Introduction
Epithelial ovarian cancer is the most deadly of the
gynecologic malignancies and the fifth leading cause of
cancer-related death among women (1). Although there has been an improvement
in the 5-year survival of patients diagnosed with advanced disease,
the long-term survival rate remains poor at 30% (1). Low survival can be attributed to the
insidious nature of ovarian cancer progression, resulting in late
diagnosis. Unfortunately, 75% of cases involve metastases to the
abdominal cavity (FIGO stages III–IV) at the time of diagnosis
(2). An additional complication
contributing to low survival is the high rate of chemoresistance
(1). The ability to predict the
patients at highest risk for rapid disease progression would allow
clinicians to optimize therapy up front using more aggressive
regimens.
The Cancer Genome Atlas (TCGA) has provided key
insight into molecular alterations that are common in ovarian
tumors (3). Of note, mutations in
a single gene, TP53, were identified in 96% of all serous
ovarian tumors (3). TP53
encodes the tumor suppressor protein p53, which acts as the major
control center in the cellular response to various stress such as
DNA-damaging chemotherapy. Once activated in response to
chemotherapy, p53 enhances cell cycle arrest and DNA damage repair,
or induces apoptosis and senescence if cellular repair is not
possible.
Although almost all serous ovarian cancer patients
harbor mutations in TP53, the mutations are extremely
heterogeneous and occur at almost every codon in the DNA-binding
domain of the gene (4). However,
the specific TP53 mutation can drastically alter the
function of the mutated protein in a myriad of different ways. For
example, studies using biochemical assays, cell models, as well as
mouse and rat models have demonstrated that some TP53
mutations abolish the wild-type (WT) function of p53 as well as
confer new oncogenic activities (5). We have termed these types of
mutations oncomorphic TP53 mutations (6). Studies in cultured cancer cell lines
and animal models of cancer demonstrate that oncomorphic
TP53 mutations can contribute to chemoresistance and cancer
progression. However, the phenomenon has not yet been convincingly
demonstrated in patients, partly due to the lack of a study
population size with sufficient power to observe significant
associations (7). This type of
analysis is now achievable through the TCGA with the availability
of clinical and genetic data from hundreds of ovarian cancer
patients. Using these data, as well as findings from patients at
the University of Iowa, we sought to test our hypothesis that
oncomorphic TP53 mutations in advanced serous ovarian tumors
are associated with worse outcomes.
Using stringent criteria to define oncomorphic
TP53 mutations, we evaluated the relationship of oncomorphic
p53 expression with progression-free survival (PFS), risk of
recurrence, and response to standard platinum and taxane
chemotherapy. Our data provide the first evidence that ovarian
cancer patients with oncomorphic TP53 mutations have worse
clinical outcomes compared to patients with unclassified
TP53 mutations, including a shorter PFS and a 60% greater
risk of recurrence. These findings have important potential
implications for all cancers characterized by mutations in
TP53.
Materials and methods
Ovarian cancer cell cultures
Eleven ovarian cancer cell lines were utilized in
these studies. ES-2, and SKOV3 cells were cultured as monolayers in
McCoy’s 5A medium. Caov3 cells were maintained in Dulbecco’s
Modified Eagle’s Medium (DMEM). Ovcar3 and UCI-107 cells were
cultured in RPMI-1640 medium. Caov4 and SW626 cells were maintained
in Leibovitz’s L-15 medium. TOV112D and OV-90 cells were cultured
in a 1:1 mixture of MCDB 105 medium containing 1.5 g/l of sodium
bicarbonate and medium 199 containing 2.2 g/l sodium bicarbonate.
UWB1.289 cells were grown in a 1:1 mixture of RPMI-1640 and Mammary
Epithelial Growth Medium (MEGM) (Clonetics/Lonza). All media
conditions were supplemented with 10% fetal bovine serum (FBS) and
1 U/ml penicillin and 10 μg/ml streptomycin and cells were
maintained in a humidified incubator with 5% CO2 at
37°C. All cell lines are available from American Type Cell Culture,
except UCI-107 cells that were generously gifted from Dr Michael J.
Goodheart.
The cell line SKOV3 has a loss of function (LOF)
TP53 mutation that results in a lack of p53 protein
expression. This cell line was used as a model to study the effects
of the most common oncomorphic TP53 mutations by stably
expressing the following mutants in TP53: R175H, R248Q,
R248Q.P72R, R248W, R273C, R273L, R273S, and Y220C as previously
described (8).
Western blot analysis
Analysis of protein expression/phosphorylation was
performed as previously described (9) for the following proteins: p53
(sc-126; Santa Cruz Biotechnology, Inc.), p21 (no. 2947), ERCC1
(no. 12345), c-Myc (no. 9402), β-catenin (no. 9582), mammalian
target of rapamycin (mTOR) (no. 2983) (all from Cell Signaling
Technology, Inc.), p-Rb S807 (no. ab47762; Abcam), and β-actin (no.
A1978; Sigma).
Clonogenic survival
Cells were trypsinized and plated in triplicate into
60 mm tissue culture dishes at 800 cells/well. Twenty-four hours
later, cells were treated with 1 μM cisplatin or 5 nM taxol for 48
h. Fresh media was added and cells were allowed to grow for 21
days. Viable clones were visualized by staining with crystal
violet, and colonies >50 cells were counted. Plating efficiency
was calculated by dividing the average number of colonies per plate
by the number of cells plated. Surviving fractions were calculated
by normalization to the plating efficiency.
Subjects
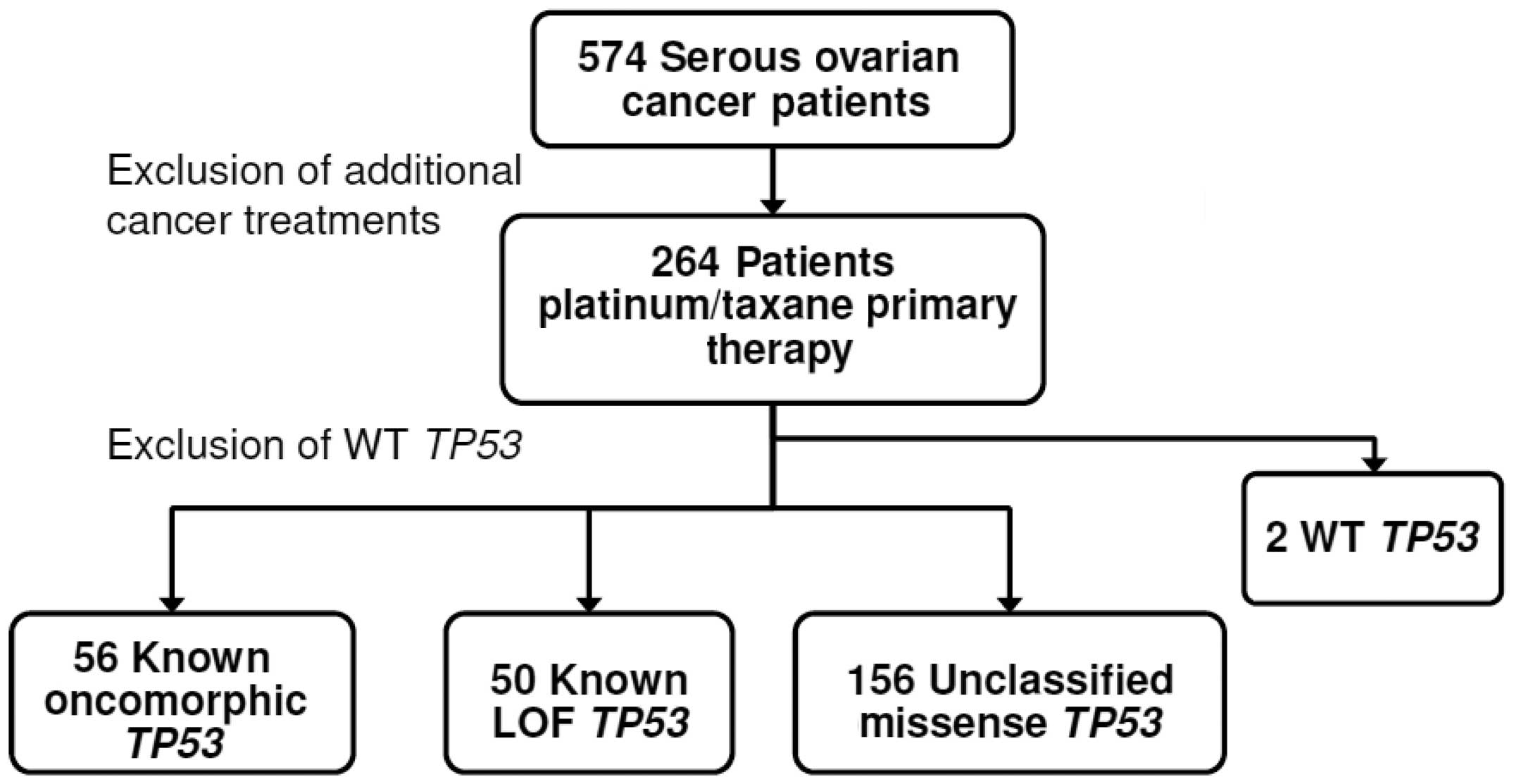
Clinical, genetic, and protein expression data from
264 advanced serous ovarian cancer patients without a previous
cancer history were downloaded from the TCGA data portal (accessed
05/06/2013). Analyses were limited to data from those patients who
received platinum (carboplatin, cisplatin, or oxaliplatin)- and
taxane (Taxotere or Paclitaxel)-based chemotherapy (Fig. 1). Clinical characteristics of the
study cohort are listed in Table
I. An independent validation patient cohort (n=32) was obtained
from the University of Iowa Gynecologic Oncology Tumor Bank. The
University of Iowa Institutional Review Board approved these
studies. The same inclusion criteria were used for both patient
cohorts: patients were of advanced stage (III or IV), specific
TP53 sequencing information was available, and clinical
outcome was known.
 | Table IClinical and pathological
characteristics of TCGA serous ovarian tumors from patients treated
with standard platinum- and taxane-based chemotherapy. |
Table I
Clinical and pathological
characteristics of TCGA serous ovarian tumors from patients treated
with standard platinum- and taxane-based chemotherapy.
| Characteristic | n | % |
|---|
| Age at
diagnosis | | |
| <60 years | 148 | 56.06 |
| ≥60 years | 116 | 43.94 |
| Vital Status | | |
| Dead | 126 | 47.73 |
| Alive | 138 | 52.27 |
| Tumor grade | | |
| G2 | 21 | 7.95 |
| G3/G4 | 236 | 89.39 |
| Unknown | 7 | 2.65 |
| FIGO stage | | |
| IIIA/B | 21 | 7.95 |
| IIIC | 197 | 74.62 |
| IV | 46 | 17.42 |
| Lymph invasion | | |
| No | 38 | 14.39 |
| Yes | 63 | 23.86 |
| Unknown | 163 | 61.74 |
| Residual
disease | | |
| ≤1 cm | 126 | 47.73 |
| >1 cm | 60 | 22.73 |
| Complete
removal | 51 | 19.32 |
| Unknown | 27 | 10.23 |
| Clinical response
to chemotherapy | | |
| Complete
response | 155 | 58.71 |
| Partial
response | 24 | 9.09 |
| Stable
disease | 19 | 7.20 |
| Progressive
disease | 12 | 4.55 |
| No data | 54 | 20.45 |
| Platinum
status | | |
| Resistant | 49 | 20.25 |
| Sensitive | 112 | 46.28 |
| Too early | 34 | 14.05 |
| Unknown | 47 | 19.42 |
| p53 mutation
type | | |
| LOF | 51 | 19.32 |
| Oncomorphic | 56 | 21.21 |
| Unclassified | 154 | 58.33 |
| WT | 2 | 0.76 |
| Unknown (no
sequence information available) | 1 | 0.38 |
Criteria for designating TP53
mutations
TP53 mutations were binned into three
categories: oncomorphic, LOF, and unclassified. Oncomorphic
mutations were designated based on previously published studies
showing that a particular mutation causes an oncogenic phenotype.
For example, Hanel et al used a knock-in mouse to determine
the function of two common mutations (10). Compared with the p53 null mouse
(p53−/−), a mouse carrying a p53 R248Q allele
(p53R248Q/−) displayed accelerated tumor onset and
shortened survival, but a mouse model carrying a p53 G245S allele
(p53G245S/−) showed no differences in survival when
compared with the p53−/− mouse (10). These are some of the first data
indicating that TP53 mutations vary in function with respect
to tumorigenicity. Eight TP53 mutations were considered
oncomorphic, and were selected based on previous in vivo and
in vitro studies [P151S (11,12),
Y163C (13), R175H (14–16),
L194R (17), Y220C (18), R248Q (10), R248W (19,20),
R273C (21,22), R273H (15,19,23),
R273L (24), R282W (13)]. LOF mutations were defined as i)
point mutations that create a stop codon (nonsense mutation); or
ii) frame shift mutations that cause significant disruptions in the
translation of the protein. WT mutations were defined as mutations
that do not alter the amino acid sequence. The remaining mutations
were single nucleotide substitutions, the function of which is not
fully known at this time, but do not meet oncomorphic criteria.
These were categorized as ‘unclassified’ mutations. Splice
mutations located at the intron-exon borders were categorized into
the ‘unclassified’ category due to conflicting studies on their
function (25–28).
Defining clinical endpoints
Clinical details available from the TCGA portal were
used to document the following clinical endpoints: PFS and platinum
status. PFS was defined as the interval between the date of initial
surgical removal of the tumor to the date of progression in
patients who were not cancer free, or date of recurrence.
Chemotherapy details were available that documented the date of
last primary platinum treatment. Platinum-free interval was defined
as the interval between last primary platinum treatment to the date
of progression or recurrence. Platinum status was defined as
resistant if the platinum-free interval was <6 months when the
patient recurred. Platinum status was defined as sensitive if the
interval to recurrence was >6 months, or the follow-up period
for those lost to contact was >6 months from the date of the
last platinum treatment. Patients who did not progress or have a
recurrence were censored in both analyses at the date of the
last-known contact.
RPPA protein data
Corrected and normalized reverse phase protein array
(RPPA) data were downloaded from the TCGA portal to analyze protein
expression differences between patients with oncomorphic, LOF, or
unclassified mutations. Detailed information on normalization has
been previously reported (3);
briefly, the raw data were converted from a log 2 value into an
arbitrary linear value and corrected based on the normalization of
means among all patient samples.
Statistical analysis
To determine if different mutations confer worse
patient outcome, plots of the Kaplan-Meier estimated cumulative
probabilities of PFS were constructed. Cox proportional hazard
regression was utilized to test for differences in PFS between
mutation types using a study endpoint of 60 months, as previously
reported (4). To assess for group
differences between the mutations on relevant clinical variables, a
χ2 test or Fisher’s exact test was utilized where
appropriate. A Kruskal-Wallis or Wilcoxon rank sum test was
performed to detect differential protein expression between all
three mutation groups, or between two groups, respectively. All
tests were two sided and tested at the 5% significance level. The
data analysis was generated using SAS software, version 9.3 (SAS
Institute, Inc.).
Results
Selection of patient population
As shown in Fig. 1,
the primary exclusion criterion was patient exposure to treatment
beyond adjuvant primary chemotherapy with platinum and taxane. The
median PFS for the study population was 13.8 months, and median
overall survival was 30.2 months, which is consistent with reported
outcomes in the full TCGA ovarian cancer data set (3).
Frequency and spectrum of TP53
mutations
Exon sequencing data were downloaded from the TCGA
portal and mutations in TP53 were annotated. Two patients
had synonymous missense mutations that retained the integrity of WT
p53 protein sequence and were designated as WT. Data for these two
patients were excluded due to insufficient sample size (Fig. 1).
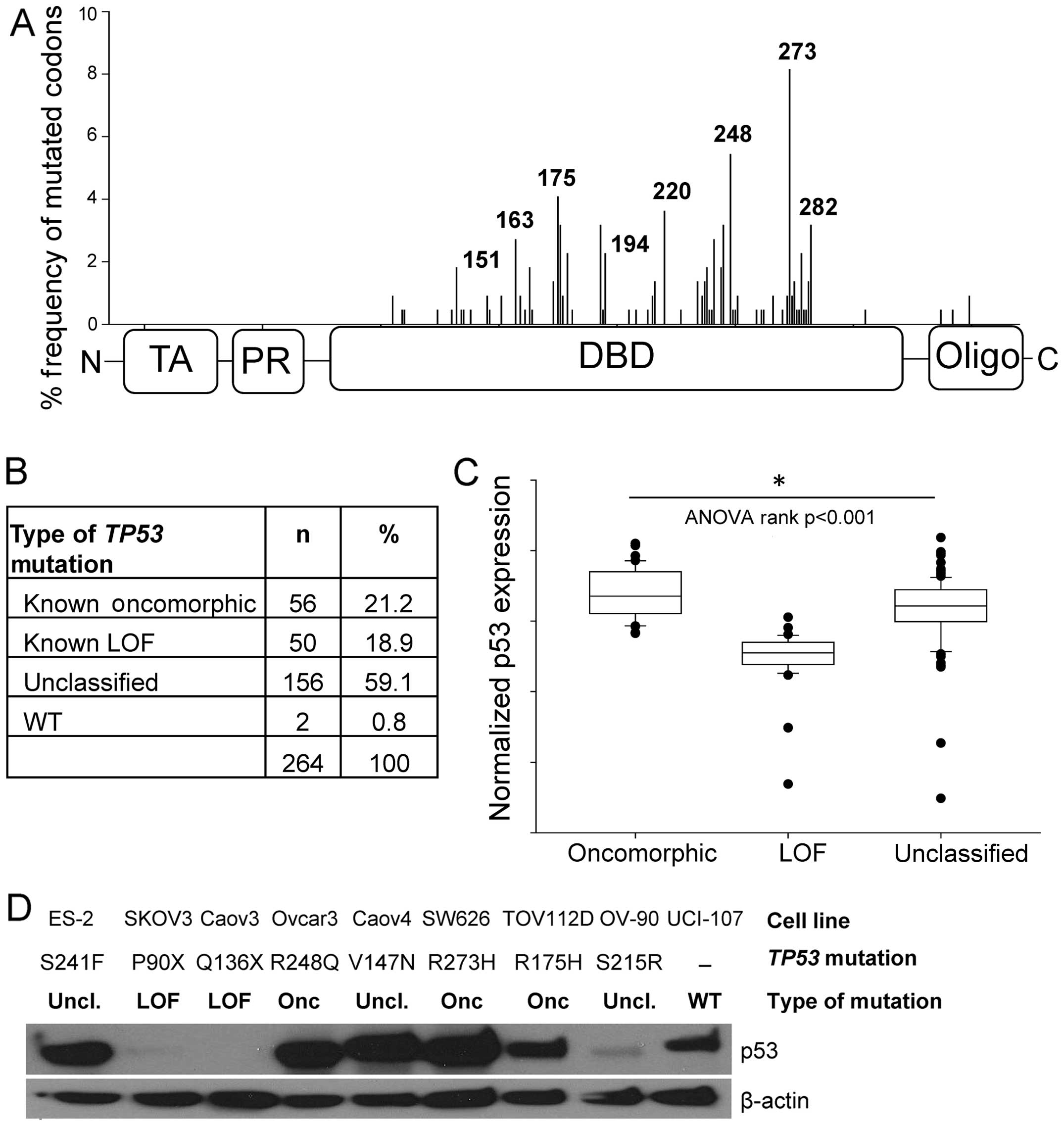
Mutations in TP53 occurred predominately in
the DNA-binding domain (Fig. 2A),
consistent with a previous report (4). The most common mutations occurred at
codons R273 (6.1%), R248 (4.6%), and R175 (3.4%). Oncomorphic
mutations comprised 21.2% of the patient population, LOF mutations
comprised 18.9%, and the remaining 59.1% were unclassified
mutations (Fig. 2B). Splice
mutations located at the intron-exon borders were categorized as
‘unclassified’ due to conflicting studies on their function
(25–28). Splice mutations occurred in 10% of
our study population, a frequency much larger than previously
reported (27). We speculate that
the advanced technology used to sequence TP53 exons is more
sensitive than used previously. The frequency of oncomorphic and
LOF mutations in this cohort is similar to that calculated from the
International Agency for Research on Cancer p53 database (4,6),
thus validating our study population.
To confirm our classification of oncomorphic and LOF
mutations, we analyzed normalized protein expression of p53 as
reported in the RPPA data set. LOF mutations result in loss of p53
protein expression, whereas oncomorphic p53 has been reported to be
hyper-stabilized (5). As expected,
we detected a significant difference in protein levels of p53 for
the oncomorphic, LOF and unclassified mutations (Fig. 2C, p<0.001). Specifically, tumors
containing oncomorphic TP53 mutations had the highest p53
protein levels, whereas tumors with LOF TP53 mutations
displayed the lowest expression of p53. Tumors with unclassified
mutations had a broad range of p53 protein expression.
We utilized a panel of nine ovarian cancer cell
lines with various TP53 mutations to characterize expression
levels of mutated p53 proteins (Fig.
2D). Three cell lines with oncomorphic TP53 mutations
displayed abundant mutated p53 protein expression. Two cell lines
with LOF TP53 mutations did not express p53 protein; and
cell lines with unclassified TP53 mutations demonstrated a
range of p53 protein expression. One cell line, UCI-107, expresses
WT TP53.
Oncomorphic mutations in TP53 confer
worse patient outcome
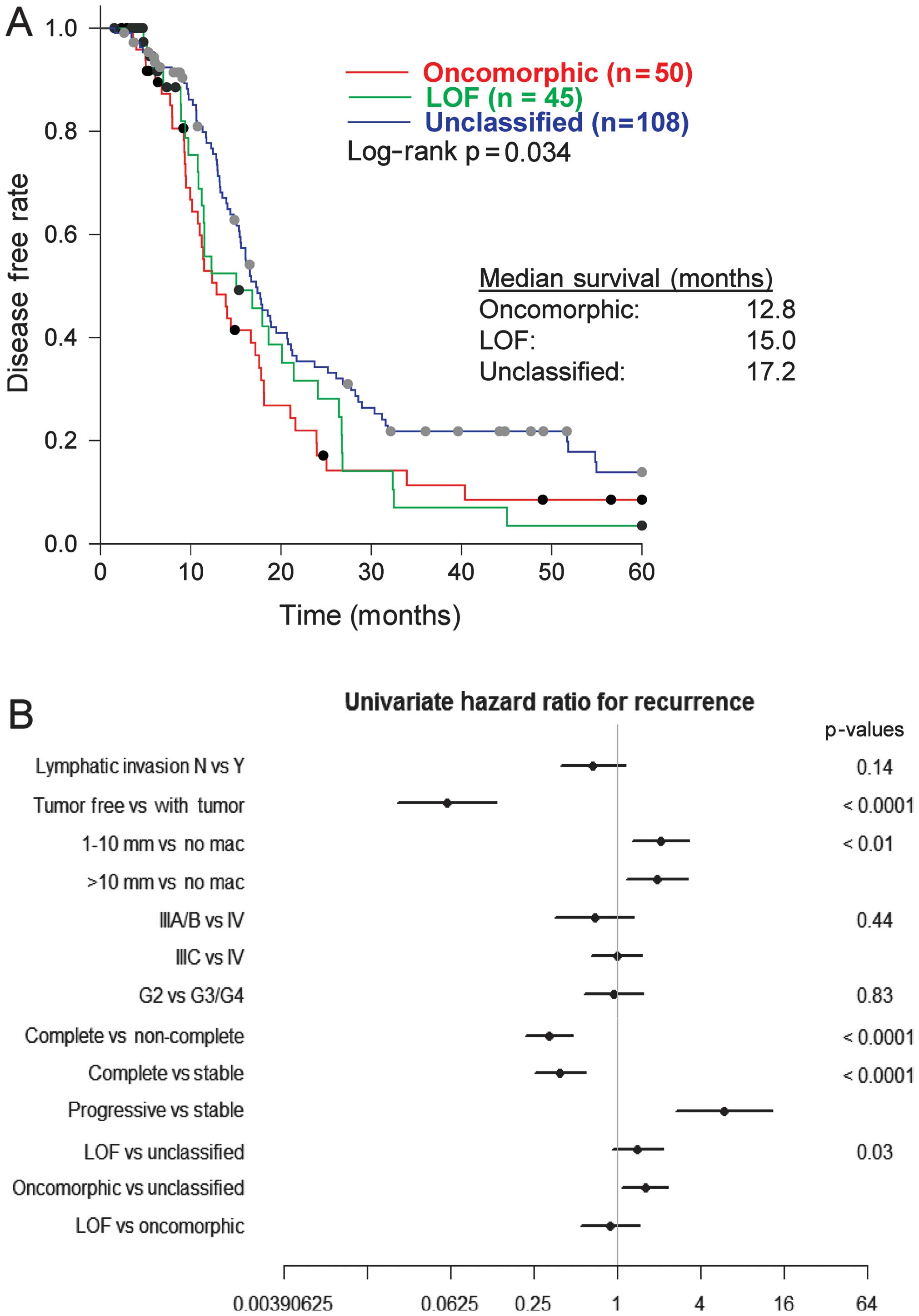
We assessed the association of oncomorphic
TP53 mutations with patient outcome, by first calculating
PFS among patients with oncomorphic, LOF, or unclassified mutations
and found a significant difference between categories (p=0.03).
Follow-up pairwise comparisons demonstrated that patients with
oncomorphic TP53 mutations showed significantly worse PFS
when compared with patients harboring unclassified mutations
(p=0.015) (Fig. 3A). The median
PFS was 12.8, 15.0, and 17.2 months for patients with oncomorphic,
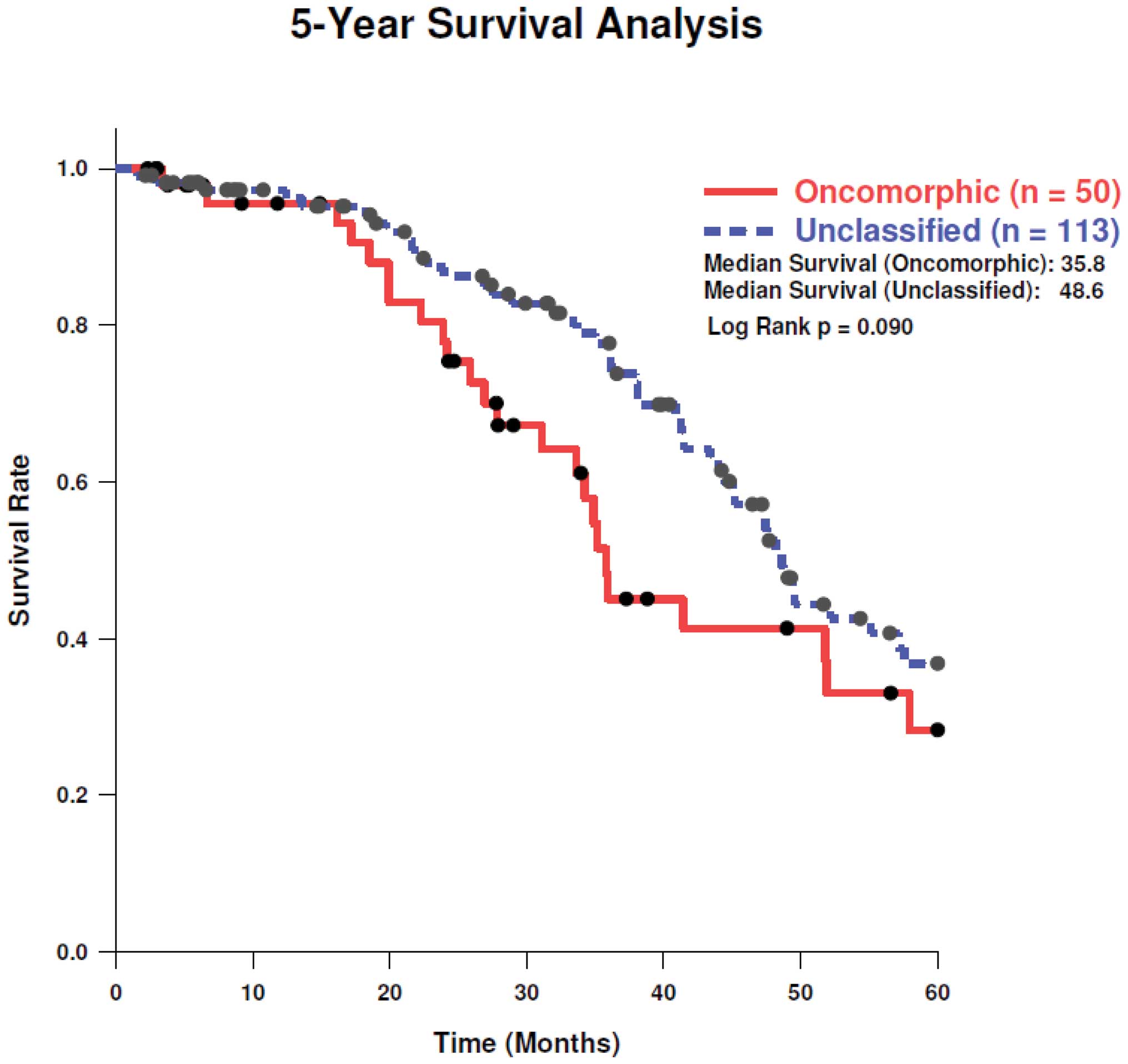
LOF, and unclassified mutations, respectively. Analysis of 5-year
survival revealed a trend towards better survival in patients with
unclassified mutations as compared to oncomorphic mutations
(Fig. 4, log-rank test
p=0.11).
To provide further insight into which clinical
factors may be contributing to the differing PFS outcomes between
mutational classifications, a univariate comparison of clinical
factors was conducted (Table II).
Patients with oncomorphic TP53 mutations displayed higher
rates of platinum resistance when compared with LOF and
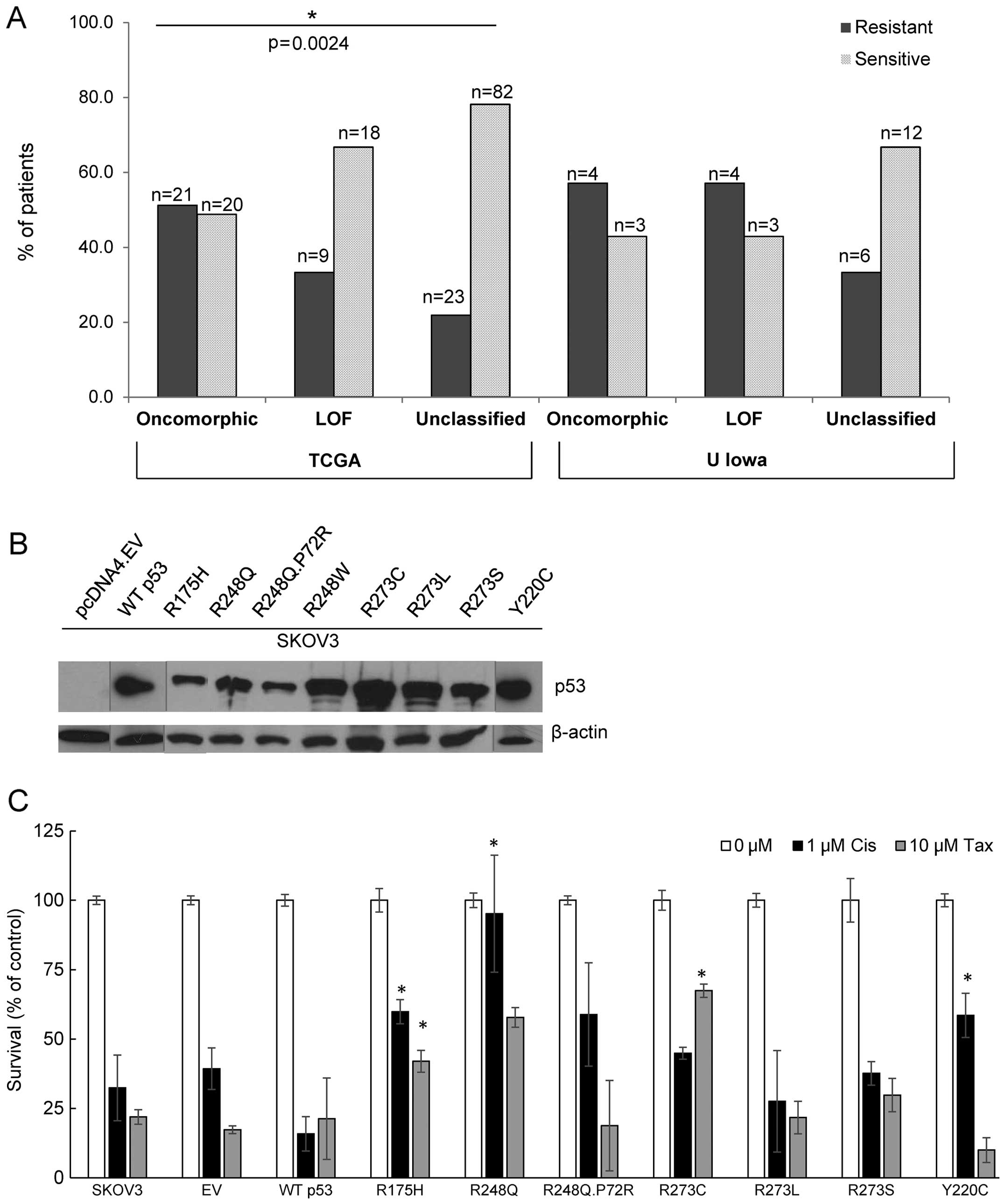
unclassified mutants (χ2 test p=0.0024). More than half
(51.2%) of patients with oncomorphic mutations displayed platinum
resistance, whereas patients with unclassified mutations had the
highest rates of platinum sensitivity (Table II). In addition, patients with
oncomorphic TP53 mutations had almost 60% higher odds of
recurrence (HR=1.60, 95% confidence intervals 1.09, 2.33, p=0.015)
when compared to patients with other unclassified mutations
(Fig. 3B). We also observed the
anticipated associations of recurrence with residual disease and
response to therapy (Fig. 3B).
| Table IIUnivariate analysis of association of
clinical factors with TP53 mutation categories (oncomorphic,
LOF, and unclassified) demonstrates that platinum status is
significantly different among the three mutation groups. |
Table II
Univariate analysis of association of
clinical factors with TP53 mutation categories (oncomorphic,
LOF, and unclassified) demonstrates that platinum status is
significantly different among the three mutation groups.
| Variable | Category | n | p-value
χ2 test |
|---|
|
|---|
| Oncomorphic | LOF | Unclassified |
|---|
| Lymphatic
invasion | No | 13 | 6 | 19 | 0.0767 |
| Yes | 9 | 13 | 39 | |
| Tumor grade | G2 | 4 | 5 | 12 | 0.8373 |
| G3/G4 | 50 | 43 | 138 | |
| Cancer status | Tumor free | 15 | 12 | 45 | 0.8439 |
| With tumor | 37 | 33 | 100 | |
| Residual tumor | ≤1 cm | 28 | 26 | 70 | 0.5075 |
| >1 cm | 11 | 13 | 33 | |
| No mac | 15 | 6 | 30 | |
| Tumor stage | IIIA/B | 6 | 4 | 11 | 0.8529 |
| IIIC | 40 | 36 | 117 | |
| IV | 10 | 10 | 25 | |
| Vital status | Dead | 28 | 22 | 73 | 0.8234 |
| Alive | 28 | 28 | 80 | |
| Platinum
status | Resistant | 21 | 9 | 23 | 0.0024 |
| Sensitive | 20 | 18 | 82 | |
| Therapy
outcome | Complete
response | 33 | 29 | 76 | 0.0970 |
| Progressive
disease | 4 | 2 | 4 | |
| Stable disease | 4 | 14 | 20 | |
To validate the clinical and genetic data obtained
from the TCGA, we determined rates of chemoresistance in patients
who were diagnosed with ovarian cancer and had banked tumors at the
University of Iowa. Sequencing information on TP53 was
available for all tumors. We observed a similar trend towards
resistance in tumors with oncomorphic TP53 (Fig. 5A). In addition, patients with
unclassified TP53 mutations demonstrated the highest
sensitivity to chemotherapy. A p53 null cell line (SKOV3) was
utilized to express the most common TP53 oncomorphic
mutations (Fig. 5B). Clonogenic
survival in response to cisplatin treatment was enhanced by cells
expressing R175H, R248Q, and Y220C oncomorphic p53 mutant proteins.
In response to taxol chemotherapy, clonogenic survival was enhanced
in cells expressing the R175H and R273C p53 mutated proteins
(Fig. 5C).
Protein expression differences between
oncomorphic mutations and unclassified mutations
We next interrogated possible mechanisms of
chemoresistance in tumors containing oncomorphic mutations by
comparing protein expression profiles between oncomorphic and
unclassified mutations. Data, which are part of TCGA dataset, were
obtained by RPPA, a high-throughput technique for simultaneous
measurement of protein expression in a large number of biological
samples using antibody-based methods. We observed differential
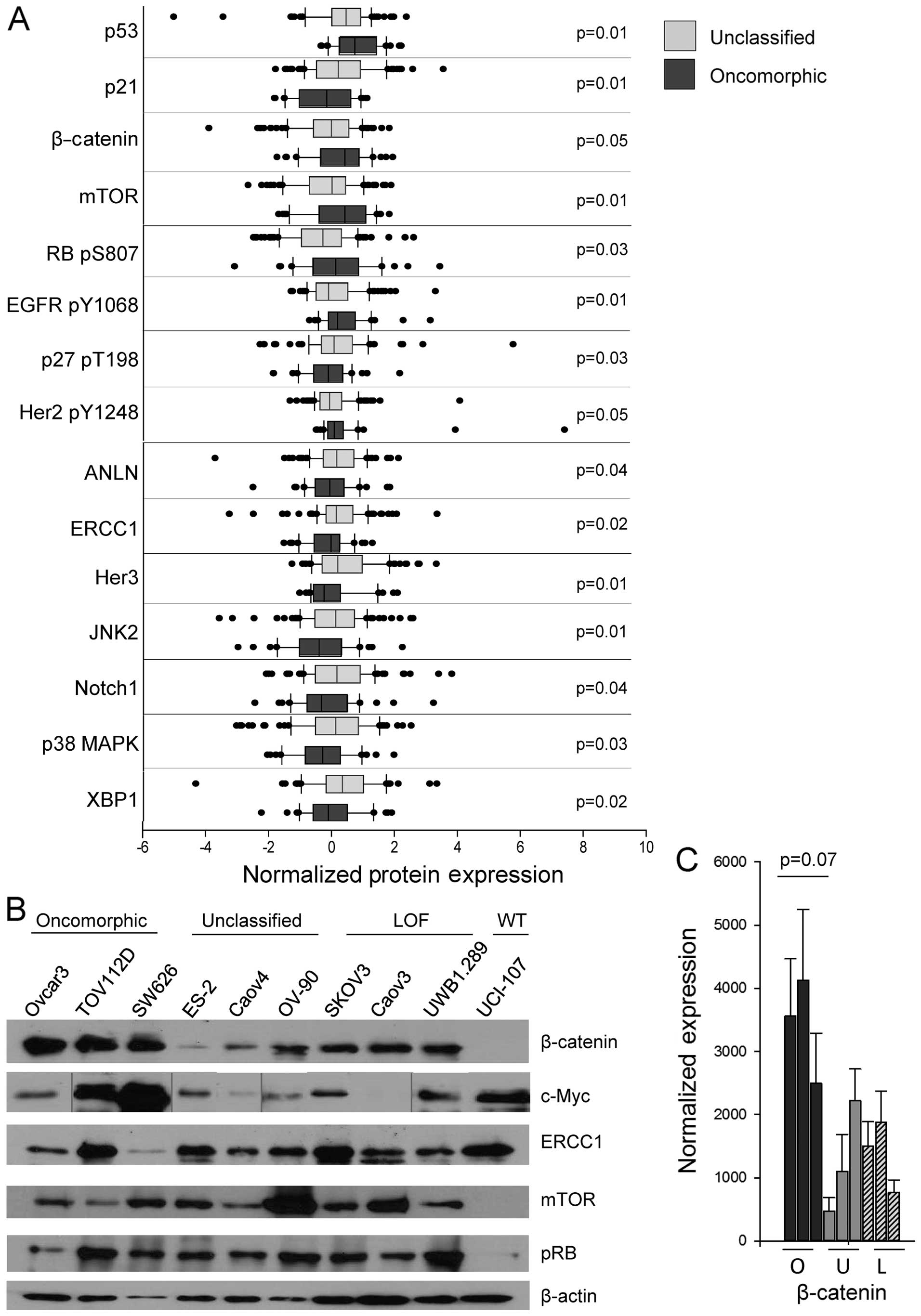
expression of 15 different proteins in tumors with either
oncomorphic or unclassified TP53 mutations (Fig. 6A, Table III). In particular, the
pro-apoptotic protein BAK and the cell cycle regulator p21
(CIP1/WAF1) were expressed at a significantly lower level in tumors
with oncomorphic TP53 mutations. β-catenin, phosphorylated
epidermal growth factor receptor (EGFR) (Y1068), and mTOR were
significantly elevated in patients with oncomorphic TP53
mutations compared with patients with unclassified mutations.
Further evaluation of the RPPA data from the three mutational
categories (oncomorphic, LOF and unclassified) revealed differences
in tumor protein expression (Table
IV). To further define the most significantly altered pathways
in cells with oncomorphic TP53, we assessed the expression
of the targets identified from the TCGA dataset in representative
cell lines (Fig. 6B). The most
highly correlated alterations were in the β-catenin pathway, known
to be associated with ovarian cancer carcinogenesis and
proliferation (29) (Fig. 6C).
| Table IIISignificant differential protein
expression between tumors with oncomorphic versus unclassified
TP53 mutations as determined by Wilcoxon rank sum test.
Median protein expression is presented. |
Table III
Significant differential protein
expression between tumors with oncomorphic versus unclassified
TP53 mutations as determined by Wilcoxon rank sum test.
Median protein expression is presented.
| Analysis
variable | Median
expression | p-value Wilcoxon
test (two sided) |
|---|
|
|---|
| Oncomorphic | Unclassified |
|---|
| ANLN | −0.08 | 0.14 | 0.042568565 |
| β-catenin | 0.40 | −0.03 | 0.047657921 |
| EGFR pY1068 | 0.16 | −0.12 | 0.007949216 |
| ERCC1 | −0.04 | 0.12 | 0.015114876 |
| Her2 pY1248 | 0.06 | −0.08 | 0.049131573 |
| Her3 | −0.26 | 0.17 | 0.010622594 |
| JNK2 | −0.42 | 0.10 | 0.00516184 |
| mTOR | 0.40 | −0.01 | 0.008801626 |
| Notch1 | −0.35 | 0.14 | 0.040835561 |
| p21 | −0.18 | 0.19 | 0.007848037 |
| p27 pT198 | −0.13 | 0.05 | 0.034859604 |
| p38 MAPK | −0.31 | 0.10 | 0.025973549 |
| p53 | 0.71 | 0.43 | 0.008155086 |
| RB pS807 S811 | 0.11 | −0.30 | 0.028065342 |
| XBP1 | −0.13 | 0.32 | 0.020961678 |
| Table IVSignificant differential protein
expression among tumors with oncomorphic, LOF and unclassified
TP53 mutations as determined by Kruskal-Wallis test. Median
protein expression is presented. |
Table IV
Significant differential protein
expression among tumors with oncomorphic, LOF and unclassified
TP53 mutations as determined by Kruskal-Wallis test. Median
protein expression is presented.
| Analysis
variable | Median
expression | p-value
Kruskal-Wallis test |
|---|
|
|---|
| Oncomorphic | LOF | Unclassified |
|---|
| ANLN | −0.08 | −0.09 | 0.14 | 0.044578193 |
| BAX | −0.11 | 0.12 | −0.13 | 0.02924444 |
| Beclin | 0.01 | −0.24 | 0.12 | 0.047399782 |
| CD31 | 0.15 | −0.23 | 0.12 | 0.019310324 |
| CMET pY1235 | −0.16 | −0.26 | 0.09 | 0.040573763 |
| EGFR pY1068 | 0.16 | −0.21 | −0.12 | 0.025684237 |
| Her3 | −0.26 | −0.23 | 0.17 | 0.003727584 |
| JNK2 | −0.42 | 0.26 | 0.10 | 0.002990478 |
| mTOR | 0.40 | 0.07 | −0.01 | 0.030649059 |
| p21 | −0.18 | −0.20 | 0.19 | 0.027093184 |
| p53 | 0.71 | −0.90 | 0.43 | 4.47E-16 |
| PCNA | 0.01 | 0.73 | −0.13 | 0.009614359 |
| RBM3 | −0.09 | 0.60 | 0.08 | 0.018983458 |
| RB pS807 S811 | 0.12 | 0.11 | −0.30 | 0.022229704 |
| XBP1 | −0.13 | −0.18 | 0.32 | 0.000881485 |
Discussion
Recent advances in cancer biology involve
understanding the effects of mutations in TP53 on the
function of the mutant protein (5). Many clinical studies have attempted
to correlate the presence of a TP53 mutation with patient
survival or the development of chemoresistance (7). The results of these studies are
conflicting primarily because of indiscriminate grouping of
TP53 mutations with different functions (oncomorphic, LOF
and unclassified). Given that 21% of all ovarian cancer patients
harbor oncomorphic TP53 mutations, studies which take into
account the functional implications of these mutations are vital.
The availability of a large cohort of ovarian cancer tumors and
corresponding clinical data through TCGA has made it possible to
address the clinical consequence of oncomorphic mutations in
TP53 for the first time and to confirm the mechanistic
implications of oncomorphic p53 expression in representative cell
models. Thus, the objective of our study was to determine if
oncomorphic TP53 mutations are associated with worse patient
outcomes. We demonstrate that oncomorphic TP53 mutations
predict for worse PFS and higher rates of chemoresistance and
recurrence. Preclinical models confirm the oncomorphic function of
the identified TP53 mutations and suggest mechanisms by
which oncomorphic TP53 drive ovarian cancer cell growth.
Although sequence similarities exist among many p53
mutant proteins, to date only stringent biological, in vivo
assays can determine oncomorphic properties (6). Accordingly, a previous study using
less stringent criteria to define ‘gain of function’ TP53
mutations did not find a significant relationship between the gain
of function mutations and chemoresistance (30). Herein we used more stringent
criteria to define oncomorphic mutations and propose that our
findings more clearly delineate the impact of these oncogenic
proteins. Our criteria required that mutations increase clonogenic
potential in vitro or increase tumorigenesis in vivo
as compared to TP53-null mice to be considered oncomorphic
(10–24,31,32).
Using these criteria, we found that the presence of a TP53
oncomorphic mutation in a patient tumor specimen predicts for
platinum resistance.
To understand the oncogenic properties of
oncomorphic p53 proteins, we analyzed differential protein
expression between the TP53 mutation groups. The cell cycle
regulator p21, which is induced by p53 and results in cell cycle
arrest, was expressed at a low level in tumors containing
oncomorphic TP53 mutations. Levels of phosphorylated p27
were also lower in these samples. Conversely, tumors with
unclassified TP53 mutations displayed higher p21 expression,
suggesting that some of the unclassified mutations may have
residual WT p53 functions. Previous studies have demonstrated that
positive p21 staining in ovarian tumor specimens correlates with an
overall survival advantage (33,34).
Our data also indicated that tumors with oncomorphic TP53
have increased expression of activated pro-growth pathways, such as
phosphorylation of EGFR, Her2, and retinoblastoma protein (Rb).
EGFR phosphorylation at Y1068 is a hallmark for activated EGFR
signaling and is the site of Grb2 and Ras binding that perpetuate
Ras activation and mitogen-activated protein kinase signaling
(35). The proteins mTOR and
β-catenin, which are commonly overexpressed in cancer, were also
significantly increased in oncomorphic TP53 tumors,
indicating enhanced pro-survival signaling, however. Recently, high
β-catenin was associated with poor ovarian cancer patient outcome
(29). This protein was the most
highly altered in our panel of ovarian cancer cell lines as well as
in patient tumors. These data correlate well with in vitro
studies showing that EGFR is a direct transcriptional target of
oncomorphic p53 proteins (36). In
addition, others have shown that oncomorphic p53 regulates
expression of key cell cycle regulators (37). Understanding the molecular
consequences of oncomorphic TP53 mutations has the potential
to identify key signaling targets that could be blocked in order to
overcome chemoresistance in tumors with these oncogenic
mutations.
Patients whose tumors expressed unclassified
TP53 mutations made up the majority of the ovarian cancer
study population. These patients represent an interesting clinical
population since our data demonstrate that patients harboring
unclassified mutations are significantly more sensitive to
chemotherapy and have lower rates of recurrence. Tumors with
unclassified TP53 mutations express the mutated p53 protein
at a fairly high level, and it is possible that these proteins have
some residual WT p53 function as evidenced by higher expression of
p21. The overall survival of patients with unclassified mutations
trended towards improved 5-year survival as compared to oncomorphic
mutations. Note, however, that overall survival data are not mature
for some patients in TCGA dataset; thus, overall survival should be
re-examined when these data are complete.
Although two patients with WT TP53 were
excluded from our study, and these patients are rare in advanced
ovarian cancers, a recent study of 11 ovarian tumors with WT p53
reported a worse overall survival and PFS as compared to a mutated
TP53 (38). The study by
Wong et al represents a step towards understanding how p53
function affects outcomes, but it remains unclear why the tumors
with functional p53 fail to respond to standard DNA-damaging
chemotherapy (38). One
possibility is that other mutations present in the tumors drive
drug resistance; another possibility is that WT p53 enforces cell
cycle checkpoints, making the cells less vulnerable to
chemotherapeutic agents which act specifically in mitosis (9).
An important aspect of p53 biology is the integrity
of the second TP53 allele. Mutant p53 proteins can exert
dominant negative activity by inhibiting DNA binding and hence, the
tumor-suppressive function of the remaining WT TP53 allele
(39). The status of both alleles
is necessary to have a complete understanding of the effect of a
particular mutation; however this is a limitation of the TCGA data.
The use of exon sequencing did not distinguish between loss of
heterozygosity (LOH) or tumor heterogeneity (3). Future studies will need to take this
into account.
In conclusion, almost all advanced serous ovarian
tumors contain TP53 mutations. Understanding the p53
mutational category, which significantly impacts function, is
critical to predicting patient outcomes. Specifically, we
demonstrate that patients with oncomorphic TP53 mutations
are significantly more resistant to chemotherapy, have shorter PFS
and a higher risk of recurrence. A recent study in Li-Fraumeni
syndrome patients analyzed the individual impact of common
TP53 missense mutations and identified a particular mutation
(R282W) that results in earlier onset of tumor formation (40). Such patients, and patients
identified in our study with oncomorphic TP53 mutations
deserve careful follow-up post-therapy and may require novel
treatment regimens to improve outcomes. In addition, when studying
the impact of new therapies in ovarian cancer, we propose that
stratification should be considered based upon p53 mutational
category.
Acknowledgements
We are grateful for the continued services provided
by the Genomics Division of the Iowa Institute for Human Genetics.
This study was supported by NIH R01CA99908 (K.K.L.) and the
Department of Obstetrics and Gynecology Research Development Fund
(K.K.L.). The agencies had no involvement in study design,
collection, analysis and interpretation of data, writing of the
report, or the decision to submit the report for publication. D.D.
and K.W.T. are owners of Immortagen, L.L.C.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cannistra SA: Cancer of the ovary. N Engl
J Med. 351:2519–2529. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cancer Genome Atlas Research Network.
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Petitjean A, Mathe E, Kato S, Ishioka C,
Tavtigian SV, Hainaut P and Olivier M: Impact of mutant p53
functional properties on TP53 mutation patterns and tumor
phenotype: lessons from recent developments in the IARC TP53
database. Hum Mutat. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Freed-Pastor WA and Prives C: Mutant p53:
one name, many proteins. Genes Dev. 26:1268–1286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brachova P, Thiel KW and Leslie KK: The
consequence of oncomorphic TP53 mutations in ovarian cancer. Int J
Mol Sci. 14:19257–19275. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hall J, Paul J and Brown R: Critical
evaluation of p53 as a prognostic marker in ovarian cancer. Expert
Rev Mol Med. 6:1–20. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Meng X, Dizon DS, Yang S, et al:
Strategies for molecularly enhanced chemotherapy to achieve
synthetic lethality in endometrial tumors with mutant p53. Obstet
Gynecol Int. 2013:8281652013. View Article : Google Scholar
|
|
9
|
Meng X, Laidler LL, Kosmacek EA, et al:
Induction of mitotic cell death by overriding G2/M checkpoint in
endometrial cancer cells with non-functional p53. Gynecol Oncol.
128:461–469. 2013. View Article : Google Scholar :
|
|
10
|
Hanel W, Marchenko N, Xu S, Xiaofeng Yu S,
Weng W and Moll U: Two hot spot mutant p53 mouse models display
differential gain of function in tumorigenesis. Cell Death Differ.
20:898–909. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gaiddon C, Lokshin M, Ahn J, Zhang T and
Prives C: A subset of tumor-derived mutant forms of p53
down-regulate p63 and p73 through a direct interaction with the p53
core domain. Mol Cell Biol. 21:1874–1887. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xie TX, Zhou G, Zhao M, et al: Serine
substitution of proline at codon 151 of TP53 confers gain of
function activity leading to anoikis resistance and tumor
progression of head and neck cancer cells. Laryngoscope.
123:1416–1423. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Scian MJ, Stagliano KE, Deb D, et al:
Tumor-derived p53 mutants induce oncogenesis by transactivating
growth-promoting genes. Oncogene. 23:4430–4443. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lang GA, Iwakuma T, Suh YA, et al: Gain of
function of a p53 hot spot mutation in a mouse model of Li-Fraumeni
syndrome. Cell. 119:861–872. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Olive KP, Tuveson DA, Ruhe ZC, et al:
Mutant p53 gain of function in two mouse models of Li-Fraumeni
syndrome. Cell. 119:847–860. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang YX and Kotlikoff MI: Inactivation of
calcium-activated chloride channels in smooth muscle by
calcium/calmodulin-dependent protein kinase. Proc Natl Acad Sci
USA. 94:14918–14923. 1997. View Article : Google Scholar
|
|
17
|
Ko JL, Chiao MC, Chang SL, et al: A novel
p53 mutant retained functional activity in lung carcinomas. DNA
Repair (Amst). 1:755–762. 2002. View Article : Google Scholar
|
|
18
|
Sproston AR, Boyle JM, Heighway J, Birch
JM and Scott D: Fibroblasts from Li-Fraumeni patients are resistant
to low dose-rate irradiation. Int J Radiat Biol. 70:145–150. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Song H, Hollstein M and Xu Y: p53
gain-of-function cancer mutants induce genetic instability by
inactivating ATM. Nat Cell Biol. 9:573–580. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Krepulat F, Löhler J, Heinlein C,
Hermannstädter A, Tolstonog GV and Deppert W: Epigenetic mechanisms
affect mutant p53 transgene expression in WAP-mutp53 transgenic
mice. Oncogene. 24:4645–4659. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bergamaschi D, Gasco M, Hiller L, et al:
p53 polymorphism influences response in cancer chemotherapy via
modulation of p73-dependent apoptosis. Cancer cell. 3:387–402.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Irwin MS, Kondo K, Marin MC, Cheng LS,
Hahn WC and Kaelin WG Jr: Chemosensitivity linked to p73 function.
Cancer cell. 3:403–410. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Duan W, Ding H, Subler MA, et al:
Lung-specific expression of human mutant p53-273H is associated
with a high frequency of lung adenocarcinoma in transgenic mice.
Oncogene. 21:7831–7838. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Morselli E, Tasdemir E, Maiuri MC, et al:
Mutant p53 protein localized in the cytoplasm inhibits autophagy.
Cell cycle. 7:3056–3061. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bourdon JC, Fernandes K, Murray-Zmijewski
F, et al: p53 isoforms can regulate p53 transcriptional activity.
Genes Dev. 19:2122–2137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hofstetter G, Berger A, Fiegl H, et al:
Alternative splicing of p53 and p73: the novel p53 splice variant
p53delta is an independent prognostic marker in ovarian cancer.
Oncogene. 29:1997–2004. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Holmila R, Fouquet C, Cadranel J, Zalcman
G and Soussi T: Splice mutations in the p53 gene: case report and
review of the literature. Hum Mutat. 21:101–102. 2003. View Article : Google Scholar
|
|
28
|
Sameshima Y, Akiyama T, Mori N, et al:
Point mutation of the p53 gene resulting in splicing inhibition in
small cell lung carcinoma. Biochem Biophys Res Commun. 173:697–703.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bodnar L, Stanczak A, Cierniak S, et al:
Wnt/β-catenin pathway as a potential prognostic and predictive
marker in patients with advanced ovarian cancer. J Ovarian Res.
7:162014. View Article : Google Scholar
|
|
30
|
Kang HJ, Chun SM, Kim KR, Sohn I and Sung
CO: Clinical relevance of gain-of-function mutations of p53 in
high-grade serous ovarian carcinoma. PloS One. 8:e726092013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Monti P, Campomenosi P, Ciribilli Y, et
al: Characterization of the p53 mutants ability to inhibit p73 beta
transactivation using a yeast-based functional assay. Oncogene.
22:5252–5260. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu G, McDonnell TJ, Montes de Oca Luna R,
et al: High metastatic potential in mice inheriting a targeted p53
missense mutation. Proc Natl Acad Sci USA. 97:4174–4179. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rose SL, Goodheart MJ, DeYoung BR, Smith
BJ and Buller RE: p21 expression predicts outcome in p53-null
ovarian carcinoma. Clin Cancer Res. 9:1028–1032. 2003.PubMed/NCBI
|
|
34
|
Schmider A, Gee C, Friedmann W, et al: p21
(WAF1/CIP1) protein expression is associated with prolonged
survival but not with p53 expression in epithelial ovarian
carcinoma. Gynecol Oncol. 77:237–242. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rojas M, Yao S and Lin YZ: Controlling
epidermal growth factor (EGF)-stimulated Ras activation in intact
cells by a cell-permeable peptide mimicking phosphorylated EGF
receptor. J Biol Chem. 271:27456–27461. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ludes-Meyers JH, Subler MA, Shivakumar CV,
et al: Transcriptional activation of the human epidermal growth
factor receptor promoter by human p53. Mol Cell Biol. 16:6009–6019.
1996.PubMed/NCBI
|
|
37
|
Di Agostino S, Strano S, Emiliozzi V, et
al: Gain of function of mutant p53: the mutant p53/NF-Y protein
complex reveals an aberrant transcriptional mechanism of cell cycle
regulation. Cancer cell. 10:191–202. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wong KK, Izaguirre DI, Kwan SY, et al:
Poor survival with wild-type TP53 ovarian cancer? Gynecol Oncol.
130:565–569. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Willis A, Jung EJ, Wakefield T and Chen X:
Mutant p53 exerts a dominant negative effect by preventing
wild-type p53 from binding to the promoter of its target genes.
Oncogene. 23:2330–2338. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu J, Qian J, Hu Y, et al: Heterogeneity
of Li-Fraumeni syndrome links to unequal gain-of-function effects
of p53 mutations. Sci Rep. 4:42232014.PubMed/NCBI
|