Introduction
TNBC is a breast cancer subtype that is negative for
estrogen and progesterone receptors and epidermal growth factor
receptor 2 (HER2; ErbB2). TNBC accounts for approximately 15–20% of
all breast cancer cases and seems to be closely related to
basal-like breast cancer (1).
Patients with TNBC have a relatively poor outcome and cannot be
treated with endocrine therapy or targeted therapies due to lack of
related receptors (2). Thus, there
is a substantial need for new therapies that can target TNBC and
the progression of this disease. EGFR is a receptor tyrosine kinase
whose function has been implicated in many biological processes;
when activated, EGFR stimulates signalling pathways involved in
cell growth, survival, and migration and its overexpression is the
primary mechanism by which it contributes to breast cancer growth
and progression (3). EGFR is
overexpressed in TNBC; indeed, EGFR expression is one of the
defining characteristics of TNBC and a predictor of poor prognosis
(4). Several small molecule TKIs
targeting EGFR have shown clinical efficacy in lung, pancreatic,
colorectal, and head and neck cancers (5–8) and
while phase II clinical studies have demonstrated that gefitinib
(Iressa®), in particular, shows antitumor activity in
patients with other breast cancer types when used as a monotherapy
or in combination with other drugs, such as docetaxel or
anastrozole (9), little benefit
has been noted in TNBC (10,11)
even though the EGFR is overexpressed (12–16).
Thus, a better comprehension of the downstream EGFR
cellular events is required for the identification of molecular
markers, which may allow the selection of patients more likely to
benefit from treatment as well as for monitoring anti-EGFR
therapies and indeed the development of novel treatment strategies
for patients positive for EGFR but resistant to gefitinib and other
anti-EGFR therapies.
In a metastatic basal like TNBC cell model, the
downregulation of HIF-1α through the EGFR signaling pathway
appeared to be necessary for inducing a positive response to
EGFR-targeted therapies and to gefitinib in particular, although it
was demonstrated that this may not be sufficient (17). Indeed EGFR subcellular distribution
to the nucleus, where it behaves as a transcription factor, has
been implicated in enhancing proliferative potential and acquired
resistance to gefitinib therapy (18).
Ligand-induced endocytosis and degradation of EGFR
play important roles in the downregulation of the EGFR signal
(19) suggesting that a way to
regulate its activity could be to target its trafficking. The
expression level of the scaffolding protein
Na+/H+ exchanger regulatory factor 1 (NHERF1)
has been demonstrated to have profound effects on the trafficking,
expression and function of the EGFR in normal cells (20,21).
NHERF1 is a 358-residue protein comprising two tandem PDZ domains
(protein-binding domains conserved in the mammalian synaptic
protein, PSD-95 Drosophila Dlg or discs large, and the
adherens junction protein, ZO-1) and a COOH-terminal ERM binding
region (22). In tumors, NHERF1 is
overexpressed (23–26) and this is associated with a more
aggressive behaviour and poor prognosis (27–29);
its expression and subcellular localization can influence breast
carcinogenesis (30,31).
In non-tumor cells, EGFR binds to the PDZ1 domain of
NHERF1 via an internal peptide motif located within the C-terminal
regulatory domain of EGFR, thus slowing its degradation and
enhancing its localization at the cell surface (21) and, in this way, modulates EGFR
biological signalling function. Molecular alterations of the PDZ1
domain that abolish the recognition of EGFR sequence enhance the
ligand-induced receptor downregulation. Interestingly, the same
effect of EGFR downregulation can be achieved with a point mutation
in the EGFR regulatory region or, at the transcriptional level,
with polymorphisms in both the coding and regulatory regions
(32). Indeed, NHERF1 can alter
EGFR function via the formation of protein complexes around EGFR
and NHERF1 has been shown to form a protein complex involving EGFR
and NF2 tumor suppressor at the adherens-junctions and this
interaction prevented EGFR from internalizing and signalling,
clustering it in different parts of the plasma membrane by
association with the actin cytoskeleton network (20).
Altogether, these data led us to hypothesize that
NHERF1 expression levels regulate EGFR trafficking and functional
expression in breast cancer cells and, in this way, modulate its
role in cancer progression and cancer response to treatment. Here,
we investigated, in the metastatic basal-like TNBC model,
MDA-MB-231, the subcellular localization of NHERF1, its interaction
with EGFR and the impact of NHERF1 overexpression on cancer cell
sensitivity to anti-EGFR treatment.
Materials and methods
Cell culture
MDA-MB-231 cells were grown in Dulbecco’s modified
Eagle’s medium high glucose (4,500 mg/l) supplemented with
NaHCO3 (3,700 mg/l), 10% (v/v) heat-inactivated fetal
bovine serum, L-glutamine (2 mM), sodium-pyruvate (1 mg/ml) and
penicillin (100 U)/streptomycin (100 mg/ml). Lines were grown in a
5% CO2/95% air humidified incubator at 37°C. For western
blotting, coimmunoprecipitation (coIP) and immunofluorescence (IF)
experiments, cells were deprived overnight and stimulated with EGF
(50 ng/ml, Calbiochem, San Diego, CA, USA) for the indicated
time.
Cells and expression vectors containing
NHERF1 mutants
Expression vectors for wild-type (wt) NHERF1 and
NHERF1 mutated in the PDZ1 domain (PDZ1mut) were developed as
described (33,34) and used for the transfection of the
breast cancer MDA-MB-231 cell line. Cells transfected with 3 μg of
DNA construct in FuGENE6 transfection reagent (Roche, Milan, Italy)
according to the manufacturer’s protocol were maintained in
complete medium containing 500 μg/ml hygromycin B (Calbiochem) and
stable clones for wtNHERF1 and PDZ1mut expressing ~3-fold NHERF1
levels and a pcDNA empty vector-expressing clone in which NHERF1
was expressed at endogenous levels were selected.
Western blotting
Samples were extracted in SDS sample buffer (6.25 mM
Tris-HCl, pH 6.8, containing 10% glycerol, 3 mM SDS, 1%
2-mercaptoethanol, and 0.75 mM of bromophenol blue), separated by
4–12% SDS-polyacrylamide gel electrophoresis and blotted to
Immobilon P (Merck Millipore, Milan, Italy). Western blotting was
performed with monoclonal antibodies (BD Biosciences Transduction
Laboratories, Milan, Italy) against NHERF1 diluted 1:250 or EGFR
diluted 1:250 against and diluted 1:2,500 against Actin
(Sigma-Aldrich, Hamburg, Germany).
Cell fractionation
After treatment, monolayers were washed with PBS and
then lysed in buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM
EDTA, 0.1 mM EGTA, 1 mM dithiothreitol and 0.5 mM
phenylmethylsulfonyl fluoride) and homogenized by five passes
through a 20-gauge needle to obtain the cell homogenate. An aliquot
was removed for the determination of total cellular protein. The
nuclear fraction was obtained by centrifuging the homogenate at 600
× g for 10 min. The resulting supernatant was centrifuged at 3500 ×
g for 10 min to obtain a pellet containing the endosomal fraction
and the supernatant was centrifuged again at 17,000 × g for 1 h to
obtain a plasma membrane pellet. The supernatant was centrifuged
again at 100,000 × g for 1.5 h, resulting in a microsomal pellet
and the soluble cytoplasmic fraction in the supernatant.
Thirty-five micrograms of each of the separated cellular fractions
was extracted in SDS sample buffer and analyzed by western
blotting.
Cross-linked gelatin layer preparation
and fractionation
Cytosol, membrane and invadopodia fractions were
obtained from cells grown on 2 mg/ml porcine skin gelatin in PBS
containing 2 mg/ml sucrose as previously described (35). Briefly, 15 ml, kept warm at 40°C,
was spread on a 150-mm diameter plastic dishes to evenly cover the
entire dish surface. Excess gelatin was removed and the layer was
maintained on ice for 10 min. Then 10 ml of ice cold, 0.5%
glutaraldhyde in PBS was added for 15 min cross-link the gelatin.
The cross-linked gelatin was then washed three times with PBS and
20 ml of 70% ethanol was added to each dish for 1 h under a sterile
bench hood to sterilize followed by two washes with sterile PBS for
5 min and two times with complete DMEM, the last wash of DMEM was
not removed and the dishes were left in a humidfied, 37°C incubator
for 1 h followed by seeding 4,000,000 cells on each dish, left for
24 h in a humidfied, 37°C, 5% CO2 incubator. Cell
fractions were isolated as follows: three washes with PBS
containing 1 mM CaCl2 and 0.5 mM MgCl2, two
times with 0.2X PBS plus 1 mM CaCl2 and 0.5 mM
MgCl2. Cells were then incubated on ice with 3 ml of
hypertonic swelling buffer [0.2X PBS supplied with 2 μl/ml protease
inhibitor cocktail (Sigma-Aldrich), PMSF 1 mM, sodium orthovanadate
1 mM] for 15 min on ice. Cell bodies were gently scraped with an
L-shaped pipette and centrifuged at 10,300 × g for 30 min.
Supernatant was collected (cytosolic soluble proteins) and placed
on ice while the pellet was resuspended with 100 μl of lysis buffer
(HEPES 5 mM, EDTA 0.5 mM, pH 7.2 supplied with protease inhibitor 2
μl/ml, PMSF 1 mM, sodium orthovanadate 1 mM, DTT 1 mM, nonidet
0.1%) and membrane proteins were extracted by 30 min of rotating at
4°C. After two PBS washes, the entire gelatin layer containing
entrapped invadopodia was scraped from the dish with 1 ml of the
lysis buffer, vortexed and protein extracted for 30 min on an
orbiting wheel at 4°C. The fractions containing membrane proteins
or invadopodia were collected in Eppendorf tubes and centrifuged at
13,000 × rpm at 4°C. The supernatant of each fraction was collected
and the pellet discarded. Protein concentration of the three
fractions were measured with Bradford reagent (Pierce, Rockford,
IL, USA). The diluted samples were precipitated with nine volumes
of −20°C acetone overnight and centrifuged at 10,300 × g for 1 h.
Proteins were resuspended in SDS sample buffer and western
blotted.
Coimmunoprecipitation
After treatment monolayers were washed two times
with ice-cold phosphate buffered saline (PBS), lysed in ice-cold
coimmunoprecipitation lysis buffer (50 mM Tris, pH 7.5, 150 mM
NaCl, 1% NP-40, 0.5% sodium deoxycholate, 100 mM
Na3VO4 and 1 mM NaF, protease inhibitors) and
homogenated by five passes through a 20-gauge needle to obtain the
total cell homogenate. An aliquot was removed for the determination
of total cellular protein concentration of which 150 mg was
incubated for 1 h at 4°C on a rotator with primary antibody
followed by the addition of protein A/G Plus-Agarose (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) and incubation at 4°C overnight
on a rotator. Immunoprecipitates were collected by centrifugation
at 2,500 rpm for 5 min at 4°C. The pellet was washed four times
with 1 ml of lysis buffer and the pellet resuspended in 40 ml of
SDS sample buffer, run on 10% SDS-PAGE and analyzed by western
blotting.
Immunofluorescence
Cells on coverslips were washed two times in sterile
PBS at RT, fixed with 3.7% ice-cold paraformaldehyde/PBS for 20
min, washed with ice-cold PBS, permeabilized with 0.1% Triton
X-100, saturated with 0.1% gelatin in PBS and then incubated with
polyclonal anti-NHERF1 primary antibody (Affinity Bio-Reagents,
Golden, CO, USA) diluted 1:300 or monoclonal anti-EGFR primary
antibody (BD Biosciences Transduction Laboratories) diluted 1:500
in 0.1% gelatin in PBS at RT for 1 h. They were then washed with
0.1% gelatin in PBS and incubated at RT for 1 h with the Alexa 488
goat anti-rabbit secondary antibody or the Alexa 568 goat
anti-mouse secondary antibody conjugate (Invitrogen, Carlsbad, CA,
USA). The coverslips were washed with ice-cold PBS, mounted with
Mowiol (Calbiochem) and observed on a BX40 microscope (Olympus,
Tokyo, Japan) with a SenSys 1401E-Photometrics CCD camera (Roper
Scientific, Tucson, AZ, USA).
MTT assay
gefitinib (Selleckchem, USA) was dissolved in DMSO
to a final concentration of 0.01–100 μM, added at these
concentrations to 1.5×103 cells in 96-bottomed well
plates and incubated for 72 h. After the incubation, MTT
(Sigma-Aldrich) was added at a concentration of 0.5 μg/ml to each
well and incubated for 1 h in a humidified atmosphere, solubilized
in 100 μl DMSO for 2 h and absorbance was measured at 570 and 655
nm in a plate reader (Packard Spectra Count, Stanford, CA, USA).
IC50 was calculated with CalculSyn software (Biosoft,
Cambridge, UK).
Degradation assay
For combined localization of gelatinolytic activity
and actin in the same section, in situ zymography using
dye-quenched (DQ)-gelatin (Molecular Probes, Eugene, OR, USA) as a
substrate for gelatinolytic activity, was performed followed by
immunolocalization of actin as described (35).
Wound healing assay
Cells were seeded in 6-well culture dishes and grown
to 80% confluence. Cells were then starved in DMEM overnight, and a
wound was introduced with a micropipette tip. The wounded cells
were washed to remove any suspended cells and further incubated in
the presence of EGF with or without gefitinib. The plates were
photographed at 0, 24 and 48 h and the exact wound width was
calculated by NIH Image J Software.
Statistical procedures
Student’s t-test was applied to analyze the
statistical significance between treatments and a p<0.05 was
considered as significant. All comparisons were performed with
InStat (GraphPad software, San Diego, CA, USA).
Results
NHERF1 is a molecular integrator of EGFR
downstream events
To demonstrate the involvement of NHERF1 in EGFR
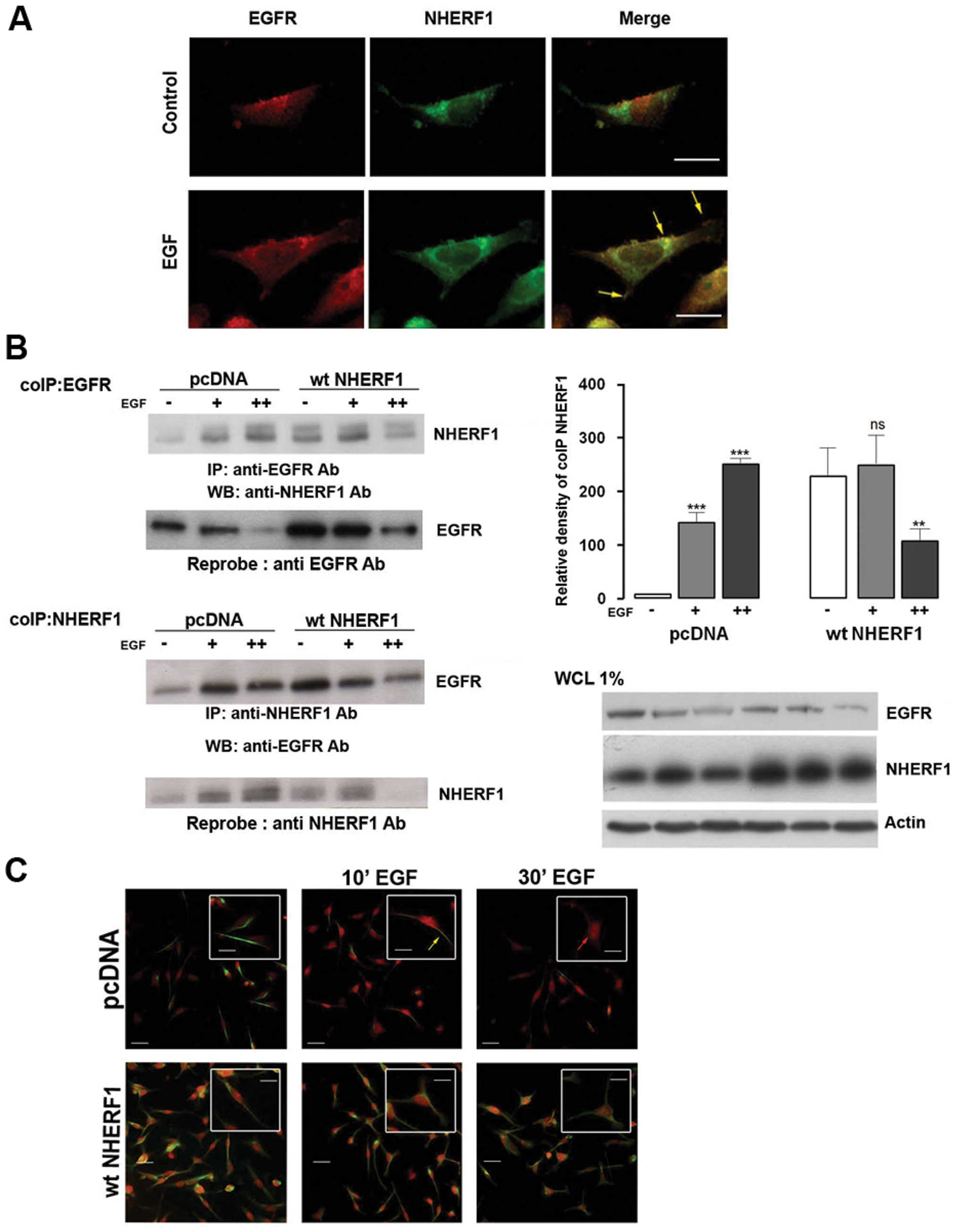
signalling, we first performed fluorescent microscopic experiments
with the metastatic basal like TNBC cell line, MDA-MB-231 (Fig. 1A). Cells were serum deprived
overnight and stimulated with EGF (100 ng/ml) for 10 min and then
immunostained with anti-EGFR and anti-NHERF1 antibodies. NHERF1
(green) did not co-localize with EGFR (red) in non-stimulated
conditions (control), while a short exposure to EGF stimulated
NHERF1 and EGFR co-localization (yellow arrow).
To biochemically validate this EGF-induced physical
interaction of NHERF1 with EGFR, we performed coimmunoprecipitation
(coIP) experiments in cells stimulated with EGF for 10 (+) or 30
(++) min and, subsequently, immunoprecipitated with an anti-EGFR
antibody (Fig. 1B). Levels of both
endogenous EGFR and NHERF1 in the EGFR complex were measured by
western blotting. In non-stimulated, empty plasmid transfected
cells expressing normal endogenous levels of NHERF1 (pcDNA
MDA-MB-231 cells), NHERF1 did not co-precipitate with EGFR and,
upon EGF stimulation, the amount of co-precipitation of NHERF1 with
EGFR increased with time. Conversely, in NHERF1 overexpressing
cells (wtNHERF1), NHERF1 and EGFR already coIPed before EGF
stimulation, and short term EGF stimulation transiently increased
this phenomenon. Immunofluorescent studies confirmed these results
(Fig. 1C). In pcDNA MDA-MB-231
cells (upper panels), NHERF1 (red) did not co-localize with EGFR
(green) in non-stimulated conditions, while after a short time
exposure to EGF, NHERF1 and EGFR co-localized in the plasma
membrane (yellow arrow). Forced ectopic overexpression of NHERF1
(lower panels) resulted in the co-localization of the two proteins
at the plasma membrane already in non-stimulated conditions and,
after short EGF exposure, the main fraction of the EGFR-NHERF1 pair
remained in the plasma membrane with only a much smaller portion
also being observed in the perinuclear compartment.
To better understand the dynamics of interaction
between NHERF1 and EGFR, we performed reciprocal NHERF1/EGFR coIP
experiments of plasma membrane, endosome and cytosol (cL) cell
fractions (Fig. 2). In the plasma
membrane fraction of pcDNA MDA-MB-231 cells (Fig. 2A), EGF stimulation increased coIPed
NHERF1 and decreased EGFR expression. When NHERF1 was
overexpressed, NHERF1 always coIPed with the receptor and EGFR
levels were not downregulated by EGF stimulation. In the endosomal
fraction (Fig. 2B), we observed
that EGF stimulation decrease the receptor level in the pcDNA
cells, while the overexpression of NHERF1 both increased the amount
of EGFR and decreased its degradation in the early endosome
fraction. NHERF1 overexpression firstly completely abrogated their
interaction in non-stimulated conditions restoring the complex
after the EGF stimulation. The EGFR was never expressed in the
cytosolic fraction (cCL) (Fig. 2C)
of either pcDNA or wtNHERF1 transfected cells and in NHERF1
overexpressing cells, NHERF1 decreased with stimulation.
Altogether, these results demonstrate that EGF
treatment in breast cells recruits NHERF1 from the cytosol to the
plasma membrane, forming a complex with EGFR. On the contrary, the
overexpression of NHERF1 results in an increased recycling of EGFR
back to and an increased stability at the plasma membrane together
with an increase of the NHERF1/EGFR complex and a reduction of this
complex in the endosomes.
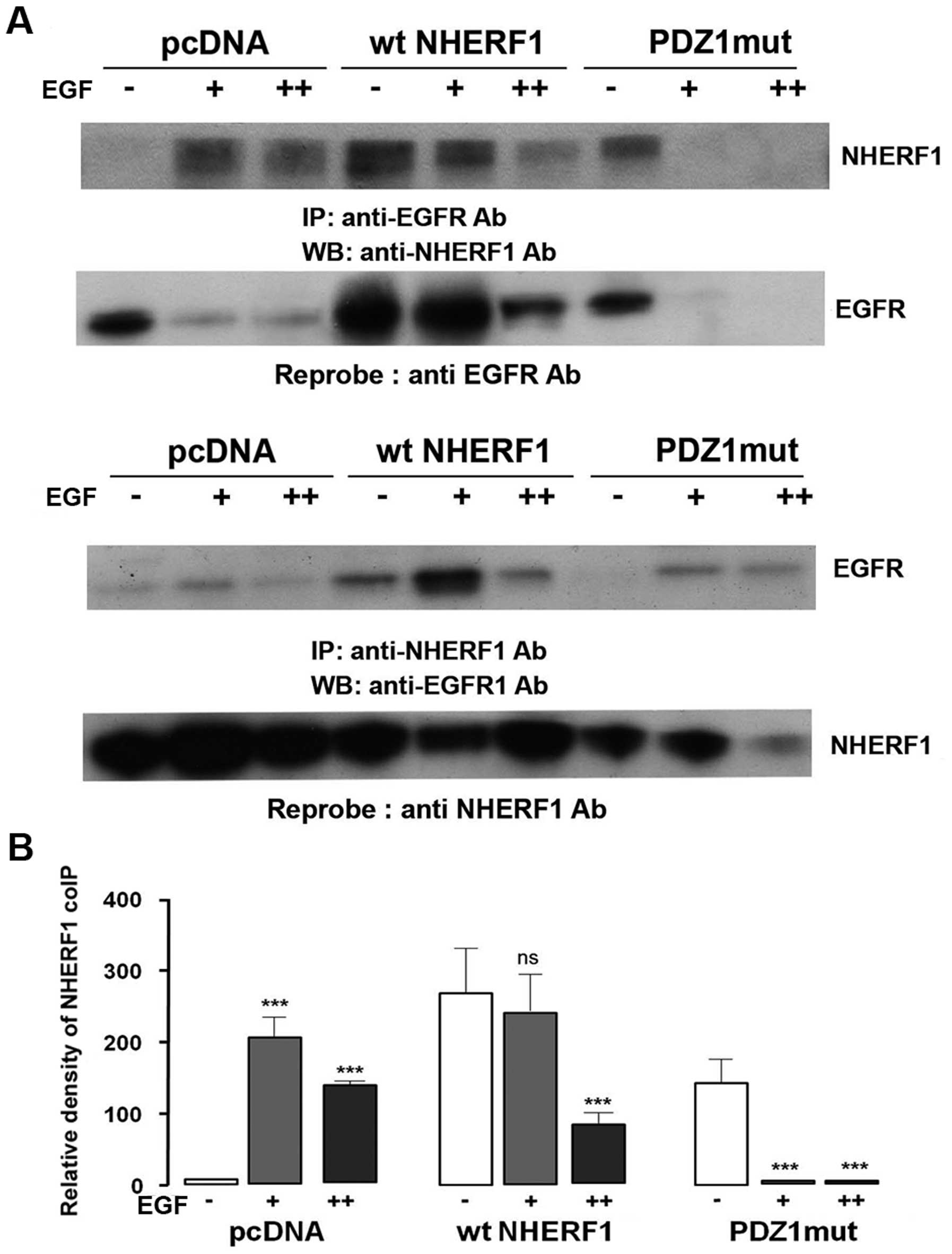
The PDZ1 domain of NHERF1 binds with
EGFR
It has been hypothesized that in non-tumor cells
EGFR binds the PDZ1 domain of NHERF1 (21) and we verified this hypothesis, in
our in vitro model, with coIP experiments of the membrane
fraction using cells transfected with PDZ1 mutated NHERF1 which can
no longer bind its protein partners (PDZ1mut). As shown in Fig. 3A, forced ectopic wild-type (wt)
NHERF1 overexpression increased NHERF1 coIPed with EGFR while
overexpressing PDZ1mut NHERF1 strongly reduced its co-precipitation
with EGFR. Moreover, overexpressing wt NHERF1 downregulated the
rate of EGFR decrease after EGF treatment, while, when the PDZ1
domain function was lost (ectopic PDZ1mut), NHERF1 overexpression
was no longer able to prevent EGFR decrease (Fig. 3B). Overall, these data agree with
Lazar et al (21) that EGFR
and NHERF1 interact through the PDZ1 domain of NHERF1, also in
breast cancer cells.
Relevance of NHERF1 expression levels in
anti-EGFR drug activity
Alterations in receptor expression and localization
have been shown to influence their reactivity to both ligands and
to inhibitors. Since EGFR inhibition is an important antitumor
therapeutic strategy we investigated the role of NHERF1 expression
in the action of one of the well known EGFR inhibitors actually in
clinical practice for anticancer therapy, the EGFR tyrosine kinase
inhibitor (TKI), gefitinib (Iressa).
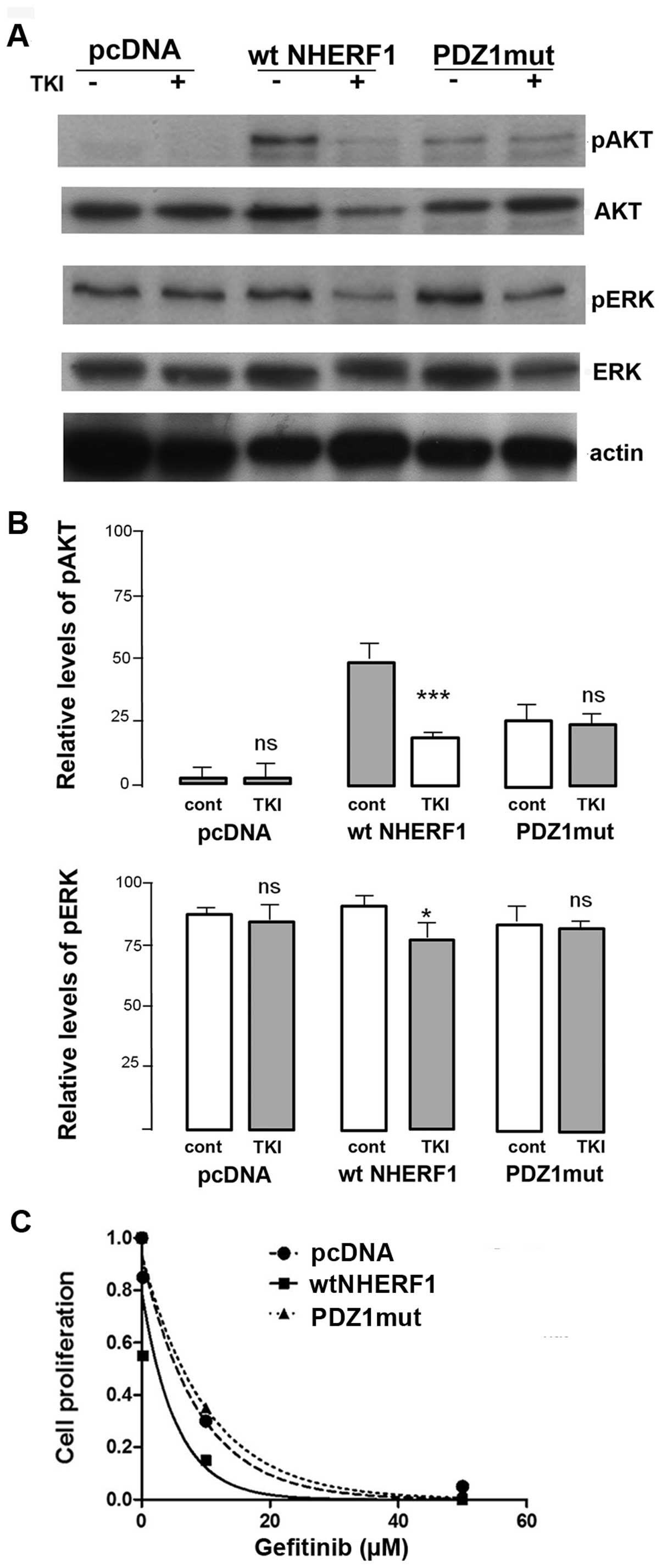
We first examined the effect of gefitinib on
signaling pathways by a series of western blot analyses (Fig. 4A) and observed that the basic
levels of phospho-AKT (pAKT) were low while significant levels of
phospho-ERK (pERK) were detected. Treatment with gefitinib had no
effect on pAKT or pERK levels in either pcDNA or PDZ1mut cells,
while the level decreased in wt-NHERF1 cells (Fig. 4B). These data indicate that
overexpression of NHERF1 may be required to render cells sensitive
to gefitinib. Indeed, analyzing the dose-response of gefitinib
inhibition of cell growth of the three cell lines (Fig. 4C), only overexpression of wtNHERF1
sensitized this resistant cell to anti-EGFR therapy with gefitinib
(shift of the IC50 from 6.36±0.52 to 2.34±0.17 μM).
However, the highest levels of cancer morbidity
depends on invasion and metastasis rather than growth (36) and the EGFR is known to be involved
in tumor cell invasion through an increase in both cell migration
and invadopodia-dependent digestion of the extracellular matrix
(ECM) (35–37). These important processes have been
rarely measured in studies on the effects of blocking EGFR activity
on tumor phenotypes. For this reason, we measured the effect of
NHERF1 overexpression on the effectiveness of anti-EGFR therapy
directed against migration and against invadopodia-dependent ECM
digestion.
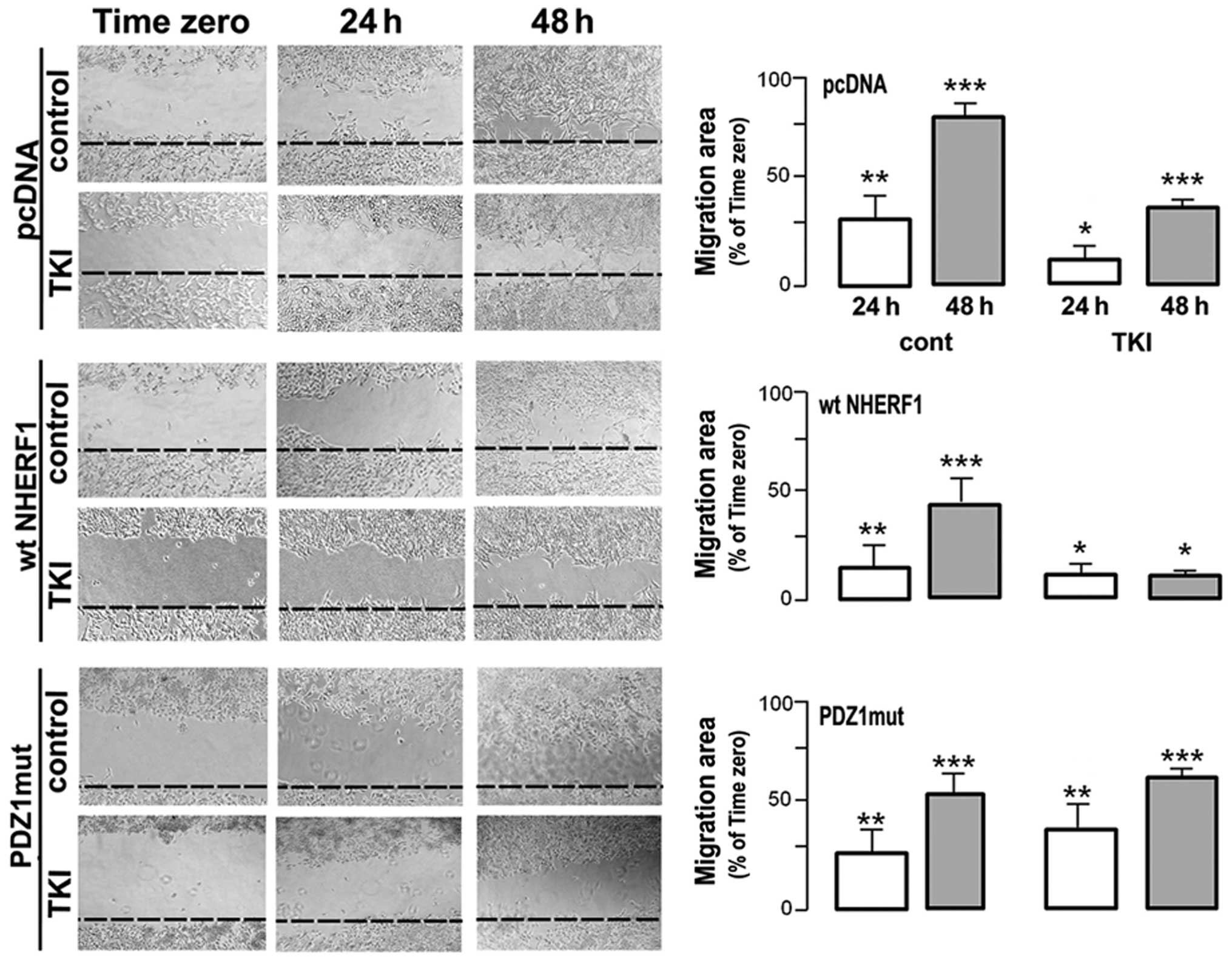
Wound healing measurements utilised to test the
ability of gefitinib to modulate MDA-MB-231 migration (Fig. 5) revealed that control monolayers
displayed rapid wound healing within 48 h that was blocked ~50% by
treatment with gifitinib while PDZ1mut overexpression abrogated the
ability of gefitinib to block motility. Importantly, overexpression
of wtNHERF1 per se slightly reduced motility in the untreated cells
but greatly sensitized the monolayers to inhibition of motility by
gefitinib, especially at 48 h (an ~75% reduction in motility).
We next measured the ability of MDA-MB-231 cells to
form invadopodia and digest the ECM. To first obtain a quantitative
comparison of the distribution of EGFR and the
Na+/H+ exchanger isoform 1 (NHE1) in
invadopodia in the different clones and the effect of EGF treatment
in pcDNA cells, cells were plated on 2% cross-linked gelatin,
fractionated for cytosol, cell membrane and invadopodia fractions
as previously described (35) and
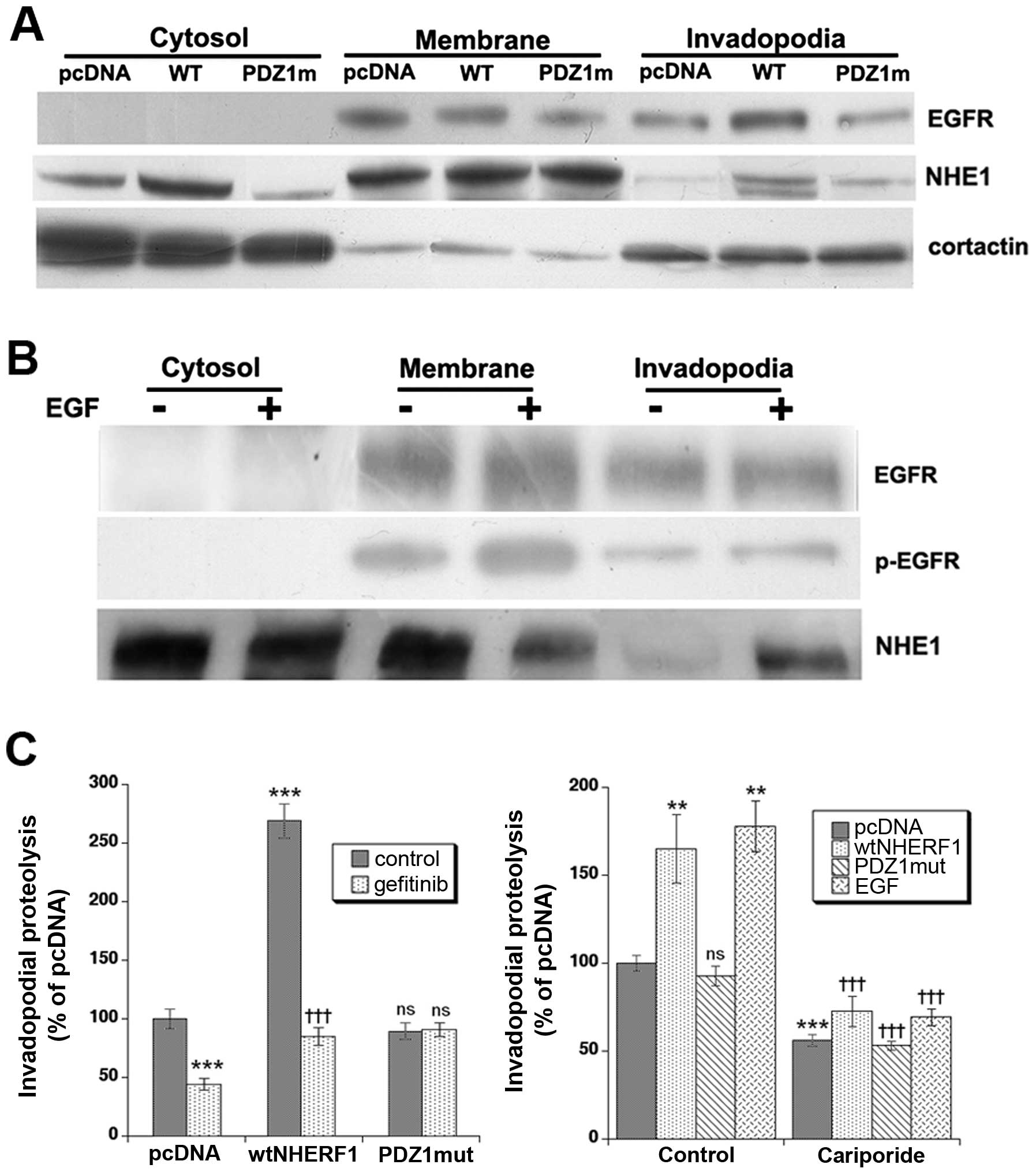
assayed by western blotting. As can be seen in Fig. 6A, overexpression of wtNHERF1 but
not PDZ1mut-NHERF1 shifted a major fraction of the EGFR and NHE1 to
the invadopodia. Cortactin was utilized as a control protein for
the purity of the fractions (35).
Interestingly, treatment of pcDNA cells with 50 ng/ml of EGF
resulted in an increase in p-EGFR in both the membrane and
invadopodia fractions and a strong increase in NHE1 expression in
the invadopodia (Fig. 6B).
We then measured invadopodia activity by
microscopically measuring the release of quenched Bodipy
fluorescence after 6-h incubation on 3D lattices of
Matrigel-DQ-gelatin as previously described (35). After 6-h incubation on
Matrigel-DQ-gelatin, the cells formed bright F-actin puncta
associated with focal matrix degradation, demonstrating functional
invadopodia formation. Measuring the proteolytic ability of the
cells via the intensity of the digestive fluorescent signal
(Fig. 6C, left panel), revealed
that wtNHERF1 overexpression greatly enhanced invadopodia-dependent
ECM digestion while PDZ1mut overexpression had no effect on the
proteolytic activity of invadopodia. Importantly, gefitinib
treatment downregulated invadopodia-dependent matrix degradation
process by ~50% in pcDNA cells and by 80% in wtNHERF1
overexpressing cells. Performing these experiments in the presence
or absence of EGF and or 1 μM of the specific NHE1 inhibitor,
cariporide, demonstrated that ~50% of basal ECM proteolysis and
65–75% of that stimulated by either wtNHERF1 transfection or 50
ng/ml EGF treatment was dependent on NHE1 activity (Fig. 6C, right panel). Altogether, these
data show that NHERF1 acts primarily at the level of the
invadopodial digestive ability, and that treatment with gefitinib
or with cariporide blocks invadopodia function. Finally, wtNHERF1
overexpression rendered the cells more sensitive to inhibition of
ECM digestion with gefitinib.
Discussion
The roles of EGFR in carcinogenesis are well known
and its signalling pathway is presently an attractive target for
therapy in a large number of tumor types (38,39).
Ligand-induced endocytosis and degradation of EGFR play important
roles in the downregulation of the EGFR signal (19) and, consequently, in both the
expression of neoplastic phenotypes and in the dynamics of
inhibitor action. In normal cells, the level of NHERF1 expression
has been demonstrated to have profound effects on the trafficking,
expression and function of the EGFR (20).
Here we find that, also in breast cancer cells,
NHERF1 is an important molecular integrator of EGFR trafficking and
that this regulation is important in determining the cells
aggressive behaviour and its response to anti-EGFR therapy. The
immunofluorescence and co-immunoprecipitation studies demonstrated
that EGFR stimulation in response to EGF drove the relocalization
of cytosolic NHERF1 first to the plasma membrane compartment
followed by a co-transport to the perinuclear region. Moreover, we
observed that, upon EGF stimulation, NHERF1 colocalized with EGFR
and our data suggest that EGF treatment first downregulates EGFR
expression probably through the internalization and degradation of
the receptor followed by, when stimulated for longer times, its
trafficking to other cell compartments as the nucleus (40). Ectopic NHERF1 overexpression
reduced EGFR degradation (Fig. 1B)
and increased its expression at the plasma membrane (Fig. 2B). This increased plasma membrane
expression linked to reduced degradation when NHERF1 was
overexpressed led us to determine if NHERF1 stabilizes EGFR at the
membrane, as previous described (21), or if there is more EGFR at the
plasma membrane through a modulation of its recycling to the
membrane. Therefore, we measured the influence of NHERF1 expression
levels and EGF stimulation on the dynamics of the EGFR/NHERF1
complex by analyzing their expression levels and interaction in
cell fractionation/coIP experiments (Fig. 2). These experiments showed that in
cells expressing endogenous NHERF1, EGF stimulation recruits
endogenous NHERF1 from the cytosol to the plasma membrane forming a
complex with EGFR that drives degradation of the receptor. When
NHERF1 is overexpressed, ectopic NHERF1 already complexes with EGFR
in non-stimulated conditions and increases the EGFR plasma membrane
residence time in EGF stimulation, through an increased recycling
of the EGFR from the endosome fraction back to the plasma membrane.
Altogether, these data suggest that the cellular NHERF1 expression
level functions to shift the cellular equilibrium from one EGFR
localization and regulatory cascade/scenario to another. The
mechanism by which the breast cancer cell can alter NHERF1
expression levels and, consequently shift the balance of recycling
endosomes, is suggested by data showing that exposure of the cancer
cell to hypoxia or low nutrients increases NHERF1 expression and
localization resulting in an increase in invasion (26).
Indeed, this increased retention time (i.e., more
stable expression) of the EGFR in the plasma membrane and the
reduction of the negative control the EGFR signalling cascade
should increase the cells aggressiveness. However, it could,
possibly, also increase its sensitivity to anti-EGFR
phosphotyrosine kinase inhibitor therapy. To test this hypothesis,
we next analysed the relevance of NHERF1 expression on the effect
of response to the tyrosine kinase inhibitor (TKI), gefitinib
(Iressa) on signalling pathways downstream the EGFR activation
(Fig. 4A) and we observed that the
overexpression of NHERF1 and its PDZ1 mutation is able per
se to increase the basal level of phosphor-AKT (Fig. 4B), but gefitinib downregulated the
AKT activation only with a functional PDZ1 domain. Gefitinib is
also able to slightly regulate the activation of ERK. We also
analysed the effect of NHERF1 expression on the effect of gefitinib
on growth and we observed that, indeed, its sensitized the cell
lines to gefitinib (Fig. 4C),
demonstrating that the TNBC cell endogenous levels of NHERF1 could
be functional but not sufficiently available for an enhanced
gefitinib efficacy. As the highest levels of cancer morbidity
depend on invasion and metastasis, we next determined the role of
NHERF1 expression levels also on motility, invasion and proteolysis
of the extracellular matrix (ECM). The wound healing motility test
(Fig. 5) revealed that, indeed,
NHERF1 overexpression sensitized the monolayer to gefitinib
treatment-dependent inhibition of in vitro cell
migration.
We next analysed in detail the presence and
proteolytic activity of invadopodia (Fig. 6) and observed that ectopic NHERF1
overexpression, as hypothesized, i) increased invadopodia formation
and the degradation of the extracellular matrix; and ii) strongly
increased the inhibitory effect of gefitinib on both the formation
of invadopodia and, more strongly, on their proteolytic activity.
These data are interesting in the context of a previous paper
showing that activation of the EGFR can initiate invadopodia
maturation via the subsequent activation of a Src-Arg-cortactin
pathway that organizes the recruitment of Arp 2/3, Nck 1 and N-WASp
proteins to the local cytoskeleton (37). Our data suggest that although the
EGFR can act to initiate invadopodia formation, its primary action
is to stimulate/regulate invadopodia-dependent ECM proteolysis. Our
observation that gefitinib had a very high capacity to block the
NHERF1-dependent increase in proteolytic activity suggests that the
EGFR, when stabilized at the membrane by NHERF1, may act primarily
at the level of invadopodia proteolytic activity. This may probably
occur through an activation of the NHE1 as it has been demonstrated
that both the overexpression of NHERF1 (26) and stimulation by EGF (35) drive an enhanced extracellular
acidification and, consequently, an enhanced protease activity by
activating the NHE1 (41). Indeed,
both EGF treatment and overexpression of wt-NHERF1, but not PDZ1mut
NHERF1, increased the level of NHE1 in the invadopodia and the
specific NHE1 inhibitor, cariporide, reduced both basal and
EGF-stimulated invadopodia-driven focal ECM proteolysis (Fig. 6).
In conclusion, a number of mechanisms have been
suggested for resistance to EGFR TKI-induced growth inhibition in
cancers, including EGFR independence, mutations in EGFR and
alterations in downstream signalling pathways. Here, we show that
the expression level of the signal transduction scaffolding protein
NHERF1, regulates EGFR recycling/degradation to stabilize the EGFR
on the plasma membrane and sensitize the cell to the TKI-dependent
inhibition of EGFR-driven motility and invadopodia-dependent ECM
proteolysis in cancer cells. The identification of the NHERF1-EGFR
network and the determination of how it is regulated may improve
our understanding of the cancer metastasis process, and the
optimization of current anticancer drugs specifically targeting
this process.
Acknowledgements
We thank Professor E. Weinman (Department of
Medicine, University of Maryland School of Medicine, Baltimore, MD,
USA) for the gift of the NHERF1 wild-type and PDZ1 domain mutated
(PDZ1MUT) constructs. S.J.R. would like to thank the Italian
Association for Cancer Research (AIRC) grant no. 11348 for
supporting this study. K.Z. is a fellow of the Marie Curie Initial
Training Network IonTraC (FP7-PEOPLE-2011-ITN Grant Agreement no.
289648). The S.J.R. laboratory is part of the Italian network
‘Istituto Nazionale Biostrutture e Biosistemi’ (INBB) and the
‘Centro di Eccellenza di Genomica in Campo Biomedico ed Agrario’ of
the University of Bari and the project BioBoP of the region
Puglia.
Abbreviations:
|
TNBC
|
triple negative breast cancer
|
|
NHERF1
|
Na+/H+ exchanger
regulatory factor 1
|
|
EGFR
|
epidermal growth factor receptor
|
|
coIP:mEGFR and coIP:mNHERF1
|
EGFR and NHERF1 coimmunoprecipitation
of the membrane fraction respectively
|
|
coIP:eEGFR and coIP:eNHERF1
|
EGFR and NHERF1 coimmunoprecipitation
of the endosome fraction respectively
|
|
WCL mCL eCL cCL whole
|
membrane, endosome and cytosolic cell
lysates
|
|
WB
|
western blotting
|
|
TKI
|
tyrosine kinase inhibitor
|
References
|
1
|
Foulkes WD, Smith IE and Reis-Filho JS:
Triple-negative breast cancer. N Engl J Med. 363:1938–1948. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Metzger-Filho O, Tutt A, de Azambuja E,
Saini KS, Viale G, Loi S, et al: Dissecting the heterogeneity of
triple-negative breast cancer. J Clin Oncol. 30:1879–1887. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Paez JG, Janne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, et al: EGFR mutations in lung cancer:
correlation with clinical response to gefitinib therapy. Science.
304:1497–1500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Korsching E, Jeffrey SS, Meinerz W, Decker
T, Boecker W and Buerger H: Basal carcinoma of the breast
revisited: an old entity with new interpretations. J Clin Pathol.
61:553–560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baker M: EGFR inhibitors square off at
ASCO. Nat Biotechnol. 22:6412004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cohen EE, Lingen MW, Martin LE, Harris PL,
Brannigan BW, Haserlat SM, et al: Response of some head and neck
cancers to epidermal growth factor receptor tyrosine kinase
inhibitors may be linked to mutation of ERBB2 rather than EGFR.
Clin Cancer Res. 11:8105–8108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Giusti RM, Shastri K, Pilaro AM, Fuchs C,
Cordoba-Rodriguez R, Koti K, et al: U.S. Food and Drug
Administration approval: panitumumab for epidermal growth factor
receptor-expressing metastatic colorectal carcinoma with
progression following fluoropyrimidine-, oxaliplatin-, and
irinotecan-containing chemotherapy regimens. Clin Cancer Res.
14:1296–1302. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sobrero AF, Maurel J, Fehrenbacher L,
Scheithauer W, Abubakr YA, Lutz MP, et al: EPIC: phase III trial of
cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin
failure in patients with metastatic colorectal cancer. J Clin
Oncol. 26:2311–2319. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Polychronis A, Sinnett HD, Hadjiminas D,
Singhal H, Mansi JL, Shivapatham D, et al: Preoperative gefitinib
versus gefitinib and anastrozole in postmenopausal patients with
oestrogen-receptor positive and
epidermal-growth-factor-receptor-positive primary breast cancer: a
double-blind placebo-controlled phase II randomised trial. Lancet
Oncol. 6:383–391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Johnston JB, Navaratnam S, Pitz MW,
Maniate JM, Wiechec E, Baust H, et al: Targeting the EGFR pathway
for cancer therapy. Curr Med Chem. 13:3483–3492. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Montemurro F, Valabrega G and Aglietta M:
Lapatinib: a dual inhibitor of EGFR and HER2 tyrosine kinase
activity. Expert Opin Biol Ther. 7:257–268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baselga J, Albanell J, Ruiz A, Lluch A,
Gascón P, Guillém V, et al: Phase II and tumor pharmacodynamic
study of gefitinib in patients with advanced breast cancer. J Clin
Oncol. 23:5323–5333. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tan AR, Yang X, Hewitt SM, Berman A,
Lepper ER, Sparreboom A, et al: Evaluation of biologic end points
and pharmacokinetics in patients with metastatic breast cancer
after treatment with erlotinib, an epidermal growth factor receptor
tyrosine kinase inhibitor. J Clin Oncol. 22:3080–3090. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Irwin ME, Mueller KL, Bohin N, Ge Y and
Boerner JL: Lipid raft localization of EGFR alters the response of
cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J
Cell Physiol. 226:2316–2328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Metro G, Finocchiaro G and Cappuzzo F:
Anti-cancer therapy with EGFR inhibitors: factors of prognostic and
predictive significance. Ann Oncol. (Suppl 2): ii42–ii45.
2006.PubMed/NCBI
|
|
16
|
She QB, Solit D, Basso A and Moasser MM:
Resistance to gefitinib in PTEN-null HER-overexpressing tumor cells
can be overcome through restoration of PTEN function or
pharmacologic modulation of constitutive phosphatidylinositol
3′-kinase/Akt pathway signaling. Clin Cancer Res. 9:4340–4346.
2003.PubMed/NCBI
|
|
17
|
El Guerrab A, Zegrour R, Nemlin CC, Vigier
F, Cayre A, Penault-Llorca F, et al: Differential impact of
EGFR-targeted therapies on hypoxia responses: implications for
treatment sensitivity in triple-negative metastatic breast cancer.
PLoS One. 6:e250802011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu YL, Chou RH, Liang JH, Chang WJ, Su KJ,
Tseng YJ, et al: Targeting the EGFR/PCNA signaling suppresses tumor
growth of triple-negative breast cancer cells with cell-penetrating
PCNA peptides. PLoS One. 8:e613622013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Curto M, Cole BK, Lallemand D, Liu CH and
McClatchey AI: Contact dependent inhibition of EGFR signaling by
Nf2/Merlin. J Cell Biol. 177:893–903. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lazar CS, Cresson CM, Lauffenburger DA and
Gill GN: The Na+/H+ exchanger regulatory
factor stabilizes epidermal growth factor receptors at the cell
surface. Mol Biol Cell. 15:5470–5480. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bretscher A, Chambers D, Nguyen R and
Reczek D: ERM-Merlin and EBP50 protein families in plasma membrane
organization and function. Annu Rev Cell Dev Biol. 16:113–143.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shibata T, Chuma M, Kokubu A, Sakamoto M
and Hirohashi S: EBP50, a beta-catenin-associating protein,
enhances Wnt signaling and is over-expressed in hepatocellular
carcinoma. Hepatology. 38:178–186. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fraenzer JT, Pan H, Minimo L Jr, Smith GM,
Knauer D and Hung G: Overexpression of the NF2 gene inhibits
schwannoma cell proliferation through promoting PDGFR degradation.
Int J Oncol. 23:1493–1500. 2003.PubMed/NCBI
|
|
25
|
Pan Y, Wang L and Dai JL: Suppression of
breast cancer cell growth by Na+/H+ exchanger
regulatory factor 1 (NHERF1). Breast Cancer Res. 8:R632006.
View Article : Google Scholar
|
|
26
|
Cardone RA, Bellizzi A, Busco G, Weinman
EJ, Dell’Aquila ME, Casavola V, Azzariti A, Mangia A, Paradiso A
and Reshkin SJ: The NHERF1 PDZ2 domain regulates
PKA-RhoA-p38-mediated NHE1 activation and invasion in breast tumor
cells. Mol Biol Cell. 18:1768–1780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Song J, Bai J, Yang W, Gabrielson EW, Chan
DW and Zhang Z: Expression and clinicopathological significance of
oestrogen-responsive ezrin-radixinmoesin-binding phosphoprotein 50
in breast cancer. Histopathology. 51:40–53. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bellizzi A, Malfettone A, Cardone RA and
Mangia A: NHERF1/EBP50 in breast cancer: clinical perspectives.
Breast Care (Basel). 5:86–90. 2010. View Article : Google Scholar
|
|
29
|
Bellizzi A, Mangia A, Malfettone A,
Cardone RA, Simone G, Reshkin SJ and Paradiso A:
Na+/H+ exchanger regulatory factor 1
expression levels in blood and tissue predict breast tumour
clinical behaviour. Histopathology. 58:1086–1095. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mangia A, Chiriatti A, Bellizzi A,
Malfettone A, Stea B, Zito FA, et al: Biological role of NHERF1
protein expression in breast cancer. Histopathology. 55:600–608.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Georgescu M, Morales FC, Molina JR and
Hayashi Y: Roles of NHERF1/EBP50 in cancer. Curr Mol Med.
8:459–468. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gebhardt F, Bürger H and Brandt B:
Modulation of EGFR gene transcription by secondary structures, a
polymorphic repetitive sequence and mutations - a link between
genetics and epigenetics. Histol Histopathol. 15:929–936.
2000.PubMed/NCBI
|
|
33
|
Weinman EJ, Steplock D, Wade JB and
Shenolikar S: Ezrin binding domain-deficient NHERF attenuates
cAMP-mediated inhibition of Na(+)/H(+) exchange in OK cells. Am J
Physiol Renal Physiol. 281:F374–F380. 2001.PubMed/NCBI
|
|
34
|
Weinman EJ, Wang Y, Wang F, Greer C,
Steplock D and Shenolikar S: A C-terminal PDZ motif in NHE3 binds
NHERF-1 and enhances cAMP inhibition of sodium-hydrogen exchange.
Biochemistry. 42:12662–12668. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Busco G, Cardone RA, Greco MR, Bellizzi A,
Colella M, Antelmi E, et al: NHE1 promotes invadopodial ECM
proteolysis through acidification of the peri-invadopodial space.
FASEB J. 24:3903–3915. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chambers AF, Groom AC and MacDonald IC:
Dissemination and growth of cancer cells in metastatic sites. Nat
Rev Cancer. 2:563–572. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mader CC, Oser M, Magalhaes MA,
Bravo-Cordero JJ, Condeelis J, Koleske AJ, et al: An
EGFR-Src-Arg-cortactin pathway mediates functional maturation of
invadopodia and breast cancer cell invasion. Cancer Res.
71:1730–1741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sebastian S, Settleman J, Reshkin SJ,
Azzariti A, Bellizzi A and Paradiso A: The complexity of targeting
EGFR signalling in cancer: from expression to turnover. Biochim
Biophys Acta. 1766:120–139. 2006.PubMed/NCBI
|
|
39
|
Grandal MV and Madshus IH: Epidermal
growth factor receptor and cancer: control of oncogenic signalling
by endocytosis. J Cell Mol Med. 12:1527–1534. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lo H-W and Hung M-C: Nuclear EGFR
signalling network in cancers: linking EGFR pathway to cell cycle
progression, nitric oxide pathway and patient survival. Br J
Cancer. 94:184–188. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Greco MR, Antelmi E, Busco G, Guerra L,
Rubino R, Casavola V, et al: Protease activity at invadopodial
focal digestive areas is dependent on NHE1-driven acidic pHe. Oncol
Rep. 31:940–946. 2014.
|