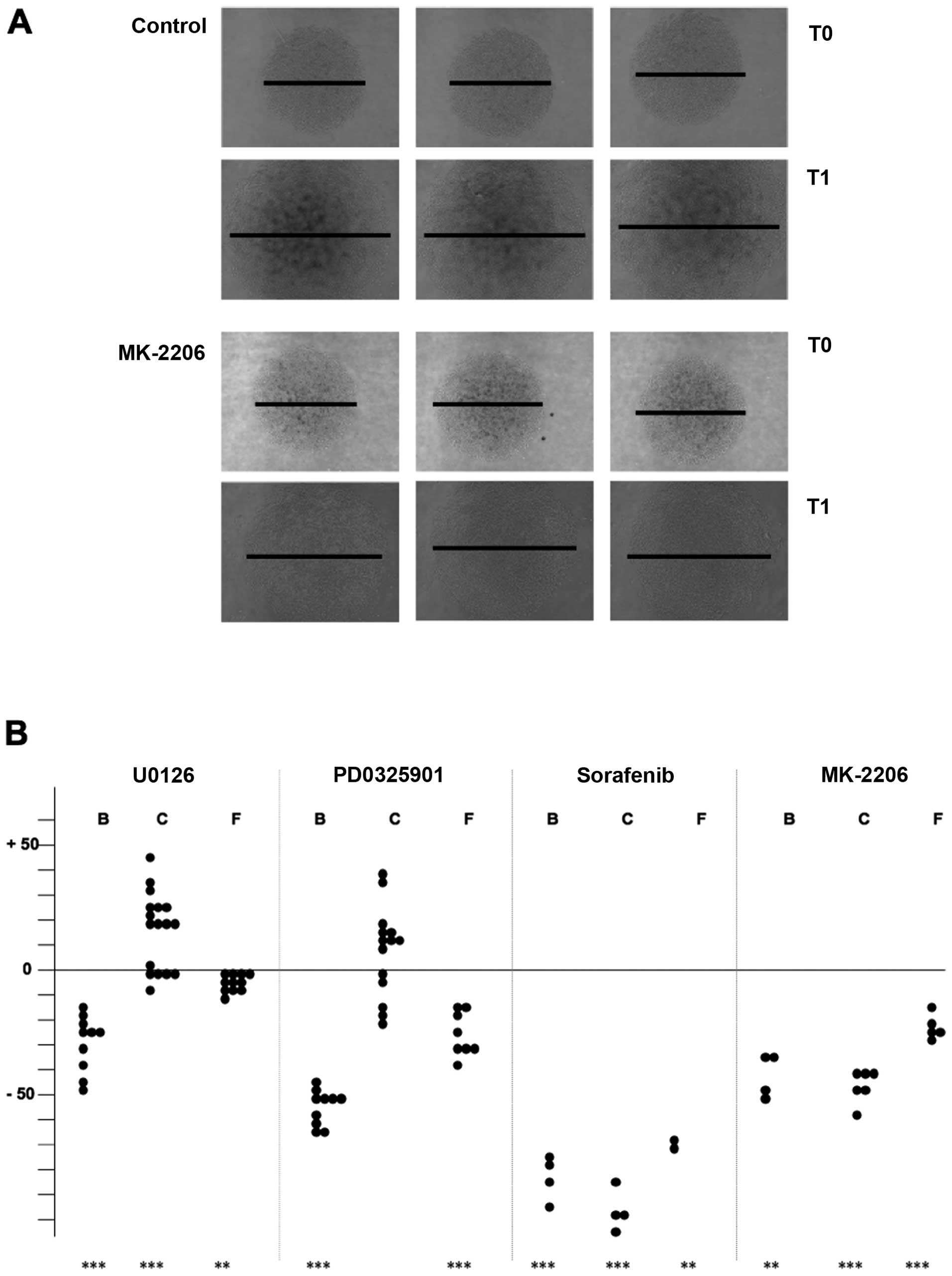
Impact of kinase inhibition on collective
cell migration
Thyroid carcinoma cells were seeded at high density
on defined areas into Petri dishes and allowed to adhere for two to
three hours to establish a circular monolayer. After floating with
medium, cells were cultured for three days in the presence or
absence of drugs (Fig. 4A for
FTC-133 cells) and the diameters of the monolayers were determined
at the beginning and the end of the culture period (Table II). In general, the migration
capacity of thyroid carcinoma cells was suppressed to a varying
extent in the presence of the different kinase inhibitors. For
B-CPAP cells, inhibition rates in the range of 30 (for U0126) up to
80% (for sorafenib) were observed. The migration capacity of
FTC-133 cells was relatively robust with regard to drug treatment
(inhibition rates in the range of 7–50%). Surprisingly,
MEK/ERK1/2-inhibitor treated Cal-62 cells showed an increased
migration rate, notwithstanding a strong inhibitory effect in the
range of 45 or 80% became visible upon MK-2206 or sorafenib
treatment. Eventhough statistical significance is only present for
U0126-treatment, the data shown in Fig. 4B demonstrate that in more than half
of the experiments performed with PD0325901 an increased migration
also took place.
| Table IIImpact of drug treatment on collective
cell migration of thyroid carcinoma cells. |
Table II
Impact of drug treatment on collective
cell migration of thyroid carcinoma cells.
| Cell line | U0126 | PD0325901 | Sorafenib | MK-2206 |
|---|
| B-CPAP | 29±9c | 53±7c | 79±12c | 39±8b |
| Cal-62 | +15±12c | +10±17 | 84±12c | 46±7c |
| FTC-133 | 7±5b | 23±9c | 53±3b | 24±6c |
We thus wondered, if the stimulating effect of U0126
and PD0325901 on the migration of Cal-62 cells could be suppressed
by a simultaneous inhibition of the PI3K/Akt pathway, because
western blot analyses revealed that the combined treatment of
thyroid carcinoma cells with U0126 and MK-2206 led to an almost
complete downregulation of pAkt and pERK1/2 expression (Fig. 5). Indeed, the migration rate of
U0126/MK-2206 dual-treated Cal-62 cells was substantially inhibited
not only in respect to U0126- or PD0325901-single-treated, but also
in comparison to untreated control cells (Table IIIA). Moreover, although
sorafenib did not affect Akt activation in U0126- or
PD0325901-treated Cal-62 cells (Fig.
5), its presence led to a substantial inhibition of cell
migration (Table IIIA). To rule
out that the inhibition observed results mainly from cytotoxic drug
effects, LDH assays were carried out. After a 24-h incubation
period none of the single or dual drug treatments induced a
noteworthy cytotoxic effect in Cal-62 cells, although dual-treated
B-CPAP cells were considerably affected (Fig. 6A). After a 72-h incubation period,
however, a moderate cytotoxic effect was observed in
sorafenib-treated Cal-62 cells. Therefore, it is possible that
under these conditions the proliferation and migration suppressing
potential of this compound is partially due to a cytotoxic effect.
Thus, in a final set of experiments, we applied sorafenib at
concentrations of 1 and 2 μM, i.e., at concentrations that did not
induce considerable cytotoxic effects in Cal-62 cells after a 72-h
incubation period neither as a single agent nor in combination with
20 μM U0126 or 2 μM PD0325901 (Fig.
6C). Under these conditions, the migration-promoting effect of
U0126 was completely compensated, but an additional inhibition, as
could be monitored for higher sorafenib concentrations, was not
observed (Table IIIB).
Altogether, these data suggest that the migration-stimulating
effect of PD0325901 and U0126 can be suppressed by Akt-dependent
and -independent mechanisms, even though an inactivation of Akt
downstream signaling molecules cannot be completely excluded.
| Table IIIImpact of single or dual drug
treatment on collective cell migration of Cal-62 cells. |
Table III
Impact of single or dual drug
treatment on collective cell migration of Cal-62 cells.
| A. |
|---|
|
|---|
| Additive | - | U0126 | PD0325901 |
|---|
| - | 0 | +23±16b | +4±18 |
| MK-2206 | 46±7c | 34±6c | 52±8c |
| Sorafenib | 84±12c | 54±7c | 33±21b |
|
| B. |
|
| Additive | - | U0126 | PD0325901 |
|
| - | 0 | +36±6b | +18±5b |
| Sorafenib 2 μM | 14±4b | 2±7 | 12±9 |
| Sorafenib 1 μM | 7±9 | 1±5 | 8±2 |
Discussion
Our study demonstrated that the migration and
proliferation of human thyroid carcinoma cells is partially
mediated via constitutive active PI3K/Akt and RAS/MAPK signaling.
Pharmacological inhibition of this signaling considerably inhibits
proliferation in all three cell lines, although to varying extent
(Table I). In general, also the
migration rate of thyroid carcinoma cells is suppressed in the
presence of the different kinase inhibitors, with the remarkable
exception of an increased migration of Cal-62 cells in the presence
of U0126- or PD0325901. This increase can be prohibited by MK-2206
treatment, i.e., via Akt inactivation, or by application of
sorafenib, a compound that does not perturb Akt activation level in
Cal-62 cells.
Crosstalk of signaling pathways in
thyroid carcinoma cells
PI3K/Akt and Ras/MAPK signaling pathways, although
first described as linear conduits were soon shown to be connected
with each other in a complicated fashion that comprises
cross-activation and -inhibition processes as well as negative
feedback loops and pathway convergence (18). In our experiments we found evidence
for the presence of cross-inhibitory circuits of PI3K/Akt on
Ras/MAPK in B-CPAP and of cross-activating circuits in FTC-133
cells (Fig. 2). As such crosstalk
depends inter alia on substrate availability that can vary between
different cell types or cell lines it is not surprising that the
interactions we observed exhibit a cell line-specific pattern that
can not be generalized (Fig. 2).
Kandil and colleagues (19) also
recently reported that cross-activation of Akt varies between
different thyroid cancer cell lines upon treatment with the MEK/ERK
inhibitor AZD6244. At the present level of knowledge, it is mainly
assumed that the PI3K/Akt and Ras/MAPK pathways can negatively
regulate each other and, in addition, the latter one can activate
the former (18). Nevertheless,
and thereby underlining our data, in a few reports it has been
shown, that the PI3K/Akt pathway can activate ERK1/2 signaling
(20–22).
Yin-yang effects in thyroid carcinoma
cells
Although we have evidence for the presence of
crosstalk between PI3K/Akt- and Ras/MAPK pathways in thyroid
carcinoma cells, it does not illuminate the yin-yang effect we
detected in Cal-62 cells treated with MEK inhibitors:
Treatment of Cal-62 cells with MEK inhibitors leads
to an almost complete inhibition of pERK1/2 expression, but leaves
pAkt expression substantially unaffected (Fig. 2). Vice versa, treatment with an Akt
inhibitor strongly decreases pAkt expression without affecting the
expression level of pERK1/2 (Fig.
2). Thus, no obvious crosstalk of the two pathways seems to be
present in Cal-62 cells.
The proliferation rate of Cal-62 cells is strongly
suppressed by MEK inhibitors, but only mildly by MK-2206 (Table I), whereas the migration rate is
significantly enhanced by MEK inhibitors and strongly suppressed by
MK-2206 (Table II). Thus, the
increased surface area observed in the collective cell migration
experiments cannot be simply the result of an increased cell mass,
or, more generally, of a growth process.
Increased migration of Cal-62 cells due to the
presence of MEK inhibitors can be suppressed by MK-2206 or
sorafenib, i.e., in the presence of low as well as high expression
levels of pAkt. To our knowledge, sorafenib treatment does not
affect Akt downstream signaling molecules. Thus, it is likely that
the stimulatory effect of MEK inhibitors can be suppressed via
Akt-dependent and -independent signaling mechanisms.
Collectively, it seems feasible that interference
with a single pathway can inhibit one cellular process, here the
cell’s proliferative capacity, and at the same time promote
another, here the cell’s migratory potential. In our example such
an effect can be interpreted by assuming the existence of an
(hyper)activated Ras/ERK1/2 pathway that suppresses cell migration
but can itself be suppressed by MEK inhibitors. Based on the above,
one could expect that Cal-62 cells migrate with a submaximal
velocity, and indeed, although we have not performed a systematic
analysis, Cal-62 shows the lowest velocity amongst the three cell
lines we have used, i.e., a 25 or 40% reduced migration rate in
comparison to FTC-133 or B-CPAP cells, respectively. This resembles
a scenario that hyperactivation of the Ras/ERK1/2 pathway can
trigger a decreased proliferation rate, i.e., cell cycle arrest
(23). An inhibitory effect of the
MAPK cascade on cell proliferation is also active in K562
erythroleukemia cells, which differentiate into megakaryocytes upon
phorbol ester stimulation, a process that is: i) accompanied by
growth retardation and ERK activation, and ii) completely
suppressed in the presence of MEK inhibitors (24). The aspect that Ras/ERK1/2 pathway
activation not only activates (25,26),
but may also suppress cell migration adds another facet to the
complex pattern of MAPK signaling. To overcome the stimulatory
effect of MEK inhibitors on Cal-62 cells, we have performed dual
treatments to interfere with more than one signaling pathway and,
thereby, successfully suppressed cell proliferation as well as
migration. Such a strategy, in clinical practice known as a subtype
of combination therapy (27), has
been used in a number of trials to attack the fatal potential of
tumor cells more efficiently. For example, in thyroid carcinoma
cells, dual Ras/MAPK and PI3K/Akt/mTOR pathway inhibition leads to
significant higher growth retardation even in a synergistic manner
when compared to single pathway inhibition (28,29).
Beside the advantage of a combinatorial treatment to increase the
inhibitory action of a single drug such a strategy also decreases
the risk of unfavorable yin-yang effects.
Acknowledgements
J.W. was supported by a grant from the German
Research Foundation and the Medical Faculty of the University of
Bonn (CRU208/TP10).
References
|
1
|
Ambrosetti MC, Colato C, Dardano A,
Monzani F and Ferdeghini M: Radioiodine ablation: when and how. Q J
Nucl Med Mol Imaging. 53:473–481. 2009.PubMed/NCBI
|
|
2
|
Hannallah J, Rose J and Guerrero MA:
Comprehensive literature review: recent advances in diagnosing and
managing patients with poorly differentiated thyroid carcinoma. Int
J Endocrinol. 2013:3174872013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dar AC and Shokat KM: The evolution of
protein kinase inhibitors from antagonists to agonists of cellular
signaling. Annu Rev Biochem. 80:769–795. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gild ML, Bullock M, Robinson BG and
Clifton-Bligh R: Multikinase inhibitors: a new option for the
treatment of thyroid cancer. Nat Rev Endocrinol. 7:617–624. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Antonelli A, Fallahi P, Ferrari SM, et al:
Dedifferentiated thyroid cancer: a therapeutic challenge. Biomed
Pharmacother. 62:559–563. 2008. View Article : Google Scholar
|
|
6
|
Nikiforov YE: Thyroid carcinoma: molecular
pathways and therapeutic targets. Mod Pathol. 21:S37–S43. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Saji M and Ringel MD: The PI3K-Akt-mTOR
pathway in initiation and progression of thyroid tumors. Mol Cell
Endocrinol. 32:20–28. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wilhelm SM, Adnane L, Newell P, Villanueva
A, Llovet JM and Lynch M: Preclinical overview of sorafenib, a
multikinase inhibitor that targets both Raf and VEGF and PDGF
receptor tyrosine kinase signaling. Mol Cancer Ther. 7:3129–3140.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Duntas LH and Bernardini R: Sorafenib:
rays of hope in thyroid cancer. Thyroid. 2:1351–1358. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fallahi P, Ferrari SM, Santini F, et al:
Sorafenib and thyroid cancer. BioDrugs. 27:615–628. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chai H, Luo AZ, Weerasinghe P and Brown
RE: Sorafenib downregulates ERK/Akt and STAT3 survival pathways and
induces apoptosis in a human neuroblastoma cell line. Int J Clin
Exp Pathol. 3:408–415. 2010.PubMed/NCBI
|
|
12
|
Carlo-Stella C, Locatelli SL, Giacomini A,
et al: Sorafenib inhibits lymphoma xenografts by targeting MAPK/ERK
and AKT pathways in tumor and vascular cells. PLoS One.
8:e616032013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nguyen TK, Jordan N, Friedberg J, Fisher
RI, Dent P and Grant S: Inhibition of MEK/ERK1/2 sensitizes
lymphoma cells to sorafenib-induced apoptosis. Leuk Res.
34:379–386. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rose A, Grandoch M, vom Dorp F, Rübben H,
Rosenkranz A, Fischer JW and Weber AA: Stimulatory effects of the
multikinase inhibitor sorafenib on human bladder cancer cells. Br J
Pharmacol. 160:1690–1698. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gedaly R, Angulo P, Hundley J, Daily MF,
Chen C and Evers BM: PKI-587 and sorafenib targeting PI3K/AKT/mTOR
and Ras/Raf/MAPK pathways synergistically inhibit HCC cell
proliferation. J Surg Res. 176:542–548. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fremin C and Meloche S: From basic
research to clinical development of MEK1/2 inhibitors for cancer
therapy. J Hematol Oncol. 3:Feb 11–2010. View Article : Google Scholar
|
|
17
|
Glassmann A, Reichmann K, Scheffler B,
Glas M, Veit N and Probstmeier R: Pharmacological targeting of the
constitutively activated MEK/MAPK-dependent signaling pathway in
glioma cells inhibits cell proliferation and migration. Int J
Oncol. 39:1567–1575. 2011.PubMed/NCBI
|
|
18
|
Mendoza MC, Er EE and Blenis J: The
Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends
Biochem Sci. 36:320–328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kandil E, Tsumagari K, Ma J, et al:
Synergistic inhibition of thyroid cancer by suppressing
MAPK/PI3K/AKT pathways. J Surg Res. 184:898–906. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hong SK, Jeong JH, Chan AM and Park JI:
AKT upregulates B-Raf Ser445 phosphorylation and ERK1/2 activation
in prostate cancer cells in response to androgen depletion. Exp
Cell Res. 319:1732–1743. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Niba ET, Nagaya H, Kanno T, et al:
Crosstalk between PI3 kinase/PDK1/Akt/Rac1 and Ras/Raf/MEK/ERK
pathways downstream PDGF receptor. Cell Physiol Biochem.
31:905–913. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang CC, Cirit M and Haugh JM:
PI3K-dependent cross-talk interactions converge with Ras as
quantifiable inputs integrated by Erk. Mol Syst Biol.
5:2462009.PubMed/NCBI
|
|
23
|
Meloche S and Pouysségur J: The ERK1/2
mitogen-activated protein kinase pathway as a master regulator of
the G1- to S-phase transition. Oncogene. 26:3227–3239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Whalen AM, Galasinski SC, Shapiro PS,
Nahreini TS and Ahn NG: Megakaryocytic differentiation induced by
constitutive activation of mitogen-activated protein kinase kinase.
Mol Cell Biol. 17:1947–1958. 1997.PubMed/NCBI
|
|
25
|
Chen H, Zhu G, Li Y, et al: Extracellular
signal-regulated kinase signaling pathway regulates breast cancer
cell migration by maintaining slug expression. Cancer Res.
69:9228–9235. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang C, Jacobson K and Schaller MD: MAP
kinases and cell migration. J Cell Sci. 117:4619–4628. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Knight ZA, Lin H and Shokat KM: Targeting
the cancer kinome through polypharmacology. Nat Rev Cancer.
10:130–137. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jin N, Jiang T, Rosen DM, Nelkin BD and
Ball DW: Dual inhibition of mitogen-activated protein kinase kinase
and mammalian target of rapamycin in differentiated and anaplastic
thyroid cancer. J Clin Endocrinol Metab. 94:4107–4112. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu D and Xing M: Potent inhibition of
thyroid cancer cells by the MEK inhibitor PD0325901 and its
potentiation by suppression of the PI3K and NF-kappaB pathways.
Thyroid. 18:853–864. 2008. View Article : Google Scholar : PubMed/NCBI
|