Introduction
Autophagy is an evolutionarily conserved catabolic
process. It can damage long-lived cellular proteins and degrade
organelles, through facilitating cytoplasmic turnover and maintains
metabolic homeostasis in double-membrane vesicles, termed
autophagosomes (1,2). When cells need to generate
intracellular nutrients and energy, for instance, during
starvation, growth factor withdrawal, or high bioenergetic demands,
autophagy can be upregulated (3).
Moreover, basal autophagy can serve as an important homeostatic
cellular recycling mechanism responsible for degrading unnecessary
or dysfunctional cellular organelles and proteins in all living
cells (4). Autophagy can promote
the survival of tumor cells in poorly vascularized and hypoxic
tumors or cytotoxic treatments (5,6).
Many preclinical studies have demonstrated that genetic or
pharmacological inhibition of autophagy can enhance drug- and
radiation-induced cytotoxicity in cell culture and in vivo
(7). Autophagy has a potent
cytoprotective survival pathway in normal and cancer cells.
Tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL) belongs to the TNF super family that can initiate
apoptosis via activating the extrinsic apoptosis pathway (8). Due to its remarkable feature of
selectively inducing apoptosis in cancer cells without causing
damage to normal cells (9), has
led to multiple clinical trials to evaluate the antitumor potential
of recombinant human TRAIL (rhTRAIL) and it emerged as a potential
therapeutic agent (10,11). TRAIL triggers typical apoptotic
signaling by binding to its receptors, death receptor 4 (DR4) and 5
(DR5), thereby recruiting the assembly of the death-inducing
signaling complex (DISC), which activates the caspase cascade
(12). Ongoing and completed phase
I and II clinical trials with TRAIL are showing clinically
promising outcomes with no apparent toxicity (13). However, recent studies have
indicated that a variety of cancer cells are resistant to the
apoptotic effects of TRAIL (14,15).
The majority of breast cancer cells are resistant to TRAIL-mediated
apoptosis (16). The mechanisms
underlying resistance to TRAIL are not fully-understood, and the
identification of TRAIL resistance factors could facilitate the
development of more effective TRAIL-based cancer therapies.
We demonstrated that autophagy may be a potential
target to overcome TRAIL resistance of breast cancer cells.
Materials and methods
Production of rhTRAIL
rhTRAIL was produced by our laboratory (17,18).
Cell culture and reagents
The human breast cancer cell line MDA-MB-231 was
obtained from the American Type Culture Collection (ATCC)
(Manassas, VA, USA), and routinely cultured in DMEM medium
(Gibco-BRL, Rockville, MD, USA)/high glucose medium supplemented
with 10% FBS (Haoyang Biological Manufacturer Co., Ltd., Tianjin,
China) containing, 100 U/ml penicillin and 100 μg/ml streptomycin.
The cells were maintained at 37°C in humidified air with 5%
CO2. Chloroquine (CQ) and monodansylcadaverine (MDC)
were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Establishment of TRAIL-acquired
autoresistance in MDA-MB-231 breast cancer cells
To establish MDA-MB-231 TRAIL-refractory cells
exhibiting resistance to the TRAIL, TRAIL-sensitive MDA-MB-231
parental cells were exposed to incremental increases of TRAIL.
TRAIL-resistance selection continued until the MDA-MB-231 cells
could sustain cell viability and proliferate when challenged with
400 ng/ml. TRAIL-refractory cells were obtained upon exposure of
MDA-MB-231 cells for a minimum of 10 months before starting any
experimental procedure. Briefly, MDA-MB-231 cells that were
initially exposed to 40 ng/ml TRAIL for 3 months, and then treated
with 400 ng/ml TRAIL for 2 months (twice weekly) resisted
continuous growth in 640 ng/ml TRAIL. The resistant cells were
maintained in medium without TRAIL for at least 10 days before each
experiment.
Cell viability assay
Cell viability was determined by colorimetric assay
using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT). Briefly, cells (2,000 cells/well) were seeded in 96-well
plates and allowed to attach overnight at 37°C. Then culture medium
containing vehicle or drugs was added to the medium in each well
and incubating for indicated time points. The cells in 96-well
plates were incubated with 20 μl MTT in growth medium at indicated
time points. After incubating at 37°C for 4 h, the supernatants
were carefully aspirated and the resulting crystals were dissolved
in 100 ml dimethyl sulfoxide (DMSO). Absorbance values at 490 nm
were determined by the Microplate Reader (Bio-Rad, Hercules, CA,
USA). Data are presented as the percentage of survival rate
relative to vehicle-treated control.
Electron microscopy assay
To detect the autophagic vacuoles directly, we
performed ultra structural analysis under electron microscopy.
Briefly, cells were fixed in a mixture of 2.5%, paraformaldehyde
and 2.0% gluteraldehyde in 0.1 M cacodylate buffer, pH 7.3, for 1
h. After fixation, the samples were post-fixed in 1%
OsO4 in the same buffer for 1 h and then subjected to
electron microscopic analysis. Representative areas were selected
for ultrathin sectioning and viewed with a JEM 1010 transmission
electron microscope (JEOL USA, Inc., Peabody, MA, USA) at an
accelerating voltage of 80 kV. Digital images were obtained with
the AMT Imaging System (Advanced Microscopy Techniques, Danvers,
MA, USA).
Immunofluorescence staining
Immunofluorescence staining was used to perform the
localization and the level of expression of LC3B and p62/SQSTM1. In
brief, cells were grown on cover slips in the 24-well plates for 3
days, for the different treatments. After fixing with 4%
paraformaldehyde for 15 min at room temperature and extensive wash
in PBS, cells were permeabilized with 0.1% Triton X-100 in PBS
(PBST) for 25 min. Then cells were blocked with 10% goat serum in
PBST for 1 h, followed by incubating with rabbit anti-LC3B or
anti-p62/SQSTM1 monoclonal antibodies (Cell Signaling Technology,
Inc., Danvers, MA, USA) and rhodamine-conjugated anti-rabbit
secondary antibodies (Kirkegaard & Perry Laboratories, Inc.,
Gaithersburg, MD, USA). The nuclear DNA was stained with
4′,6-Diamidino-2-phenylindole (DAPI) for 5 min. Finally, antifading
medium was added and the cover slips were immediately observed
under a DP71 fluorescence microscope (Olympus, Tokyo, Japan).
MDC staining
MDC staining of autophagic vacuoles was performed
for autophagy analysis. Autophagic vacuoles were labeled with MDC
(50 μM) in PBS at 37°C for 10 min. And then, the cells were washed
three times with PBS. Autophagic vacuoles were observed immediately
under DP71 fluorescence microscope (excitation wavelength, 380 nm;
emission filter, 525 nm).
Immunoblotting analysis
Vehicle- or drug-treated cells were lysed in a lysis
buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Nonidet
P-40, 0.25% sodium deoxycholate, 0.1% SDS with protease inhibitors.
Lysates were centrifuged at 12,000 rpm for 15 min. Supernatants
were collected, subjected to electrophoresis on 12% (for LC3B
antibody) or 10% (for other primary antibody) SDS-polyacrylamide
gels and transferred to polyvinylidene fluoride membranes
(ImmobilonP; Millipore, Bedford, MA, USA). The membrane was blocked
with 5% non-fat dry milk for 1 h, then incubated with indicated
primary antibody (Cell Signaling Technology, Inc.) overnight at
4°C. The membrane was treated with horseradish peroxidase
conjugated secondary antibodies. Signals were detected by enhanced
chemiluminescence.
siRNA transfection
The synthetic small interfering RNA (siRNA)
oligos-specific to light chain 3 (LC3) was purchased from Shanghai
GenePharma, Ltd. (Shanghai, China) with the corresponding sequence:
5′-AAAUCCCGGUGAUAAUAGA-3′. Cells were seeded in 6-well plates at a
density of 6×105 cells/well in antibiotic-free medium
and allowed to attach overnight. siRNA and transfection reagent
were diluted, respectively, in a separate tube containing 200 μl of
serum-free medium. Following 5 min incubation, siRNA-containing
medium was added to DharmaFect-containing medium. This mixture was
allowed to incubate for 20 min to allow liposome formation and
siRNA loading. Antibiotic-free complete medium was then added to a
final volume of 2 ml and plated onto cells. Transfected cells were
split 1:2 48 h later into new 6-well plates and allowed to attach
overnight. The next day, cells were retransfected exactly as
before, so as to achieve more efficient and sustained knockdown for
an extended period of time. Cells were re-seeded 24 h later for
subsequent experimentation.
Statistical analysis
SPSS software version 18.0 was used for statistical
analysis. Variance analysis was used to determine significance. All
error bars represent the SD of three experiments. Differences with
p<0.05 were considered significant.
Results
TRAIL reduced cell viability and induced
autophagy in breast cancer
To assess the effects of TRAIL on breast cancer cell
viability and to examine whether TRAIL-refractory cells were
resistant to TRAIL, MDA-MB-231 cells and MDA-MB-231
TRAIL-refractory cell lines were treated with varying doses of
TRAIL (0, 10, 20, 40, 80, 160 ng/ml and 0, 20, 40, 80, 160, 320,
640, 1280 ng/ml, respectively) for 72 h. Drug-induced cell death
was measured on MTT assay. TRAIL resistance was demonstrated in the
TRAIL-refractory cells (Fig.
1A).
The TRAIL-refractory cells were separately examined
for the morphological changes of MDA-MB-231 cells and MDA-MB-231
TRAIL-refractory cells by light microscopy. We observed that the
cells were larger after treatment with TRAIL (Fig. 1B).
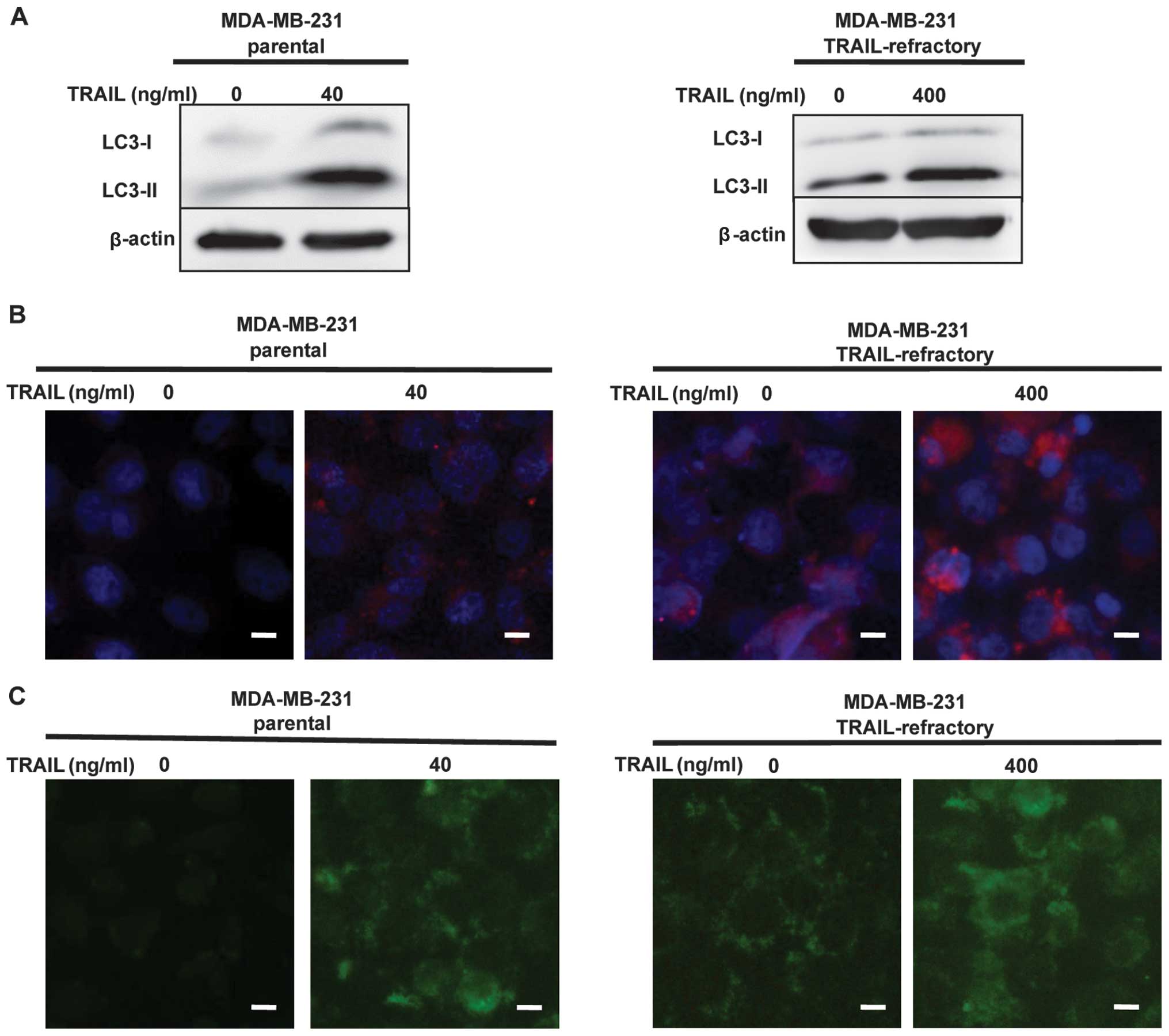
To find the mechanisms involved in TRAIL-mediated
cell death, we investigated the autophagic pathway.
The MDA-MB-231 and MDA-MB-231 TRAIL-refractory cells
were treated with TRAIL (40 or 400 ng/ml) for 72 h. The percentages
of autophagic vacuoles in tested cell lines following treatment
with TRAIL were analyzed by LC3B conversion by immunoblotting and
immunofluorescence staining. As shown in Fig. 2A, LC3B immunoblotting revealed that
LC3B-I (16 kDa) and LC3-II (14 kDa), LC3B-lipidated form was
increased in a dose-dependent manner in MDA-MB-231 and MDA-MB-231
TRAIL-refractory cells following TRAIL treatment. High levels of
LC3-lipidated form indicated impairment in autophagosome
maturation. Fluorescence micrographs (Fig. 2B) revealed the number and intensity
of punctuate LC3B fluorescence increased after treatment with
TRAIL, we utilized this property of LC3B to initially monitor
changes in the dynamics of the autophagic process. The results
showed, LC3B-lipidated form increased in a dose-dependent manner in
breast cancer cells following TRAIL treatment.
To detect the development of autophagic vacuoles,
non-treated and treated MDA-MB-231 or MDA-MB-231 TRAIL-refractory
cells were stained with MDC. MDC is a specific marker for
autophagic vacuoles (19). The
result showed TRAIL-induced autophagic vacuoles formation in breast
cancer cells when compared with control cells (Fig. 2C).
TRAIL resistance correlated with an
accumulation of autophagosomes
Autophagic activity might represent a previously
unrecognized pro-survival pathway underlying acquired
autoresistance to TRAIL. Since it is a dynamic, multi-step process
that can be modulated at several steps, both positively and
negatively, we examined autophagy in several ways. Firstly, the
striking accumulation of autophagosomes was measured by LC3
lipidation on western blotting and fluorescent staining. As shown
in Fig. 3A, immunoblotting
exhibited distinct patterns of LC3B expression between
TRAIL-sensitive and TRAIL-refractory cell lines. In TRAIL-sensitive
cell lines, LC3B existed primarily in its cytosolic form, LC3B-I.
By contrast, TRAIL-refractory cells were characterized by the
upregulating of the lipidated form, LC3B-II. Using fluorescence
microscopy, we confirmed the high basal levels of autophagosomes in
TRAIL-resistant cell lines. As shown in Fig. 3B, TRAIL-refractory cells exhibited
a significant increase of LC-3B per individual cell when compared
with MDA-MB-231 cells, indicating that MDA-MB-231 TRAIL-refractory
cells exhibited punctuate structures that are typical features of
autophagosomes.
Moreover, we performed two complementary
experimental strategies by measuring p62/SQSTM1 protein expression
to further distinguish the level of autophagy. As shown in Fig. 3A and C, immunoblotting and
immunofluorescence staining detected a slight but significant
reduction in the total p62/SQSTM1 protein content in
TRAIL-refractory-derived whole cell lysates when compared with
p62/SQSTM1 protein expression status in TRAIL-naïve MDA-MB-231
parental cells.
We used MDC staining and electron microscopy to
further confirm that autophagosome formation was increased in
TRAIL-refractory cells. We demonstrated that TRAIL-refractory cells
exhibited higher fluorescent density and more MDC-labeled particles
compared with TRAIL-naïve MDA-MB-231 cells, indicating that TRAIL
resistance correlates with the increase of MDC recruitment to
autophagosomes in the cytoplasm of cells (Fig. 3D). Further, electron microscopy
images clearly showed the presence of a large number of
autophagosomes in TRAIL-refractory cells but not in MDA-MB-231
cells (Fig. 3E). These results
suggested that TRAIL resistance was related to an accumulation of
autophagosomes.
Blocking autophagosome function enhances
TRAIL efficacy in TRAIL-refractory cells
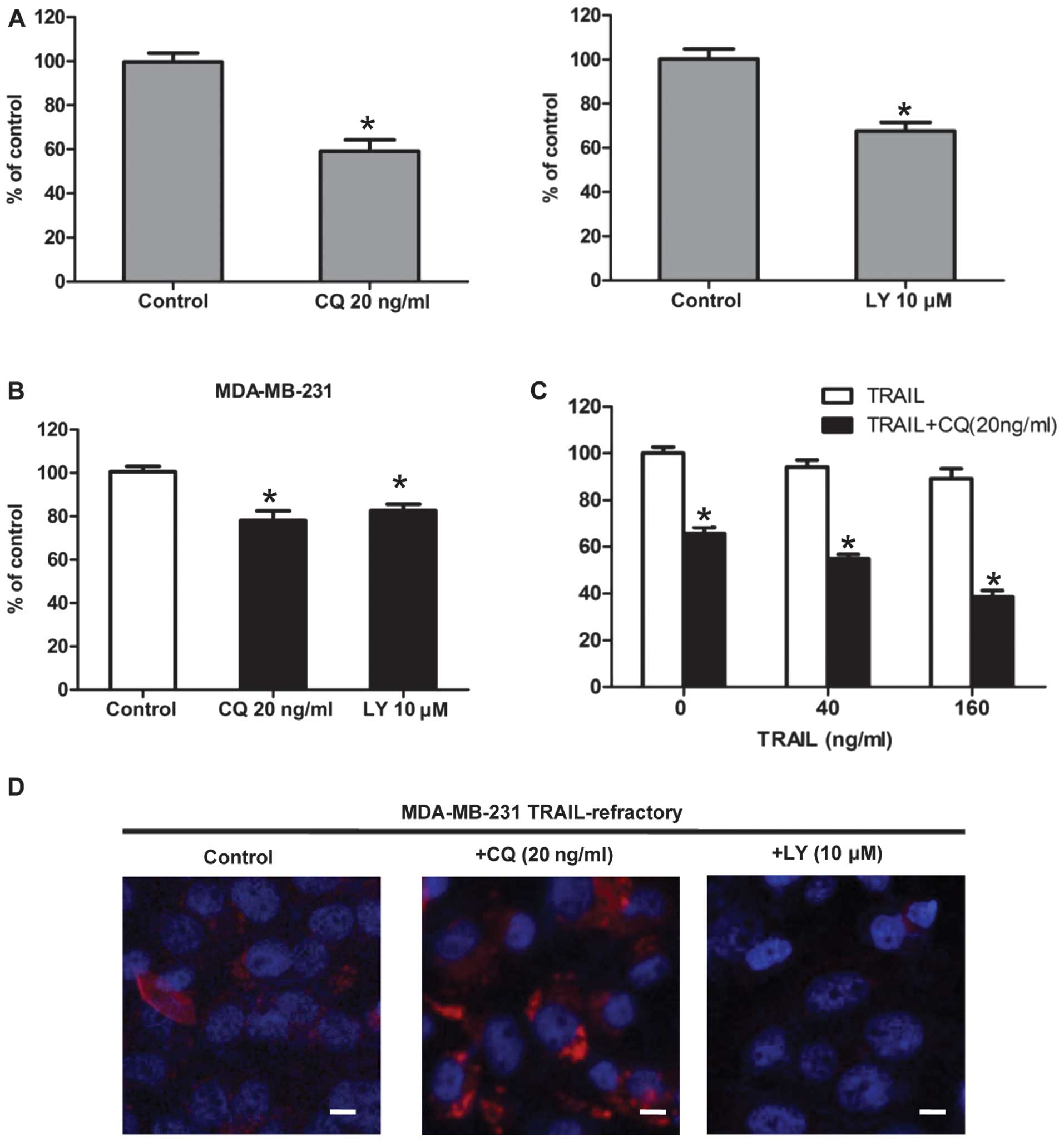
To pharmacologically evaluate whether the basal
autophagy was actively involved in the development of TRAIL
resistance, we assessed the growth inhibitory effects of autophagy
inhibitors (20). Firstly,
TRAIL-resistant MDA-MB-231, TRAIL-refractory and TRAIL-naïve
MDA-MB-231 cells were pretreated with CQ. The cytotoxic effect of
CQ treatment was measured by MTT. The result showed that CQ
effectively reduced cell viability in TRAIL-refractory cells
(Fig. 4A). This was further
supported when similar studies were carried out in the presence of
2-(4-morpholinyl)-8-phenylchromone (LY294002). In terms of cell
viability, TRAIL-refractory cells were exquisitely sensitive to
this agent that blocks phosphatidylinositol 3-kinase activity and
prevents autophagic sequestration (Fig. 4A). Pharmacologically-induced loss
of autophagosome formation is highly cytotoxic to TRAIL-refractory
cells (Fig. 4A) compared with cell
viability effects in TRAIL-naïve MDA-MB-231 parental cells
(Fig. 4B). Representative
immune-confocal images of MDA-MB-231 TRAIL-refractory cells
cultured in the absence or presence of CQ (20 ng/ml) and LY294002
(10 μM) for 72 h are shown in Fig.
4D. We further confirmed that LY294002 treatment was efficient
at reducing the number of LC3-positive autophagosomes while CQ was
able to increase the number of LC3-positive autophagosomes in
TRAIL-refractory cells. Collectively, these findings strongly
suggested that increased macroautophagy actively provided a
survival function to TRAIL-refractory cells.
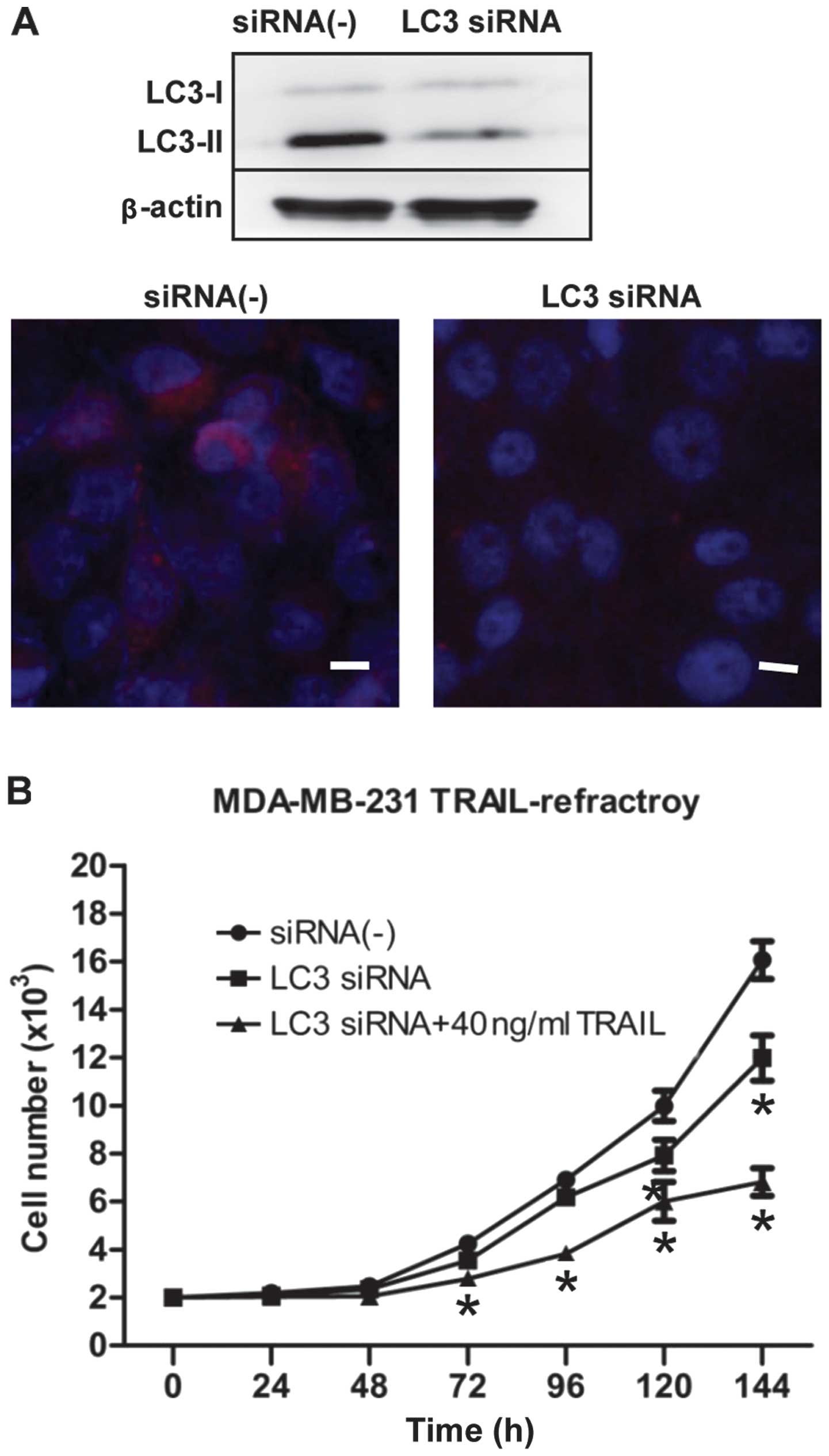
To find additional evidence that autophagy plays a
critical survival role in enabling TRAIL-refractory cell
proliferation and to avoid any off-target side-effects that may
confound interpretation of the results obtained with autophagy
inhibitors, we tested the potent and highly sequence-specific
mechanism of RNA interference (RNAi) to block LC3-dependent
autophagosome formation. TRAIL-refractory cells transiently
transfected with siRNA targeting Atg8/LC3 gene (the key
autophagy regulatory gene). As shown in Fig. 5A, LC3 siRNA blocked the expression
of LC3B by immunoblotting and immunofluorescence staining.
Moreover, we further confirmed that transfection of siRNA
knock-down of LC3 was followed by exposure to 40 ng/ml TRAIL, an
ineffective low-dose of TRAIL to TRAIL-refractory cells, obviously
inhibited cell proliferation (Fig.
5B). Collectively, these data demonstrated clearly that
hyperactivation of basal autophagy is actively involved in the
development of resistance of breast cancer cells to TRAIL.
Combined TRAIL and autophagy inhibition
acts synergistically to reduce TRAIL-refractory breast cancer cell
viability
Thus far, we have demonstrated that TRAIL-refractory
cells are exquisitely more sensitive to lysosomotropic inhibition
of autophagy when compared to MDA-MB-231 parental cells. To
determine whether lysosomotropic inhibition of autophagy could
reverse TRAIL-refractory breast cancer cell resistance to TRAIL,
TRAIL-refractory cells were treated with TRAIL (40 and 160 ng/ml),
CQ (20 ng/ml) for 48 h, and their viability was assessed by MTT.
The result showed that treatment with TRAIL alone led to reduction
in viable cells of 3–8%; their combination, however, yielded a more
pronounced decrease in viability of 40–70% (Fig. 4B). Data analysis revealed that the
decrease in cell viability was statistically significant (p≤0.001;
Fig. 4B). Therefore, CQ-mediated
autophagy blockade, on its own or in combination with TRAIL
significantly decreased cell viability. Co-treatment of
TRAIL-refractory cells with TRAIL and autophagy blockade reduced
cell viability to a greater extent than either alone.
Discussion
Since TRAIL is preferentially cytotoxic to tumor
cells but not normal cells, it is considered to have strong
potential as an anticancer agent. However, ~50% of tumor cell lines
and the majority of primary tumors derived from cancer patients
have shown resistance to TRAIL and most human breast cancer cell
lines are highly resistant to TRAIL-induced apoptosis (14). Thus, the key to success of
development of TRAIL receptor-targeted therapies for cancer
treatment is overcoming tumor resistance.
Our laboratory has previously shown that MTDH could
contribute to TRAIL resistance in breast cancer cells both in
vitro and in vivo, suggesting that MTDH inhibition could
be used to restore TRAIL sensitivity in TRAIL-resistant breast
cancers (21). We also
demonstrated that MTDH could enhance resistance to TRAIL-induced
death by MTDH may overlap mechanisms: Akt activation, upregulation
of Bcl-2 mediated by miR-16, downregulation of caspase-8, and
decreased recruitment of caspase-8 into the DISC.
Autophagy is an evolutionarily conserved catabolic
process. It is characterized by the appearance of autophagic
vesicles and their content degraded by the cellular lysosomal
system and the cell death process (22). In the cancer cells, it is still
unclear if autophagy represents a survival mechanism or is involved
in type II programmed cell death (PCD) (23). In addition, the common clinical
anticancer drug CQ, inhibited autophagy and counteracted the
cytotoxic effect of TRAIL.
We investigated whether the formation of
autophagosomes was further enhanced in the presence of TRAIL in
breast cancer cells by immunoblotting, immunofluorescence staining
and MDC staining. Results from analysis indicated that the
induction of autophagy was dose-dependent in MDA-MB-231 and
MDA-MB-231 TRAIL-refractory cells (Fig. 2). Our results suggested that
autophagy might represent a general mechanism responsible for
circumventing and/or delaying TRAIL-induced cell death.
To unambiguously demonstrate that enhanced basal
autophagy causally protected MDA-MB-231 TRAIL-refractory cells from
cell death upon chronic exposure to TRAIL, we measured
autophagosome accumulation by fluorescence microscopy of LC3B-II
immunoblotting (Fig. 3).
Microtubule associated protein 1 LC3 protein, the first-known
mammalian protein that is specifically associated with the
autophagosomal membrane is involved in the formation of
autophagosomes and its alteration from a cytosolic form LC3B-I to a
lipidated form LC3B-II. Thus it has been widely used as a molecular
marker of autophagosomes. The typical punctate staining that
accompanied the translocation of LC3B-II from the cytosol to the
autophagosome membrane was detected at high levels in MDA-MB-231
TRAIL-refractory cells, suggesting that MDA-MB-231 TRAIL-refractory
cells are uniquely characterized by their ability to sustain high
levels of TRAIL-induced macroautophagy without induction of cell
death. Autophagy flux was also confirmed by fluorescence microscopy
and immunoblotting of p62/SQSTM1 protein (Fig. 3A and C). p62/SQSTM1 itself is
degraded by autophagy. It was shown that p62/SQSTM1 protein
expression was reduced in TRAIL-refractory cells supporting the
notion that the catabolic function of activated basal autophagy
plays a pro-survival role in TRAIL-refractory cells. When high
levels of LC3B-lipidated form associate with impairment in
autophagosome maturation, this phenomenon is accompanied by a
marked increase in the level of p62/SQSTM1. Conversely, increased
LC3B-II levels together with a reduction of p62/SQSTM1 protein
levels characterized the occurrence of autophagic flux increase
(24).
MDC autophagy-specific fluorescence staining
analyses and electron microscopy also confirmed this point
(Fig. 3D and E). Briefly, using
complementary approaches, we showed an upregulated formation of
autophagosomes in the MDA-MB-231 TRAIL-refractory breast cancer
cells.
Further, to determine whether autophagy induced by
TRAIL could provide an indispensable role in cell survival and
facilitate the development of acquired resistance to TRAIL, we
pharmacologically impaired function of macro-autophagosomes by
using the small-molecule autophagy inhibitors LY294002 and CQ, by
MTT assays (Fig. 4). The results
showed that they significantly reduced cell viability in
TRAIL-refractory cells. Further, combination treatment with TRAIL
and CQ synergistically reduced the viability of MDA-MB-231
TRAIL-refractory cancer cells (Fig.
4), suggesting CQ was able to reverse the resistance to TRAIL
and interestingly even played synergistic action with TRAIL.
LY294002 is in many cases a very effective inhibitor
of class I PI3K/Akt in autophagy (25). CQ and hydroxy-chloroquine (HCQ) are
often used in combination with chemotherapeutic drugs to enhance
the efficacy of tumor cell killing. Moreover, they can suppress
autophagy by inhibiting the lysosomal protease activity via
neutralization of the lysosomal pH. HCQ and CQ are commonly used in
suppression of autophagy (26). CQ
acts on autophagosome maturation and blocks autophagic flux, as
well as increases the expression of autophagic markers, including
the number of autophagosomes and level of LC3-II. In fact, CQ is
probably the only autophagy inhibitor currently used in clinical
trials (phase I and II) in combination with various cancer
therapeutic agents in different tumor types, including pancreatic,
breast, colon and prostate cancer, as well as advanced solid tumors
(27).
We finally used RNAi to specifically inhibit
autophagy formation (Fig. 5). The
knocking down of LC3 (the autophagosome membrane protein) by siRNA
similarly decreased TRAIL-R cell viability as measured by MTT. In
contrast to either agent alone, TRAIL and autophagy LC3 siRNA
showed a profound combinatorial effect, greatly inhibited the
proliferation of TRAIL-refractory cells. These combined studies not
only demonstrate that development of acquired resistance to TRAIL
was related to the activation of autophagy but further confirmed an
active role of chemoresistance and cancer cell survival in the
maintenance of TRAIL refraction. We will further explore the
mechanism of autophagy in TRAIL-refractory breast cancer cells.
In summary, we have shown for the first time that
autophagy plays a potent cytoprotective mechanism in the resistance
of TRAIL. In addition, effectively blocking autophagosome formation
could enhance TRAIL efficacy in MDA-MB-231 TRAIL-refractory cells.
Clinical trials are currently ongoing which explore the combination
of anti-autophagy strategies with standard therapies in human
cancers (28). Our data indicate
that caution is necessary in the selection of autophagy inhibitors
for combination with TRAIL receptor-targeted therapies, which
possibly could enhance the pro-apoptotic effect of TRAIL at doses
well-tolerated by patients.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (Beijing, China) (nos. 30772133,
81072150, 81172529, and 81272903) and the Shandong Science and
Technology Development Plan (no. 2012GZC22115). We thank Cunzhong
Yuan and Shi Yan for technical support with experiments. We also
thank Yang Wang, Qiang Huo and Xia Ding for critical discussing and
substantial help.
References
|
1
|
Mizushima N: Physiological functions of
autophagy. Curr Top Microbiol Immunol. 335:71–84. 2009.PubMed/NCBI
|
|
2
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eskelinen EL: The dual role of autophagy
in cancer. Curr Opin Pharmacol. 11:294–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Degenhardt K, Mathew R, Beaudoin B, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Amaravadi RK, Lippincott-Schwartz J, Yin
XM, et al: Principles and current strategies for targeting
autophagy for cancer treatment. Clin Cancer Res. 17:654–666. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qadir MA, Kwok B, Dragowska WH, et al:
Macroautophagy inhibition sensitizes tamoxifen-resistant breast
cancer cells and enhances mitochondrial depolarization. Breast
Cancer Res Treat. 112:389–403. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wiley SR, Schooley K, Smolak PJ, et al:
Identification and characterization of a new member of the TNF
family that induces apoptosis. Immunity. 3:673–682. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ashkenazi A, Pai RC, Fong S, et al: Safety
and antitumor activity of recombinant soluble Apo2 ligand. J Clin
Invest. 104:155–162. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Walczak H, Miller RE, Ariail K, et al:
Tumoricidal activity of tumor necrosis factor-related
apoptosis-inducing ligand in vivo. Nat Med. 5:157–163. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mahalingam D, Szegezdi E, Keane M, de Jong
S and Samali A: TRAIL receptor signalling and modulation: are we on
the right TRAIL? Cancer Treat Rev. 35:280–288. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abdulghani J and El-Deiry WS: TRAIL
receptor signaling and therapeutics. Expert Opin Ther Targets.
14:1091–1108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Menke C, Bin L, Thorburn J, Behbakht K,
Ford HL and Thorburn A: Distinct TRAIL resistance mechanisms can be
overcome by proteasome inhibition but not generally by synergizing
agents. Cancer Res. 71:1883–1892. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Keane MM, Ettenberg SA, Nau MM, Russell EK
and Lipkowitz S: Chemotherapy augments TRAIL-induced apoptosis in
breast cell lines. Cancer Res. 59:734–741. 1999.PubMed/NCBI
|
|
15
|
Yoshida T, Zhang Y, Rivera Rosado LA and
Zhang B: Repeated treatment with subtoxic doses of TRAIL induces
resistance to apoptosis through its death receptors in MDA-MB-231
breast cancer cells. Mol Cancer Res. 7:1835–1844. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rahman M, Davis SR, Pumphrey JG, et al:
TRAIL induces apoptosis in triple-negative breast cancer cells with
a mesenchymal phenotype. Breast Cancer Res Treat. 113:217–230.
2009. View Article : Google Scholar :
|
|
17
|
Bossen C, Tardivel A, Willen L, et al:
Mutation of the BAFF furin cleavage site impairs B-cell homeostasis
and antibody responses. Eur J Immunol. 41:787–797. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim SH, Kim K, Kwagh JG, et al: Death
induction by recombinant native TRAIL and its prevention by a
caspase 9 inhibitor in primary human esophageal epithelial cells. J
Biol Chem. 279:40044–40052. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Biederbick A, Kern HF and Elsässer HP:
Monodansylcadaverine (MDC) is a specific in vivo marker for
autophagic vacuoles. Eur J Cell Biol. 66:3–14. 1995.PubMed/NCBI
|
|
20
|
Mariño G, Ugalde AP, Salvador-Montoliu N,
et al: Premature aging in mice activates a systemic metabolic
response involving autophagy induction. Hum Mol Genet.
17:2196–2211. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang N, Wang X, Huo Q, et al: The
oncogene metadherin modulates the apoptotic pathway based on the
tumor necrosis factor superfamily member TRAIL (Tumor Necrosis
Factor-related Apoptosis-inducing Ligand) in breast cancer. J Biol
Chem. 288:9396–9407. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kelekar A: Introduction to the review
series Autophagy in Higher Eukaryotes - a matter of survival or
death. Autophagy. 4:555–556. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pankiv S, Clausen TH, Lamark T, et al:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Valentim L, Laurence KM, Townsend PA, et
al: Urocortin inhibits Beclin1-mediated autophagic cell death in
cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol
Cell Cardiol. 40:846–852. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sternberg CN, Donat SM, Bellmunt J, et al:
Chemotherapy for bladder cancer: treatment guidelines for
neoadjuvant chemotherapy, bladder preservation, adjuvant
chemotherapy, and metastatic cancer. Urology. 69(Suppl 1): S62–S79.
2007. View Article : Google Scholar
|
|
28
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: therapeutic implications. Mol
Cancer Ther. 10:1533–1541. 2011. View Article : Google Scholar : PubMed/NCBI
|