Introduction
Lung cancer is one of the most common cancers in the
world and accounts for ~28% of all cancer deaths, and non-small
cell lung cancer (NSCLC) accounts for ~80% of lung cancers
(1). Currently, surgery,
chemotherapy and radiotherapy are still the main conventional
treatment of lung cancer. While chemotherapy significantly improves
symptoms and the quality of life of patients with lung cancer,
however, its efficacy and safety remain a primary concern as
toxicity and other side effects of chemotherapy remains a key
obstacle (2). Therefore, there is
an urgent need for novel strategies or reagents of lung cancer.
Alternative medicines, especially herbal therapies (3,4), are
becoming more and more attractive. Among these therapies,
traditional Chinese medicine (TCM) is probably the best
established. Recently, to discover and develop specific herbal
extracts and combinations formulas in TCM that can preferentially
kill cancer cells without significant toxicity has become an
important area in lung cancer therapy.
Triptolide (TP), a diterpenoid triepoxide compound
(Fig. 1A), is one of the major
biologically active components extracted originally from the
Chinese herb Tripterygium wilfordii Hook F (5). Numerous studies have revealed that TP
has a myriad of biological properties, including immunosuppression,
anti-inflammation, and has been applied to the treatment of
autoimmune diseases such as nephritis and rheumatoid arthritis
(6). TP has been reported to exert
anti-cancer activity in diverse tumor cell types in vitro,
including colon cancer (7),
ovarian cancer (8), myeloma
(9), myeloid leukemia (10), adrenal cancer (11) and pancreatic cancer cells (12), and to prevent tumor growth in
vivo via inhibiting cell proliferation and inducing apoptosis
(13). Therefore, with its
broad-spectrum anticancer activity, TP has a considerable potential
as a chemotherapeutical agent.
The natural product camptothecin (CPT) is a
pentacyclic alkaloid, first isolated in 1966 from the extract of a
Chinese plant Camptotheca accuminata. CPT and its analogues
have received increasing attention as a promising class of
antitumor agents, which act by a unique mechanism inhibiting DNA
topoisomerase (14). Among the CPT
family, 10-hydroxycamptothecin (HCPT), as shown in Fig. 1B, displays more potent antitumor
effects and is less toxic in experimental animals and in human
clinical evaluations as compared with CPT. Due to its special cell
killing mechanism, that is different from that of other cytotoxic
agents, it is difficult to induce cross drug resistance when HCPT
is applied together with other anticancer agents. Similarly to TP,
HCPT also shows a broad spectrum of antitumor activity against
various types of cancers, such as gastric carcinoma, hepatoma,
leukemia, bladder carcinoma, and lung cancer (15–18).
A wealth of data now implicate that TP in
combination with other anticancer agents, such as idarubicin
(19), sorafenib (20), oxaliplatin (7), 5-FU (21), AraC (22), TRAIL (23) and ionizing radiation (24), synergistically increase their
cytotoxic effects, suggesting that, next to its potential as a
single drug, TP also appears to have potential as a combinatorial
drug for the treatment of malignancies. Similarly to the
aforementioned findings, our previous study demonstrated that TP
combined with HCPT can synergistically suppress the proliferation
of pancreatic cancer cell line PANC-1, and induces cell apoptosis
(25). However, the detailed
mechanism for the synergistic effect of these two herbal medicines
remains unclear. The present study was designed to determine the
combined efficacy of TP and HCPT on A549 human lung adenocarcinoma
cells, and to investigate whether the synergistic cytotoxicity on
cell growth may be attributed to induction of apoptosis.
Furthermore, to uncover the molecular mechanisms, the involvement
of mitochondria-dependent biochemical markers, as well as tumor
suppressor protein phosphatase 2A (PP2A), Akt and MAPKs signaling
pathways in mediating TP/HCPT-triggered apoptosis were investigated
to help us to better use TCMs in cancer therapy.
Materials and methods
Chemicals
Triptolide (≥98%) was obtained from Beijing
Fan-China Biotechnology Co. Ltd. (Beijing, China).
7-ethyl-10-hydroxycamptothecin (HCPT) was obtained from Sigma (NY,
USA). These drugs were stored in a stock solution in
dimethylsulfoxide (DMSO) at −20°C and diluted to various
concentrations with serum-free culture medium when used. Okadaic
acid (OA) was purchased from Sigma-Aldrich (St. Louis, MO, USA).
SB203580, PD98059 and LY294002 were obtained from Calbiochem (San
Diego, CA, USA). The
3-(4.5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-
(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) was from Promega
Co. Ltd. (Southampton, UK), and phenazine methosulfate (PMS)
(1×10−4 mol/l) was purchased from Sigma. Annexin V/FITC
apoptosis detection kit was obtained from BD Pharmingen (San Diego,
CA, USA). Serine/threonine phosphatase assay kit was purchased from
Promega (Madison, WI, USA). Mitochondria/cytosol fractionation kit
was from Biovision (Mountain View, CA, USA). Antibodies against
caspase-3, -9, cytochrome c, Bax, Bcl-2, PP2A-A subunit, PP2A-C
subunit, Akt and all MAPK family (ERK1/2, p38 and JNK) were all
obtained from Cell Signaling Technology (Beverly, MA, USA).
Antibodies against caspase-12, GRP78 and CHOP were purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phospho-PP2A
Cα (Tyr307) was from Abcam (Cambridge, MD, USA). Antibodies against
β-actin and all of the secondary antibodies were obtained from
Kangcheng (Shanghai, China).
Cell culture
Human lung cancer A549 cells (American Type Culture
Collection; ATCC CCL185) were maintained in monolayer culture at
37°C in a humidified atmosphere with 5% CO2 in RPMI-1640
medium (Gibco-BRL, USA) supplemented with 10% fetal bovine serum
(FBS) (Sijiqin Biotechnology Co. Ltd., China), 1%
penicillin-streptomycin (100 U/ml penicillin and 100 μg/ml
streptomycin).
Cytotoxicity assay
To determine the optimal concentration of the
combination of TP and HCPT, A549 lung cancer cells
(1×104 cells/well in 96-well plates) were exposed to
increasing concentrations of TP (0, 6.25, 12.5, 25, 50, 100, 200
and 400 ng/ml) or HCPT (0, 0.25, 0.5, 1, 2, 4, 8 and 16 μg/ml) for
24 h, respectively. For the cytotoxicity assay of TP/HCPT
combination treatment, A549 cells were treated with TP (25 ng/ml)
and variable concentrations of HCPT (0, 0.5, 1, 2, 4 and 8 μg/ml)
for 24 h, and cell viability was evaluated by the MTS assay using
tetrazolium compound MTS and the electron coupling reagent PMS.
After incubation for the indicated time, 20 μl MTS/PMS mixture was
added to each well; then, the cells were incubated for 1 h and
absorbance was measured at 490 nm. The background absorbance from
the control wells was subtracted from the actual absorbance value.
Three duplicate studies were performed for each experimental
condition. Dose-response curves were plotted on the basis of the
data derived from the MTS assay.
Combination index and dose reduction
index analyses
The multiple drug effect analysis of Chou and
Talalay (26), which is based on a
median-effect principle, was used to evaluate the nature of the
interaction between TP and HCPT. The combination index (CI) is a
parameter that indicates whether the interaction of two or more
drugs is synergistic, additive, or antagonistic. Briefly,
synergism, additivity and antagonism are indicated by CI<1, CI=1
and CI>1, respectively. The dose reduction index (DRI) is a
parameter that indicates the degree to which a drug dose can be
reduced when used in combination with another drug and maintain an
equivalent effect level. The CI and DRI for TP/HCPT were calculated
based on the data from the MTS assay as described previously
(25,27).
Determinations of cell morphology and
cell apoptosis
A549 cells (1.5×105 cells/well) were
seeded in 6-well culture plates for 24 h and then were treated with
TP and HCPT alone or in combination for 24 h, and then photographed
under phase-contrast microscope and harvested by centrifugation.
For apoptosis determination, we used the Annexin V/FITC kit as
described by the manufacturer. The collected cells were resuspended
in binding buffer at a concentration of 1×106 cells/ml.
After incubation, 100 μl of the solution was added by 5 μl of
Annexin V-FITC and 5 μl of propidium iodide (PI), and then
incubated for 15 min at room temperature in the dark. At the end of
incubation, 400 μl of binding buffer was added, and the cells were
analyzed immediately by flow cytometry. Flow cytometry analysis was
performed with untreated cells as a control.
Western blot analysis
Total protein extracts were prepared as described
previously (28). Cells were
suspended in cell lysis buffer containing 150 mM NaCl, 50 mM Tris
(pH 7.6), 15 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 mM
Na3VO4, and 1 mM NaF, and complete protease
inhibitor tablet (Roche, Indianapolis, IN, USA) on ice for 30 min.
The lysate solution was then centrifuged at 20,000 g for 20 min at
4°C, and the supernatant protein extract was stored at −80°C.
For cytosolic cytochrome c analysis,
cytosolic fractions were prepared by using Mitochondria/Cytosol
fractionation kit according to the manufacturer’s instructions.
Briefly, the cells were collected and suspended in cytosolic
extraction buffer containing 0.1% DTT and protease inhibitor on ice
for 10 min. The mixture was homogenized in an ice-cold dounce
tissue grinder (40–50 strokes) and centrifuged at 700 g for 10 min.
The supernatants were then transferred into fresh tubes,
centrifuged at 10,000 g for 30 min, and the supernatants were
collected as cytosolic fraction and stored at −80°C.
The concentration of protein in each cell lysate was
determined by using a BCA protein assay kit (Pierce, Rockford, IL,
USA) with bovine serum albumin (BSA) as the standard. Subsequent
western blot analysis was performed as described (reference?)
previously. All blots were developed using enhanced
chemoluminescence reagents (Super signal dura kit, Pierce)
following the manufacturer’s instructions.
PP2A phosphatase activity assay
PP2A activity was determined using a
serine/threonine phosphatase assay system in accordance with the
manufacturer’s protocols. Cells were briefly lysed with a
phosphatase lysis buffer (50 mM Tris-HCl pH 7.5, 10% glycerol,
0.05% β-mercaptoethanol, 0.1 mM EDTA, 0.05% Triton X-100, 0.5 mM
PMSF, phosphatase inhibitor cocktail) and measured for phosphatase
activity using a PP2A-type specific buffer (250 mM imidazole pH
7.2, 1 mM EGTA, 0.1% β-mercaptoethanol, 0.5 mg/ml bovine serum
albumin). Free phosphate, generated from a synthetic
phosphothreonine peptide RRA(pT)VA specific for PP2A, was
quantified by measuring molybdate/malachite green/phosphate complex
at 630 nm. EGTA and EDTA were included in the lysis buffer to
inhibit PP2B and PP2C, respectively. The effective range of the
assay is 100–4,000 pmol of phosphate.
Statistics
Results are expressed as mean ± SE. Statistical
significance between groups was determined using one-way ANOVA and
Dunnett’s comparison. p<0.05 were considered statistically
significant.
Results
Combination treatment of TP and HCPT
induced growth inhibition of A549 cells
The effect of TP or HCPT as a single agent on the
growth of the A549 lung adenocarcinoma cells was first assessed. As
shown in Fig. 2A, either TP or
HCPT individually caused a markedly dose-dependent reduction in
cell viability, with 50% growth inhibition (IC50) of
273.2 ng/ml and 8.62 μg/ml, respectively. Next, we adopted a
combination treatment by keeping the concentration of TP constant
at IC10 value (25 ng/ml), together with varying
concentrations of HCPT (0–8 μg/ml). We found that the combined
treatment of TP and HCPT (TP/HCPT) substantially suppressed A549
cell growth as compared to single drug alone. Fig. 2B showed a 3.7-fold decrease of
IC50 for HCPT when added in presence of 25 ng/ml TP
(IC50=2.34 μg/ml), compared to HCPT treatment alone,
indicating that the combination treatment with TP and HCPT is more
effective in inhibition of A549 cell growth than single drug
treatment.
 | Figure 2Combinatorial effects of TP and HCPT
on A549 cells. (A) Individual effect of TP or HCPT on cellular
growth. A549 cells were treated with graded concentrations of TP
(6.25, 12.5, 25, 50, 100, 200 and 400 ng/ml, respectively) or HCPT
(0.25, 0.5, 1, 2, 4, 8 and 16 μg/ml, respectively) for 24 h.
Cellular growth was measured by MTS assay. Data are expressed as
the means ± standard error (SE) (n=4). (B) The combined effects of
TP and HCPT on cellular growth. A549 cells were treated with HCPT
(0–8 μg/ml) combined with or without TP (25 ng/ml) for 24 h, and
the cell proliferation was monitored. Data are expressed as the
means ± SE (n=4). |
In order to assess their synergistic effect on A549
cells, we evaluated synergy using CalcuSyn software to evaluate the
combination index (CI) which was originally described by Chou and
Talalay (26,27), when synergism, additivity and
antagonism are defined by CI<1, CI=1 and CI>1, respectively.
The CI value was nearly 1.20 when A549 cells were exposed to the
combination of 25 ng/ml TP and 0.5 μg/ml HCPT, indicating a slight
antagonism. However, when A549 cells were exposed to the
combination of 25 ng/ml TP and variable concentrations of HCPT (1–8
μg/ml), it demonstrated clear evidence of synergy since the CI
value ranged from 0.4 to 0.7 (<1) (Table I). Similar to our recent findings
in pancreatic cancer cell line PANC-1 (25), a higher synergistic effect (lower
CI value) was observed for lower dose combination. Once the
interaction between the two agents was found to be synergistic, we
next sought to determine the dose reduction index (DRI) values for
TP and HCPT in A549 cancer cells. The DRI analysis further
indicated that TP/HCPT combination has the potential to reduce both
the doses of TP (ranging from 1.10- to 20.97-fold dose reduction)
and HCPT (ranging from 1.60- to 3.74-fold dose reduction) in A549
cells (Table I). In all the
subsequent experiments, we treated the cells with the combination
of TP (25 ng/ml) and HCPT (1–4 μg/ml) and TP/HCPT exhibited higher
synergistic effect in inhibiting the growth of A549 lung
adenocarcinoma cells.
 | Table ICombination index (CI) and dose
reduction index (DRI) analysis for triptolide and
hydroxycamptothecin combination therapy in A549 cells. |
Table I
Combination index (CI) and dose
reduction index (DRI) analysis for triptolide and
hydroxycamptothecin combination therapy in A549 cells.
| Combination
therapy | | | |
|---|
| | | |
|---|
| HCPT (μg/ml) | TP (ng/ml) | CI | DRI for HCPT | DRI for TP |
|---|
| 0.5 | 25 | 1.18 | 3.74 | 1.10 |
| 1 | 25 | 0.46 | 4.51 | 4.21 |
| 2 | 25 | 0.40 | 3.52 | 8.34 |
| 4 | 25 | 0.41 | 2.84 | 17.39 |
| 8 | 25 | 0.67 | 1.60 | 20.97 |
Treatment with TP and HCPT
synergistically induced apoptosis of A549 cells
To investigate the effect of TP/HCPT on apoptosis in
A549 cells, Annexin V/propidium iodide (PI) staining-based FACS
analysis was performed to detect the externalization of
phosphatidylserine on the cell membrane, a hallmark of early
apoptosis. Cells undergoing early-stage apoptosis are stained with
Annexin V-FITC-positive and PI-negative. Fig. 3A quantifies the increase in
apoptotic cells labeling with Annexin V+/PI−,
which increases from 0.4% in the control group to 6.2 and 7.2% in
the TP- and HCPT-treated group, respectively. Various combinatorial
treatments resulted in more apoptotic cells than either single-drug
treatment alone, by increasing the apoptosis rate up to 12, 17.8
and 26.7%. It revealed that various combination treatments induced
significantly higher percentage of apoptosis as compared to either
TP or HCPT treatment alone (Fig.
3B), indicating TP/HCPT can promote apoptosis.
Synergistic effect of TP and HCPT on cell
apoptotic pathways
In order to determine whether TP/HCPT induced
apoptosis is mediated via the mitochondria-dependent and/or the ER
stress-triggered signaling pathways, we examined the effects of
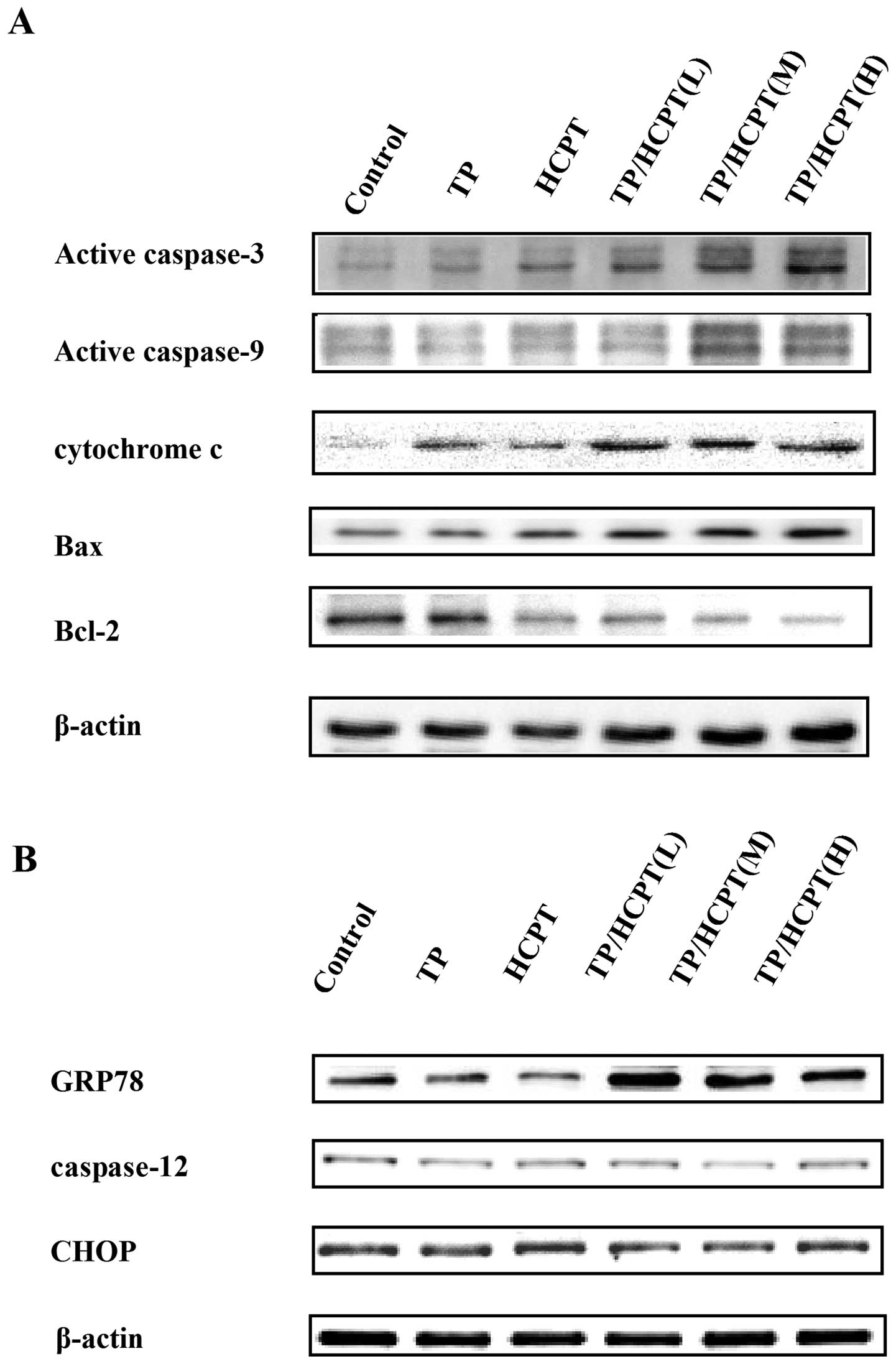
TP/HCPT on the related protein level changes by western blot assay.
Initially, the expression of caspase-9, -3, cytochrome c
(cytosolic), Bax and Bcl-2 were examined to evaluate the
mitochondrial apoptotic pathways. Caspase-9 is known to be involved
in the activation of the caspase cascades for cleaving and
activating caspase-3, which is an integral step in most apoptotic
events. The results indicated that TP or HCPT each only resulted in
a slight upegulation of active (cleaved) caspase-3 and -9
production, whereas the combinatorial TP/HCPT treatment caused
pronounced upegulation of active caspase-3 and -9 (Fig. 4A), suggesting that enhanced cell
apoptosis is mediated through activation of caspase-3 and -9
pathway. In addition, the combined treatment also augmented the
release of cytochrome c from mitochondria, diminution of
Bcl-2, and marked increase in Bax expression (Fig. 4A), which displayed features of
mitochondria-dependent apoptotic signals.
Next, we further determined the expression of three
hallmarks of endoplasmic reticulum stress (ERS), including glucose
regulated protein 78 (GRP78) and CHOP which are known to promote ER
stress-induced apoptosis (29,30),
as well as ER-specific caspase-12 (31). It revealed that TP/HCPT combination
treatment upegulated the GRP78 expression, which is a key regulator
in ER stress signaling that has a dynamic capacity to regulate the
balance between cell survival and apoptosis (Fig. 4B). However, CHOP and caspase-12,
the critical executioners involved in the ER stress-mediated
apoptosis, were not affected by the treatments. These results
suggest that the apoptotic action of TP/HCPT is mainly mediated by
caspase-9 and -3 mitochondrial apoptotic pathways.
Synergistic effect of TP and HCPT on PP2A
activity as well as MAPKs and Akt signaling pathways
Multiple signaling pathways, such as
mitogen-activated protein kinases (MAPKs) family and protein kinase
B (Akt) signal transduction pathways, are essential for cell
survival and important for the regulation of apoptosis (32–34).
To begin elucidating the signaling events that mediate
TP/HCPT-induced apoptosis, we first analyzed the activation of
three MAPK effectors (p38, JNK and ERK1/2) and Akt in A549 cells
using phospho-specific antibodies. As shown in Fig. 5A, TP/HCPT treatment synergistically
evoked a dramatic phosphorylation of ERK1/2 and p38 (i.e.,
activation), in a similar pattern that a maximum response occurred
when TP was combined with HCPT at the highest concentration (4
μg/ml). However, no significant effect of TP/HCPT combination on
JNK phosphorylation was observed. The levels of non-phosphorylated
p38, JNK and ERK were unaffected by either mono-therapy or
combinatorial treatment. On the other hand, we observed that while
the phosphorylated Akt was slightly stimulated by TP alone,
however, combination treatment of TP and HCPT significantly
inhibited TP-induced activation of Akt (Fig. 5A). In particular, cells exposed to
TP combined with the highest concentration of HCPT underwent
remarkable decrease in Akt phosphorylation.
Protein phosphatase 2A (PP2A) is one of the major
protein serine/threonine phosphatases that regulate diverse
cellular functions such as cell division and transcription
(35), and has attracted
considerable attention due to its apoptosis-inducing effect and
tumor-suppressing function (36–38).
Hence, we evaluated the contribution of PP2A by accessing the
phosphatase activity, and the levels and modification of PP2A
composition subunits. We noted that the activity of PP2A was
stimulated by both TP and HCPT mono-therapies. Moreover,
combinatorial TP/HCPT treatment markedly enhanced the activity of
PP2A in a dose-dependent manner, with the maximum response
occurring when TP was combined with the highest concentration of
HCPT (Fig. 5B). Furthermore,
alteration of PP2A composition subunit levels was observed. As
shown in Fig. 5C, structural A
subunit was dramatically enhanced by TP/HCPT combination treatment.
In contrast, TP/HCPT caused a marked decrease in the level of total
PP2A-C. Subsequently, the phosphorylation at Tyr307 (Y307) of the
PP2A-C subunit was investigated because it contributes to decrease
in PP2A activity (39). It was
noteworthy that TP/HCPT treatment induced the downregulation of
PP2Ac (Y307) phosphorylation, which was in accordance with PP2A
activation.
TP/HCPT induced apoptosis is mediated
through PP2A-regulated ERK, p38 MAPK and Akt signaling
pathways
PP2A has been demonstrated to act as a deactivator
of several kinases as its substrates, such as MAPKs and Akt
(40–42). To confirm this role, we used
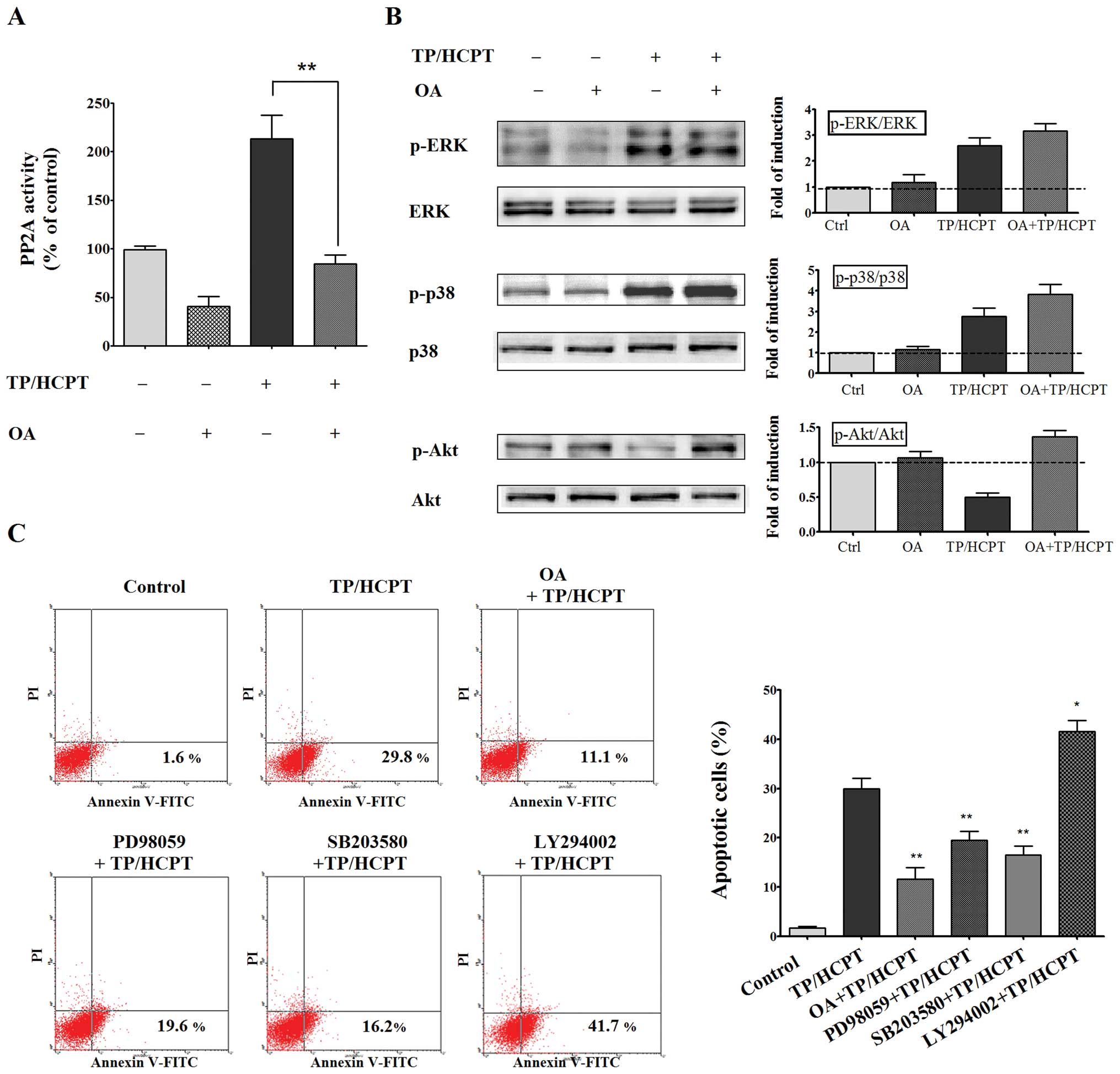
okadaic acid (OA), a selective inhibitor of PP2A, and to examine
whether inhibition of PP2A by OA modulates the phosphorylation
status of ERK1/2, p38 MAPKs and Akt in TP/HCPT-treated A549 cells.
Notably, pretreatment of A549 cells with 50 nM of OA effectively
abolished both the basal level as well as the TP/HCPT-induced
increase in PP2A activity (Fig.
6A). After verifying the inhibition of PP2A activity by OA, we
investigated the role of PP2A in the dephosphorylation of ERK1/2,
p38 and Akt signaling cascades. As shown in Fig. 6B, OA on its own had no effect on
the activation of either ERK1/2 MAPK or p38 MAPK. However,
inhibition of PP2A by OA enhanced the ability of TP/HCPT to
increase the phosphorylation of ERK1/2 and p38, indicating that the
dephosphorylation of the ERK and p38 MAPK signaling pathways might
be PP2A-dependent. Despite differential involvement of MAPKs and
Akt survival signaling cascades upon treatment with TP and HCPT, OA
also effectively prevented Akt dephosphorylation induced by TP/HCPT
(Fig. 6B), indicating all the
ERK1/2 MAPK, p38 MAPK and Akt signaling cascades might be
downstream of PP2A.
Next, to further identify the role of PP2A and its
substrates in mediating TP/HCPT-induced apoptosis, we used specific
pharmacological inhibitors of the different pathways to evaluate
the apoptotic rates. As shown in Fig.
6C, OA pretreatment greatly abrogated the apoptotic effect of
TP/HCPT, implying a role for PP2A in TP/HCPT-stimulated apoptosis.
PD98059, an inhibitor of MEK1, which is the kinase responsible for
the activation of ERK1/2 MAPK, significantly reduced the
synergistic effect of TP and HCPT on apoptosis. Similarly,
pretreatment of cells with SB203580, a well-established inhibitor
of p38 MAPK, also inhibited the apoptotic effect of TP/HCPT,
suggesting the activation of ERK and p38 are required for apoptosis
caused by TP/HCPT. We then employed LY294002, a well-known PI3K
inhibitor, to examine whether the PI3K/Akt signaling pathway also
plays a key role in mediating apoptosis. Our data showed that,
contrary to the effect observed with the ERK and p38 inhibitors,
LY294002 dramatically potentiated TP/HCPT-stimulated apoptosis by
40%, indicating that inhibition of the PI3K/Akt signaling pathway
was critical to the combinational effect of TP and HCPT on
apoptosis. Together these results indicate that TP/HCPT trigger
apoptosis plausibly by activation of the p38 and ERK MAPK cascades,
and inhibition of the Akt survival pathway through a mechanism
involving activation of PP2A.
Discussion
Recently, the interest in exploiting traditional
Chinese medicine (TCM) for prevention or treatment of cancer has
been greatly increased (4). Among
TCMs, two well-known herbal medicines triptolide (TP) and
hydroxycamptothecin (HCPT), that are produced mainly in regions of
China (as well as some other Asian areas such as Japan and North
Korea), have been found to exhibit anticancer potential both in
vitro and in vivo (16,43,44).
Our previous study, which determined the effects of TP and HCPT on
pancreatic cancer, demonstrated that TP combined with HCPT
synergistically increase their cytotoxic effect in pancreatic
cancer cells PANC-1, suggesting that the combination of TP and HCPT
may possess clinical potential for the treatment of cancers
(25). As yet, however, the
molecular basis underlying the synergistic cytotoxicity of these
two herbal medicines has remained poorly understood. We attempted
to investigate the efficacy of combinatorial TP/HCPT treatment in
human non-small cell lung cancer (NSCLC) cell line A549 in
vitro. In the present study, the results present an extension
of our prior findings by showing that the synergistic TP/HCTP
anticancer effect in A549 lung adenocarcinoma cells is mediated by
apoptosis induction via activation of mitochondria-dependent
apoptotic pathway, along with enhanced PP2A activity as well as
activation of ERK1/2 and p38 MAPKs cascades and inhibition of Akt
survival pathway, which provide new insight into the mode of action
of the traditional Chinese medicine TP together with HCPT in cancer
therapy.
A wealth of data indicate that TP, with its
broad-spectrum anticancer activity, can be used as a single agent
to treat different tumors. Recently, several pieces of evidence
have been reported indicating the possibility of using TP in
combination with other anticancer drugs to improve efficacy; also
as a single drug, TP has been found to enhance the action of other
anticancer agents or therapies, such as idarubicin (19), sorafenib (20), 5-FU (21) and ionizing radiation (24), making the combination superior to
mono-therapy alone. In addition to the aforementioned agents, our
previous study also demonstrated TP combined with HCPT have
therapeutic potential for pancreatic cancer (25). Thus, elucidating the mode of action
of TP together with HCPT in killing cancer cells will help us to
understand and to better use TCMs in cancer therapy. Our present
study by keeping TP concentration at its IC10 value
combined with variable concentrations of HCPT demonstrated a
significant decrease in IC50 of HCPT which indicated the
increased cytotoxicity of HCPT by TP, suggesting that the
combinatorial TP/HCPT drug regimens substantially suppressed A549
cell growth as compared to either mono-therapy. Further CI analyses
revealed a synergistic interaction between the two agents in most
of the combination doses tested. Similar to our recent findings in
pancreatic cancer cell line PANC-1 (25), a higher synergistic effect (lower
CI value) was observed for lower dose combination. Moreover,
combinatorial TP/HCPT treatment yielded favorable DRI values which
allowed for both TP and HCPT dose reduction. Taken together, such
synergistic interactions between TP and HCPT provide an opportunity
to reduce the doses of the individual drug and thereby reducing
their adverse toxicities.
Apoptosis (programmed cell death), is characterized
by several morphological and biochemical events (45). To determine whether the decrease in
A549 cells growth is attributed to apoptosis seems vital, as the
induction of apoptosis in cancer cells is the major indicator of
anticancer effects. Hereby, Annexin V/PI staining-based FACS
analysis was employed to evaluate the proportion of apoptotic
cells. Apoptotic cells are characterized by a series of
morphological alterations such as shrinkage of the cells and the
nuclei, loss of adhesion to adjacent cells, membrane blebbing, DNA
fragmentation and chromatin condensation (46). In the present study, A549 cells
treated with combinatorial TP/HCPT treatment exhibited evidence of
apoptotic morphology, i.e., cellular shrinkage, cytoplasmic
blebbing and condensation of nuclei, which are characteristics of
apoptosis (data not shown). These observations were confirmed by
flow cytometry analysis which clearly revealed the number of
apoptotic cells dramatically increased after combined exposure,
suggesting that TP acts synergistically with HCPT in promoting
apoptosis which greatly contributed to the inhibition of NSCLC A549
cell growth.
Three major distinct pathways have been reported to
mediate TP-induced apoptosis in various cell lines, including the
death receptor-mediated (extrinsic), mitochondrial-mediated
(intrinsic) and the endoplasmic reticulum (ER) stress pathway
(11,12,23,47).
Mitochondria are known to play a crucial role in the apoptotic cell
death induced by anticancer agents (48–50).
In the present study, we did observe that the combined treatment of
TP and HCPT induced activation of caspase-3, -9 and release of
cytochrome c from mitochondria into the cytosol, which are
regarded as hallmarks of the mitochondria-mediated apoptotic
pathway. Moreover, we found that combinatorial TP/HCPT drug
regimens resulted in a dramatic increase in expression of
pro-apoptotic protein Bax, while the expression of anti-apoptotic
protein Bcl-2 was markedly inhibited, suggesting a shift in the
dynamic balance between the outputs of pro-apoptotic and
anti-apoptotic pathways. Based upon the fact that the Bcl-2/Bax
ratio plays a crucial role in cancer cell apoptosis (51), we reasoned that the reduction in
Bcl-2/Bax ratio by TP/HCPT would allow less Bcl-2-Bax complex. The
net effect would be the release of more free Bax, which then
translocated into the mitochondrial membrane and induced the
opening of the mitochondrial permeability transition pore to allow
the release of cytochrome c and ultimately triggered the
caspase cascade activation. There is emerging evidence for a close
ER-mitochondria relationship (52), and cross-talk between mitochondrial
and ER plays an essential role in determining cell commitment to
apoptosis (53,54). Therefore, we further investigated
the hallmarks of ER stress-mediated apoptosis, including
glucose-regulated protein 78 (GRP78) and its downstream
pro-apoptotic proteins CHOP and caspase-12. GRP78 has been
documented as a key regulator in ER stress signaling that has a
dynamic capacity to regulate the balance between cell survival and
apoptosis in ER-stressed cells (29). TP mono-therapy has been reported to
downregulate GRP78 and leads to ER stress-mediated apoptosis in
pancreatic cancer cells (12).
Intriguingly, in stark contrast, our study presented herein
revealed that TP/HCPT combination treatment upegulated the
expression of GRP78, while the downstream proteins CHOP and
caspase-12, as the critical executioners involved in ER
stress-mediated apoptosis (30,31),
were not affected. It is possible that in the early stage of ER
stress, the GRP78 was activated by TP/HCPT to alleviate the stress,
thereby restoring the ER homeostasis and leading to prevention of
the induction of downstream CHOP and caspase-12. Taken together,
these findings suggest that combinatorial TP/HCPT drug regimens
induce caspase-dependent apoptosis in A549 lung cancer cells mainly
through modulating the Bax- and Bcl-2-triggered mitochondrial
pathway.
Many studies have been reported that the
pro-apoptotic activity of TP is due to its modulation of
apoptosis-activating proteins (10,47,55).
It appears from these studies that TP influences multiple proteins
and pathways associated with cell growth and survival. Likewise,
many studies demonstrate that HCPT can induce apoptosis of various
cancer cells by influencing the expression of cancer suppression
genes. The direct targets of TP together with HCPT, however, remain
unidentified. Numerous studies have reported that MAPKs and Akt/PKB
signal transduction pathways play crucial roles in a variety of
chemotherapeutic agent-induced apoptotic signaling (33,34).
The three distinct sets of MAPKs are known to be activated
differentially depending on the cell type and the nature of the
stimuli administered (56,57). Our study demonstrated that the
combined treatment of TP and HCPT exhibited an enhancing effect on
both the phosphorylation of ERK and p38. Of the MAPKs, ERK1/2
behaves as a mitogen-activated factor that is believed to mediate
both cell proliferation and survival, whereas p38 and JNK are
activated in response to cellular stresses and appear to exert both
protective as well as pro-apoptotic effects (57–59).
Intriguingly, in the present study, pretreatment of the cells with
either ERK-inhibitor PD 98059 or p38-inhibitor SB 203580 could
effectively abolished the apoptotic effect of TP/HCPT, indicating
that the activation of mitogenic stimuli-activated ERK and
stress-induced p38-MAPK both contribute to TP/HCPT-induced
apoptosis. Next, we tried to characterize the phosphorylation
status of Akt which is known as an anti-apoptotic kinase (34). In the present study, we found that
TP/HCPT triggered remarkable downregulation of Akt pathway.
Moreover, contrary to the effect observed with the ERK and p38
inhibitors, inhibition of the Akt pathway with the PI3K inhibitor
LY294002 potentiated TP/HCPT-induced apoptosis in A549 cells, which
is consistent with the notion that the Akt cascade provides
survival signals that counter-balance the apoptotic response
induced by TP/HCPT. Thus, despite differential involvement of MAPKs
and Akt survival signaling cascades upon treatment with TP and
HCPT, the activation of ERK1/2 and p38, and inhibition of Akt
signal transduction pathway could plausibly all contribute to
eliciting apoptosis in A549 cells. However, the potential of
cross-talk between the MAPKs and Akt pathways still needs further
investigation.
Recently, protein phosphatase 2A (PP2A), an
important protein serine/threonine phosphatase, is attracting more
and more attention due to its apoptosis-inducing effect and
tumor-suppressing function (36–38).
In the present study, we observed that TP/HCPT stimulated a
significant enhancement of PP2A activity, along with alterations of
its composition subunits, including down-regulated PP2A catalytic C
subunit, up-regulated PP2A structural A subunit, and decreased
phosphorylation at PP2A-C Tyr307 (Y307). PP2A-A subunit, a
scaffolding protein for the holoenzyme, is reported to
allosterically modulate the enzymatic properties of PP2A-C
(60), we thus reasoned that
enhanced expression of PP2A-A might contribute to the induction of
PP2A activity. Previous studies have revealed that
post-translational modifications of the highly conserved
carboxy-terminal sequence of PP2A-C subunit, especially the
phosphorylation at Tyr307 can affect PP2A phosphatase activity
(35,39). In the present study, we found the
depression of phosphorylation at PP2A-C Y307 closely matched the
enhancement of PP2A activity.
PP2A is one of the most studied regulators of MAPKs
and Akt by dephosphorylating the threonine or the tyrosine residue
and thereby downregulate their activities (32,40),
and is known to directly interact with these kinases (41). Our previous study demonstrated that
PP2A inhibition, by its specific inhibitor MC-LR, leads to a
dramatic activation of p38-MAPK (28). In agreement with the prior
findings, the present study demonstrated the downregulation of p38
and ERK MAPKs by PP2A, as a specific PP2A inhibitor of OA resulted
in a significant increase in TP/HCPT-induced phosphorylation of
ERK1/2 and p38. On the other hand, OA dramatically reversed the
down-regulation of Akt phosphorylation, suggesting the involvement
of a phosphatase 2A in TP/HCPT-induced dephosphorylation of Akt.
However, our present study also showed that a significant increase
in PP2A activity triggered by TP/HCPT was surprisingly accompanied
by abnormal phosphorylation of ERK and p38, implying that the
modulation of the MAPK signaling pathway is not all in a
phosphatase-dependent manner. Actually, because the phosphorylation
of MAPKs is a reversible process which is regulated by a
coordinated balance between protein kinase-mediated phosphorylation
and protein phosphatase-mediated dephosphorylation, it might be
possible that, in addition to PP2A activation, TP/HCPT stimulate
the activation of ERK- and p38-MAPK also by acting on the MAPKs
upstream kinases, for example, MAPK kinases (MKKs) such as MKK1/2
(ERK upstream kinases) and MKK3/6 (p38 upstream kinases), and even
the MKK upstream kinase MAPK kinase kinases (MKKKs) (61), which requires further research.
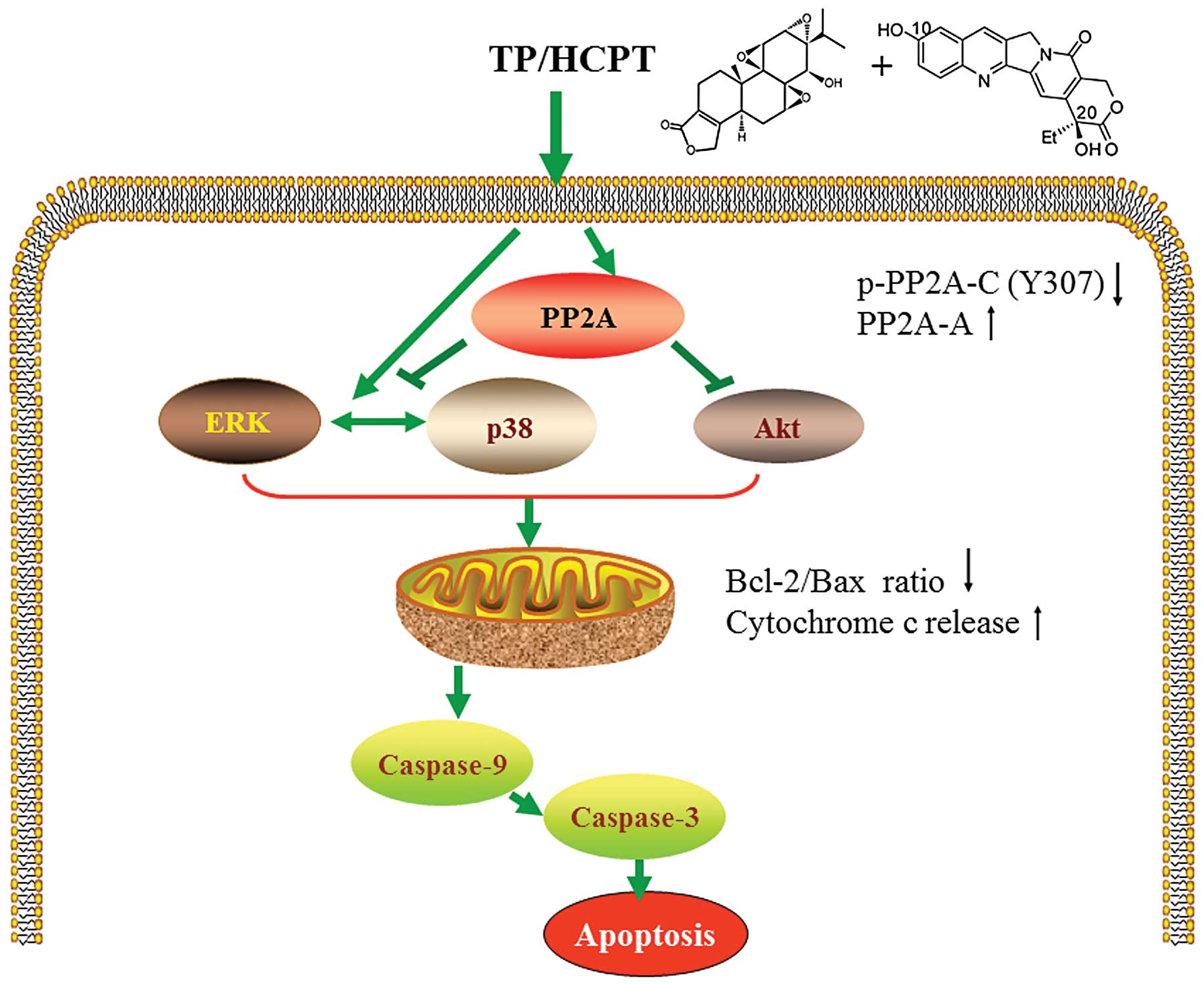
In conclusion, our study discovers a new mode of
action of TP together with HCPT in anticancer therapy. To our
knowledge, this is the first report demonstrating that TP and HCPT
synergistically exert in vitro anticancer activity in human
NSCLC A549 lung adenocarcinoma cells through induction of
apoptosis. Importantly, our findings suggest that TP/HCPT trigger
apoptosis in human lung cancer cells by activation of p38 and ERK
MAPK cascades, and inhibition of the Akt survival pathway through a
mechanism involving activation of PP2A, which uncover a molecular
mechanism that may underlie this combinatorial therapy (Fig. 7). Additionally, such synergistic
interactions, which allow for both TP and HCPT dose reduction,
raise the interesting possibility that TP/HCPT may have a
clinically beneficial effect on anticancer dose reduction and,
thereby leading to a decrease in cytotoxicity during chemotherapy.
Since our present conclusions were primarily based on in
vitro studies, in vivo assessment of the anticancer
effect of TP/HCPT in e.g., xenograft models is warranted. Further
studies are required to improve our understanding of the
synergistic anticancer action of TP/HCPT and to develop new
combinatorial therapies for cancer.
Acknowledgements
This study was supported by Science Foundation from
the Natural Science Foundation of Zhejiang Province (nos.
LY12H29010 and LQ14H290002), Public Technology Applied Research
Projects from Science and Technology Department of Zhejiang
Province (no. 2013C33199) and Zhejiang Provincial Administration of
Traditional Chinese Medicine (no. 2014ZB012).
References
|
1
|
He YY, Zhang XC, Yang JJ, et al:
Prognostic significance of genotype and number of metastatic sites
in advanced non-small-cell lung cancer. Clin Lung Cancer.
15:441–447. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rajeswaran A, Trojan A, Burnand B and
Giannelli M: Efficacy and side effects of cisplatin- and
carboplatin-based doublet chemotherapeutic regimens versus
non-platinum-based doublet chemotherapeutic regimens as first line
treatment of metastatic non-small cell lung carcinoma: a systematic
review of randomized controlled trials. Lung Cancer. 59:1–11. 2008.
View Article : Google Scholar
|
|
3
|
Chen Y, Zhu J and Zhang W: Antitumor
effect of traditional Chinese herbal medicines against lung cancer.
Anticancer Drugs. 25:983–989. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang S, Wu X, Tan M, et al: Fighting fire
with fire: poisonous Chinese herbal medicine for cancer therapy. J
Ethnopharmacol. 140:33–45. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kupchan SM, Court WA, Dailey RG, Gilmore
CJ and Bryan RF: Triptolide and tripdiolide, novel antileukemic
diterpenoid triepoxides from Tripterygium wilfordii. J Am Chem Soc.
94:7194–7195. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Corson TW and Crews CM: Molecular
understanding and modern application of traditional medicines:
triumphs and trials. Cell. 130:769–774. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu Y, Xiao E, Yuan L and Li G: Triptolide
synergistically enhances antitumor activity of oxaliplatin in colon
carcinoma in vitro and in vivo. DNA Cell Biol. 33:418–425. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao H, Yang Z, Wang X, et al: Triptolide
inhibits ovarian cancer cell invasion by repression of matrix
metalloproteinase 7 and 19 and upregulation of E-cadherin. Exp Mol
Med. 44:633–641. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nakazato T, Sagawa M and Kizaki M:
Triptolide induces apoptotic cell death of multiple myeloma cells
via transcriptional repression of Mcl-1. Int J Oncol. 44:1131–1138.
2014.PubMed/NCBI
|
|
10
|
Chen F, Liu Y, Wang S, et al: Triptolide,
a Chinese herbal extract, enhances drug sensitivity of resistant
myeloid leukemia cell lines through downregulation of HIF-1α and
Nrf2. Pharmacogenomics. 14:1305–1317. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu PP, Liu KC, Huang WW, et al: Triptolide
induces apoptosis in human adrenal cancer NCI-H295 cells through a
mitochondrial-dependent pathway. Oncol Rep. 25:551–557. 2011.
|
|
12
|
Mujumdar N, Banerjee S, Chen Z, et al:
Triptolide activates unfolded protein response leading to chronic
ER stress in pancreatic cancer cells. Am J Physiol Gastrointest
Liver Physiol. 306:G1011–G1020. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li J, Liu R, Yang Y, Huang Y, Li X and
Shen X: Triptolide-induced in vitro and in vivo cytotoxicity in
human breast cancer stem cells and primary breast cancer cells.
Oncol Rep. 31:2181–2186. 2014.PubMed/NCBI
|
|
14
|
Hsiang YH, Hertzberg R, Hecht S and Liu
LF: Camptothecin induces protein-linked DNA breaks via mammalian
DNA topoisomerase I. J Biol Chem. 260:14873–14878. 1985.PubMed/NCBI
|
|
15
|
Zaki NM: Augmented cytotoxicity of
hydroxycamptothecin-loaded nanoparticles in lung and colon cancer
cells by chemosensitizing pharmaceutical excipients. Drug Deliv.
21:265–275. 2014. View Article : Google Scholar
|
|
16
|
Hu W, Zhang C, Fang Y and Lou C:
Anticancer properties of 10-hydroxycamptothecin in a murine
melanoma pulmonary metastasis model in vitro and in vivo. Toxicol
In Vitro. 25:513–520. 2011. View Article : Google Scholar
|
|
17
|
Fu YR, Yi ZJ, Yan YR and Qiu ZY:
Hydroxycamptothecin-induced apoptosis in hepatoma SMMC-7721 cells
and the role of mitochondrial pathway. Mitochondrion. 6:211–217.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kotoh S, Naito S, Yokomizo A, et al:
Increased expression of DNA topoisomerase I gene and collateral
sensitivity to camptothecin in human cisplatin-resistant bladder
cancer cells. Cancer Res. 54:3248–3252. 1994.PubMed/NCBI
|
|
19
|
Liu Y, Chen F, Wang S, et al: Low-dose
triptolide in combination with idarubicin induces apoptosis in AML
leukemic stem-like KG1a cell line by modulation of the intrinsic
and extrinsic factors. Cell Death Dis. 4:e9482013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alsaied OA, Sangwan V, Banerjee S, et al:
Sorafenib and triptolide as combination therapy for hepatocellular
carcinoma. Surgery. 156:270–279. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tang XY, Zhu YQ, Tao WH, Wei B and Lin XL:
Synergistic effect of triptolide combined with 5-fluorouracil on
colon carcinoma. Postgrad Med J. 83:338–343. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pigneux A, Mahon FX, Uhalde M, et al:
Triptolide cooperates with chemotherapy to induce apoptosis in
acute myeloid leukemia cells. Exp Hematol. 36:1648–1659. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen Z, Sangwan V, Banerjee S, et al:
Triptolide sensitizes pancreatic cancer cells to TRAIL-induced
activation of the death receptor pathway. Cancer Lett. 348:156–166.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang W, Yang S, Su Y, et al: Enhanced
antitumor effect of combined triptolide and ionizing radiation.
Clin Cancer Res. 13:4891–4899. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang SW, Wang W, Xie XY, Zhu WP and Li FQ:
In vitro synergistic cytotoxic effect of triptolide combined with
hydroxycamptothecin on pancreatic cancer cells. Am J Chin Med.
39:121–134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: the combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Meng G, Sun Y, Fu W, Guo Z and Xu L:
Microcystin-LR induces cytoskeleton system reorganization through
hyperphosphorylation of tau and HSP27 via PP2A inhibition and
subsequent activation of the p38 MAPK signaling pathway in
neuroendocrine (PC12) cells. Toxicology. 290:218–229. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao S, Xiong Z, Mao X, et al: Atmospheric
pressure room temperature plasma jets facilitate oxidative and
nitrative stress and lead to endoplasmic reticulum stress dependent
apoptosis in HepG2 cells. PLoS One. 8:e736652013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han J, Back SH, Hur J, et al:
ER-stress-induced transcriptional regulation increases protein
synthesis leading to cell death. Nat Cell Biol. 15:481–490. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nakagawa T, Zhu H, Morishima N, et al:
Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and
cytotoxicity by amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Komatsu M, Furukawa T, Ikeda R, et al:
Involvement of mitogen-activated protein kinase signaling pathways
in microcystin-LR-induced apoptosis after its selective uptake
mediated by OATP1B1 and OATP1B3. Toxicol Sci. 97:407–416. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cowan KJ and Storey KB: Mitogen-activated
protein kinases: new signaling pathways functioning in cellular
responses to environmental stress. J Exp Biol. 206:1107–1115. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Downward J: Mechanisms and consequences of
activation of protein kinase B/Akt. Curr Opin Cell Biol.
10:262–267. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sontag E: Protein phosphatase 2A: the
Trojan Horse of cellular signaling. Cell Signal. 13:7–16. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen W, Wang Z, Jiang C and Ding Y:
PP2A-mediated anti-cancer therapy. Gastroenterol Res Pract.
2013:6754292013. View Article : Google Scholar
|
|
37
|
Perrotti D and Neviani P: Protein
phosphatase 2A: a target for anticancer therapy. Lancet Oncol.
14:e229–e238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Janssens V and Rebollo A: The role and
therapeutic potential of Ser/Thr phosphatase PP2A in apoptotic
signalling networks in human cancer cells. Curr Mol Med.
12:268–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen J, Martin BL and Brautigan DL:
Regulation of protein serine-threonine phosphatase type-2A by
tyrosine phosphorylation. Science. 257:1261–1264. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY
and Chiang CW: Regulation of phosphorylation of Thr-308 of Akt,
cell proliferation, and survival by the B55alpha regulatory subunit
targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol
Chem. 283:1882–1892. 2008. View Article : Google Scholar
|
|
41
|
Lee ER, Kim JH, Choi HY, Jeon K and Cho
SG: Cytoprotective effect of eriodictyol in UV-irradiated
keratinocytes via phosphatase-dependent modulation of both the p38
MAPK and Akt signaling pathways. Cell Physiol Biochem. 27:513–524.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen L, Liu L, Yin J, Luo Y and Huang S:
Hydrogen peroxide-induced neuronal apoptosis is associated with
inhibition of protein phosphatase 2A and 5, leading to activation
of MAPK pathway. Int J Biochem Cell Biol. 41:1284–1295. 2009.
View Article : Google Scholar
|
|
43
|
Li XJ, Jiang ZZ and Zhang LY: Triptolide
progress on research in pharmacodynamics and toxicology. J
Ethnopharmacol. 155:67–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tan W, Lu J, Huang M, et al: Anti-cancer
natural products isolated from chinese medicinal herbs. Chin Med.
6:272011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen YW, Huang CF, Yang CY, Yen CC, Tsai
KS and Liu SH: Inorganic mercury causes pancreatic beta-cell death
via the oxidative stress-induced apoptotic and necrotic pathways.
Toxicol Appl Pharmacol. 243:323–331. 2010. View Article : Google Scholar
|
|
47
|
Carter BZ, Mak DH, Schober WD, et al:
Triptolide induces caspase-dependent cell death mediated via the
mitochondrial pathway in leukemic cells. Blood. 108:630–637. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang Q, Du H, Geng G, et al: Matrine
inhibits proliferation and induces apoptosis via BID-mediated
mitochondrial pathway in esophageal cancer cells. Mol Biol Rep.
41:3009–3020. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li Y, Zhang S, Geng JX and Hu XY: Curcumin
inhibits human non-small cell lung cancer A549 cell proliferation
through regulation of Bcl-2/Bax and cytochrome C. Asian Pac J
Cancer Prev. 14:4599–4602. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Qin R, Shen H, Cao Y, et al: Tetrandrine
induces mitochondria-mediated apoptosis in human gastric cancer
BGC-823 cells. PLoS One. 8:e764862013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yip KW and Reed JC: Bcl-2 family proteins
and cancer. Oncogene. 27:6398–6406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Giorgi C, De Stefani D, Bononi A, Rizzuto
R and Pinton P: Structural and functional link between the
mitochondrial network and the endoplasmic reticulum. Int J Biochem
Cell Biol. 41:1817–1827. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Deniaud A, Sharaf el dein O, Maillier E,
et al: Endoplasmic reticulum stress induces calcium-dependent
permeability transition, mitochondrial outer membrane
permeabilization and apoptosis. Oncogene. 27:285–299. 2008.
View Article : Google Scholar
|
|
54
|
Pinton P, Giorgi C, Siviero R, Zecchini E
and Rizzuto R: Calcium and apoptosis: ER-mitochondria
Ca2+ transfer in the control of apoptosis. Oncogene.
27:6407–6418. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shao H, Ma J, Guo T and Hu R: Triptolide
induces apoptosis of breast cancer cells via a mechanism associated
with the Wnt/β-catenin signaling pathway. Exp Ther Med. 8:505–508.
2014.PubMed/NCBI
|
|
56
|
Whitmarsh AJ and Davis RJ: Transcription
factor AP-1 regulation by mitogen-activated protein kinase signal
transduction pathways. J Mol Med (Berl). 74:589–607. 1996.
View Article : Google Scholar
|
|
57
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lee ER, Kang YJ, Kim JH, Lee HT and Cho
SG: Modulation of apoptosis in HaCaT keratinocytes via differential
regulation of ERK signaling pathway by flavonoids. J Biol Chem.
280:31498–31507. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Leung HW, Hour MJ, Chang WT, et al:
P38-associated pathway involvement in apoptosis induced by
photodynamic therapy with Lonicera japonica in human lung squamous
carcinoma CH27 cells. Food Chem Toxicol. 46:3389–3400. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Turowski P, Favre B, Campbell KS, Lamb NJ
and Hemmings BA: Modulation of the enzymatic properties of protein
phosphatase 2A catalytic subunit by the recombinant 65-kDa
regulatory subunit PR65alpha. Eur J Biochem. 248:200–208. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Muslin AJ: MAPK signalling in
cardiovascular health and disease: molecular mechanisms and
therapeutic targets. Clin Sci (Lond). 115:203–218. 2008. View Article : Google Scholar
|