Introduction
ST6Gal1 is known to be a regulator for interaction
between galectin-1 and N-glycans which possess α-2,6-sialic acid
(1) and α-2,6-sialylation plays an
important role in many biological processes (2–4).
Galectin-8 plays a regulatory role in cell adhesion as an
extracellular matrix (ECM) (5–7). We
demonstrated that cell surface glycosylation appeared to regulate
cell adhesive or invasive properties to galectin-1 in human
anaplastic large cell lymphoma (H-ALCL) cell line (1). Cell surface sialic acid or N-glycans
regulate adhesive or invasive properties to galectin-1 and linkage
of sialic acid is essential to cell adhesive capacity to
galectin-1. Furthermore, cell adhesion or invasion through
galectin-1 is regulated by phosphatidylinositol 3-phosphate kinase
(PI3K), mitogen-activated protein kinase (MAPK), Rho and
cytoskeleton (1). Previously,
galectin-8 was reported to promote cell movement which is regulated
by cytoskeleton (8). Therefore,
galectins are needed for cell movement in extracellular space. But
biological roles of galectin-8 in human malignant lymphoma are not
yet fully understood. In this report we discuss the fundamental
roles of galectin-8 in human malignant lymphoma.
Materials and methods
Cell line
H-ALCL cell line was established in our laboratory.
The cell line derived from a patient has been approved for use by
the Bioethics Committee in Fukushima Medical University (Fukushima,
Japan). The H-ALCL cells were grown in the culture medium of
RPMI-1640 containing 15% fetal calf serum in 5% CO2 at
37°C. The H-ALCL cell line expresses the galectin-1 receptors,
CD45RA [leukocyte common antigen (LCA), no. 422071; Nichirei Corp.,
Tokyo, Japan] and CD45RO (UCHL-1, N1520; Dako Japan Co., Ltd.,
Kyoto, Japan) on the flow cytometric analysis (FACSCalibur;
Becton-Dickinson, Tokyo, Japan) (data not shown).
Cell surface lectin array analysis
We applied the cell surface lectin array analysis to
detect the cell surface glycosylations according to Landemarre
et al with several modifications (9). The H-ALCL cells were treated with or
without neuraminidase from Arthrobacter ureafaciens (AU)
(no. 10269611001; Roche Diagnostics GmbH, Mannheim, Germany) at 0.2
U/ml, at 37°C for 30 min. Then, the cells were cytospun and
cytospin cell preparations were stained by lectins. The
Phaseolus vulgaris-L(L-PHA) lectin was from L-1801-5, EY
Laboratories, Inc. (San Mateo, CA, USA). The 96-well plate was
coated by each lectin and air-dried. The neuraminidase-treated or
non-treated H-ALCL lymphoma cells (1×106/2 ml) were
applied to each well (100 μl/well) and incubated at 37°C for 60
min. After aspiration of the medium, PBS was added to each well and
then aspirated to remove non-adhered cells. Then 100 μl of 3.7%
formaldehyde was added to each well to fix the adhesive cells at RT
for 40 min. After aspiration of formaldehyde, 100 μl of 0.1%
crystal violet was added to each well and the plates were incubated
at RT for 40 min. After washing twice, 100 μl of 10% acetic acid
was added to each well and the absorbance at 570 nm was determined
using an ELISA plate reader (1).
In our previous, unpublished data, PNA and HPA lectin (B-2301-2,
L-3601-1, respectively; EY Laboratories, Inc.) reactive
oligosaccharides were 2,3-sialylated and L-PHA lectin reactive
oligosaccharides were 2,6-sialylated. To analyze the effect of
O-glycosylation cells were treated with O-glycosylation inhibitor,
benzyl 2-acetamido-2-deoxy-α-D-galactopyranoside (Bz-α-GalNAc
B5019; Sigma, St. Louis, MO, USA) at a concentration of 2 mM in
culture medium for 72 h at 37°C. Then, Bz-α-GalNAc-treated or
non-treated H-ALCL lymphoma cells (1×106/2 ml) were
applied to each well (100 μl/well) and incubated as described
above. In cell surface lectin array desialylation of O-glycans
(PNA, HPA lectin reactivitiy, PNA, B-2301-2 and HPA, L-3601-1; EY
Laboratories, Inc.) was validated in our recent report (1). To analyze the effect of
N-glycosylation cells were treated with N-glycosylation inhibitor,
tunicamycin (TM) (T7765; Sigma), at a concentration of 1.6 μg/ml,
for 48 h at 37°C. Inhibition of N-glycans by TM was validated by
inhibition of cell adhesion to L-PHA lectin (L-1801-5; EY
Laboratories, Inc.) on cell surface L-PHA lectin array
analysis.
Cell adhesion assay
Tissue culture plates with 96 wells were coated with
human recombinant galectin-8 (ATGP0385; ATGen, Ltd., Gyeonggi,
South Korea) (10 μg/well) and matrix proteins (fibronectin 4305-FN,
0.5, 1.0 μg/well, R&D Systems, Minneapolis, MN, USA; laminin
L2020, 10 μg/well, collagen 4 C5533, 50 μg/well and collagen 1
C5983, 10 μg/well, all from Sigma) and dried at room temperature.
H-ALCL cells with or without neuraminidase (from AU final
concentration 0.2 U/ml, at 37°C for 30 min, and recombinant
α-2,3-neuraminidase 1.0, 2.0 and 4.0 U/ml, at 37°C for 30 min, from
New England BioLabs, Inc., Ipswich, MA, USA) treatment were added
to each well and incubated at 37°C for 1 h. After aspiration of the
medium, PBS was added to each well and then aspirated to remove
non-adhered cells. Then 100 μl of 3.7% formaldehyde was added to
each well to fix the adhesive cells at RT for 5 min. After
aspiration of formaldehyde, 100 μl of 0.1% crystal violet
(038-17792; Wako Pure Chemical Industries, Ltd., Osaka, Japan) was
added to each well and the plates were incubated at RT for 5 min.
After washing with PBS, 100 μl of 10% acetic acid was added to each
well and the absorbance at 570 nm was determined using an ELISA
plate reader (iMark™ Microplate Reader; Bio-Rad, Hercules, CA, USA)
(1). For adhesion assay of ECM,
laminin (10 μg/well), fibronectin (1 μg/well), collagen type 1 (10
μg/well) and collagen type 4 (50 μg/well) were coated to wells and
air-dried.
Invasion assay
The invasion assay (haptotaxis) was performed based
on the methods previously reported (1) with several modifications. The 24-well
culture plate was filled with 600 μl the culture medium RPMI-1640
containing 15% BSA and 15% FCS. The lower surfaces of the membranes
of Transwell chamber (Chemotaxicell; Kurabo, Osaka, Japan) with 8
μm pore membrane were coated with 10 μl human recombinant
galectin-8 (0.125 μg/μl, ATGP0385; ATGen, Ltd.) and air-dried at
RT. Then coated Chemotaxi-cells were inserted into each well. The
100 μl of 2.4×106/ml H-ALCL cells were inserted into
each Chemotaxi-cell and incubated at 37°C for 24 h. After
incubation, the invaded cells of each well were counted by trypan
blue exclusion methods. The cell count was performed using
triplicate wells with at least two independent experiments. To
analyze the effect of cell surface sialylation, H-ALCL cells were
treated with neuraminidase from AU (final concentration 0.2 U/ml)
at 37°C for 30 min. For analysis of PI3K inhibitor, wortmannin
(681675) and MAPK inhibitor, PD98059 (513000) (both from
Calbiochem, Darmstadt, Germany) or Rho inhibitor (C3 transferase,
CYO-CT04; Funakoshi Co., Ltd., Tokyo, Japan) cells were
pre-incubated with wortmannin 1.7 μM or PD98059 25 μM for 1 h, or
C3 transferase 2.0 μg/ml, 2 h. Then the cell adhesion or invasion
assay was performed. We confirmed the expression of PI3K, MAPK and
Rho in the tumor cells of H-ALCL on immunohistochemical staining
(data not shown).
The knock-down of ST6Gal1
In order to analyze the regulatory mechanism of cell
surface sialylation by ST6Gal1, siRNA transfection was performed as
described previously with several modifications (1). For transfection, INTERFERin (Polyplus
Transfection, New York, NY, USA) was used according to the
instructions of the manufacturer. For knock-down experiments, siRNA
[cat. no. s12842 (called type 42) sense, AGACAGUUUGUACAAUGAAtt and
anti-sense, UUCAUUGUACAAACUGUCUtt; or cat. no. s12843 (type 43)
sense, ACCACUCAGAUAUCCCAAAtt and antisense, UUUGGGAUAUCUGAGUGGUat;
Ambion, Tokyo, Japan] was used. For control experiments, Ambion
Silencer™ Select Negative Control no. 1 siRNA (cat. no. 4390843;
Ambion) was applied. After 24 h incubation, the immunohistochemical
staining was performed by anti-ST6Gal1 antibody (dilution ×100,
AF5924; R&D Systems) and knock-down effect was validated by
inhibition of ST6Gal1 protein expression in the cytoplasm of H-ALCL
cells as shown in our recent reports (1).
Cell growth inhibitory effects
The H-ALCL cells were grown with or without
neuraminidase treatment and then cells were treated with galectin-8
(7.5 μM, ATGP0385; ATGen, Ltd.). After 7 days, the number of viable
cells was counted by trypan blue exclusion methods.
Results
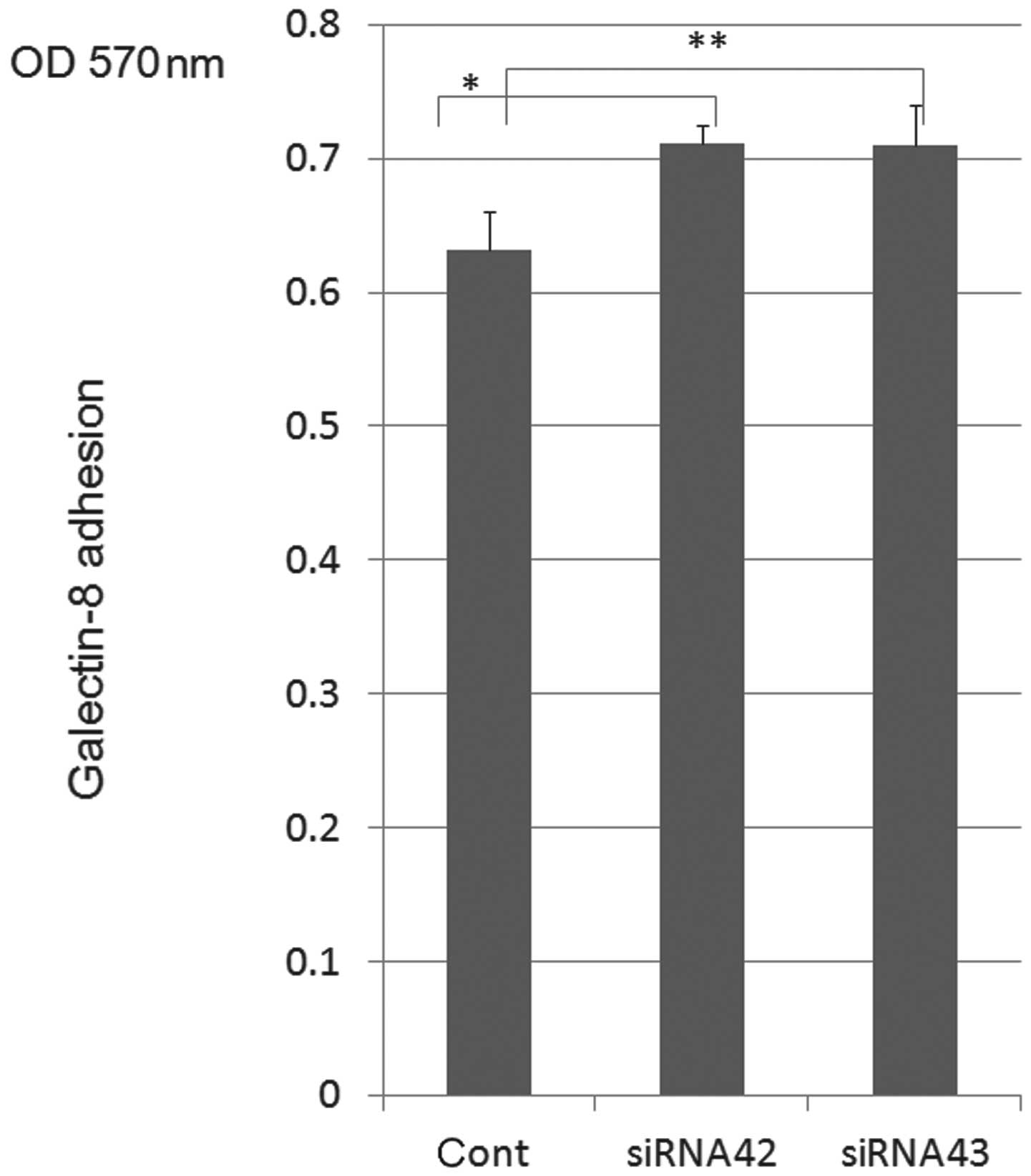
On the knock-down assay for ST6Gal1, ST6Gal
decreased ST6Gal1 protein expression in the cytoplasm of H-ALCL
cells [data shown in (1)] and
showed enhancement of cell adhesion to galectin-8 (Fig. 1). Galectin-8-mediated cell adhesion
was dramatically enhanced by Bz-α-GalNAc treatment (Fig. 2A). Treatment with TM resulted in
inhibition of cell binding activity to L-PHA lectin and inhibition
of cell adhesion to galectin-8, laminin and fibronectin (Fig. 2Ba and b). The N-glycosylation
inhibitor, swainsonine did not inhibit cell adhesion to galectin-8
(Fig. 2C). Galectin-8-mediated
cell adhesion was inhibited by treatment of neuraminidase (Fig. 3). Treatment with
α-2,3-neuraminidase resulted in enhancement of cell adhesion to
galectin-8 (Fig. 4). Neuraminidase
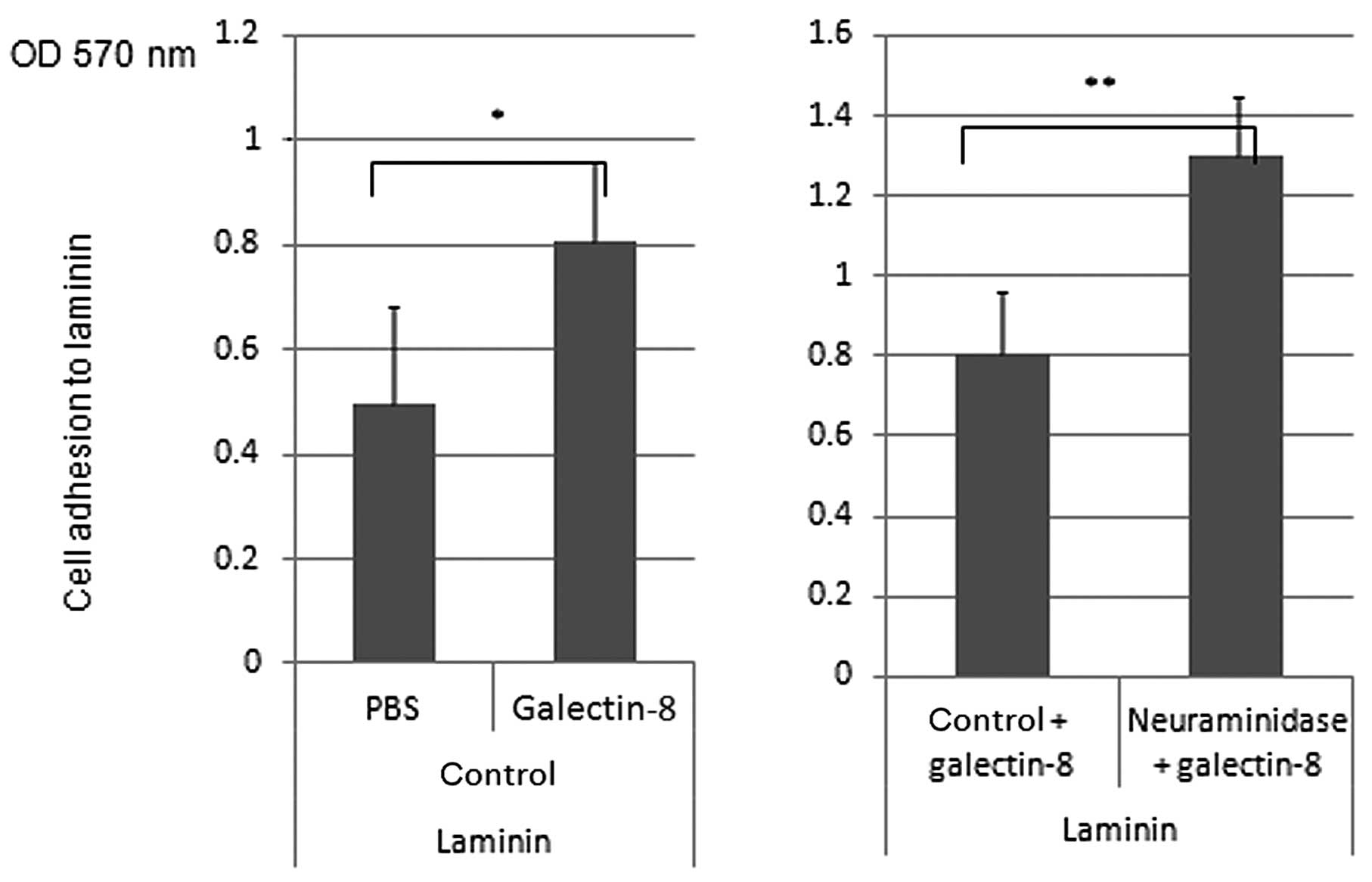
treatment slightly enhanced cell adhesion to laminin (Fig. 5A), and knock-down of ST6Gal1
resulted in enhancement of cell adhesion to laminin, but not to
fibronectin, collagen type 1 or 4 (Fig. 5B). Galectin-8 pre-treatment
dramatically enhanced cell adhesion to laminin and neuraminidase,
treatment also enhanced cell adhesion to laminin in combination
with galectin-8 (Fig. 6).
Neuraminidase treatment induces growth inhibition of lymphoma cells
by galectin-8 (0.5 μM, 7 days, Fig.
7). Cell adhesion to galectin-8 was not altered by treatment of
Rho inhibitor, C3-transferase, but Rho inhibitor pre-treatment
resulted in dramatic inhibition of cell invasion to galectin-8
(Fig. 8). The presence of PI3K
inhibitor, wortmannin resulted in inhibition of cell invasion to
galectin-8 (Fig. 9).
Discussion
Based on our previous study, cell surface
sialylation inhibits cell death in human lymphoma (10). Oversialylation of cell surface by
uridine diphosphate-N-acetylglucosamine 2-epimerase
(UDP-GlcNAc2-epimerase), which is a key enzyme in biosynthesis of
sialic acid, protects lymphoma cells and promotes cell growth.
Neuraminidase treatment or knockdown of UDP-GlcNAc2-epimerase
resulted in reduction of cell surface sialylation and showed
enhancement of cell death by ceramide. This phenomenon is closely
associated with protection of a membrane permeability death by cell
surface-masking effect due to sialylation. In our previous study
α-2,6-sialylation of L-PHA reactive oligosaccharides is known to be
closely associated with a worse prognosis of the patients in human
diffuse large B-cell lymphoma (DLBCL) (11). However, biological roles of
α-2,6-sialylation of L-PHA reactive oligosaccharides still remain
unclear. α-2,6-sialylation of N-glycans may inhibit cell adhesion
to galectin-1-facilitating cell invasiveness based our previous
research (12). Analysis using
patient cases in human DLBCL α-2,6-sialylation of L-PHA reactive
oligosaccharides showed a worse prognosis of the patients (11). The data suggested that
α-2,6-sialylation of L-PHA reactive oligosaccharide may possess a
significant biological role. From the present data
α-2,6-sialylation of N-glycans by ST6Gal1 appeared to modulate cell
adhesion to galectin-8. This biological phenomenon may be related
to lymphoma cell adhesion and motility in vivo, because cell
adhesion to ECM is known to be closely associated with cell
motility based on biological analysis (13). Furthermore, galectin-8-mediated
cell adhesion and invasion may play a significant role in lymphoma
metastasis, because cell adhesion or invasion which is regulated by
cell surface sialylation is known to be related to metastasis, and
inhibition of cell adhesion to ECM by cell surface sialylation is
known to facilitate release of tumor cells from primary site
resulting in a frequent tumor cell distant metastasis in
vivo (13). This mechanistic
model of modulation of cell adhesion to galectin-1 by ST6Gal1 may
provide a new concept in understandings the regulatory mechanisms
of lymphoma metastasis. Galectin-1 and -8 deposit to extracellular
space and H-ALCL cells migrate through interaction between cell
surface glycans and galectin-1 or -8. The balance between
galectin-1-mediated and galectin-8-mediated adhesion may be
associated with movement of lymphoma cells through ECM.
α-2,6-sialylation of glycans on CD45, which is one
of the candidates of galectin-1 receptors, is reported to regulate
apoptosis induced by galectin-1 with interaction between galectin-1
and CD45 glycans (14). These data
suggested that the galectin-1-mediated apoptosis is inhibited by
cell surface α-2,6-sialylation, which is regulated by ST6Gal1. In
our previous data cell surface N-glycans on CD45 interact with
galectin-1 resulting in induction of cell apoptosis (15). Taken together, sialic acid of
N-glycans on CD45 may regulate cell apoptosis on human lymphoma
cells. The α-2,6-sialylation inhibited tumor cell apoptosis induced
by galectin-1 resulting in a more aggressive behavior of human
malignant lymphoma.
In the present study treatment of Bz-α-GalNAc
enhanced cell adhesion to galectin-8. The lectin array analysis of
Bz-α-GalNAc treatment resulted in enhancement of the reactivity of
PNA and VVA lectins suggesting that O-glycans were sialylated
(1). Bz-α-GalNAc treatment can
desialylate the β-galactose residue, which is recognized by PNA
lectin, and is a candidate for galectin-8 receptor. On the other
hand GalNAc residue which is recognized by VVA lectin is not a
galectin-8 receptor. Thus, the findings are complex as to how
treatment of neuraminidase inhibits cell adhesive properties to
galectin-8. In the other aspects several reports suggested that
sialylated β-galactose residue can be a receptor for galectin-8.
The cleavage of sialic acid from this structure by treatment of
neuraminidase abrogated the binding capacity of galectin-8 to these
glycans.
In the present investigation we clarified the
effects of TM in cell adhesion to galectin-8, laminin and
fibronectin. Treatment of TM resulted in inhibition of elongation
of L-PHA reactive oligosaccharides (Fig. 10) (showing inhibitory effects of
cell adhesion to L-PHA lectin) and also inhibition of cell adhesion
to galectin-8, laminin and fibronectin. This result suggested that
L-PHA reactive oligosaccharides are needed for recognition of
galectin-8 on cell adhesion and may regulate cell adhesion to
galectin-8 in ALCL. Previously, loss of L-PHA reactive
oligosaccharides appeared to be associated with a worse prognosis
of the patients in DLBCL (11).
Taken together loss of L-PHA reactive oligosaccharides may be
related to biological phenomena, such as cell adhesion to
galectin-8, laminin and fibronectin, leading to alteration of cell
adhesion, invasion and metastasis in vivo.
Rho G-protein family is well known to regulate cell
motility, especially formation of stress fiber (8). Galectin-1 and -8-mediated cell
adhesion is regulated by Rho (8).
In the present study treatment of C3-transferase, a Rho inhibitor,
resulted in dramatic inhibition of cell invasion to galectin-8. In
our speculation galectin-8-mediated cell motility is regulated by
Rho and its effector protein, actin cytoskeleton which contributes
to formation of stress fiber and contraction of a posterior portion
of moving cells. In our recent study PI3K and MAPK are involved in
cell motility of ALCL cells (1).
The present data indicate that PI3K may be involved in cell
invasion to galectin-8 in ALCL. In future investigation Rac1 and
Cdc42, which are members of cell motility-regulating proteins will
be analyzed in galectin-8-mediated cell invasion in anaplastic
large cell lymphoma.
In previous analysis, galectin-1 induced cell death
(17–20). Galectin-3-induced apoptosis
regulated cell surface sialylation in human DLBCL (21) and galectin-8 induces cell death as
reported previously (6,22). In the present study desialylation
by neuraminidase treatment induced galectin-8-mediated growth
inhibition of lymphoma cells. Based on these data sialic acid may
control growth of lymphoma cells by modulation of sensitivity to
galectins. In our specultion ALCL cells may escape from
galectin-mediated growth inhibition due to masking effects of
sialic acid resulting in a more aggressive phenotype and these
speculations suggest that oversialylated lymphoma cells are more
aggressive and may explain a reason why α-2,6-sialylated L-PHA
reactive oligosaccharides are related to a worse prognosis of the
patients of DLBCL.
Acknowledgements
We are grateful to Ms. M. Satoh and Mrs. H. Kaneko
for their technical assistant and advice.
References
|
1
|
Suzuki O and Abe M: Galectin-1-mediated
cell adhesion, invasion and cell death in human anaplastic large
cell lymphoma: Regulatory roles of cell surface glycans. Int J
Oncol. 44:1433–1442. 2014.PubMed/NCBI
|
|
2
|
Zhuo Y and Bellis SL: Emerging role of
alpha2,6-sialic acid as a negative regulator of galectin binding
and function. J Biol Chem. 286:5935–5941. 2011. View Article : Google Scholar :
|
|
3
|
Dall’Olio F, Chiricolo M, Ceccarelli C,
Minni F, Marrano D and Santini D: Beta-galactoside alpha2,6
sialyltransferase in human colon cancer: contribution of multiple
transcripts to regulation of enzyme activity and reactivity with
Sambucus nigra agglutinin. Int J Cancer. 88:58–65. 2000. View Article : Google Scholar
|
|
4
|
Dall’Olio F, Chiricolo M and Lau JT:
Differential expression of the hepatic transcript of
beta-galactoside alpha2,6-sialyltransferase in human colon cancer
cell lines. Int J Cancer. 81:243–247. 1999. View Article : Google Scholar
|
|
5
|
Levy Y, Arbel-Goren R, Hadari YR, Eshhar
S, Ronen D, Elhanany E, Geiger B and Zick Y: Galectin-8 functions
as a matricellular modulator of cell adhesion. J Biol Chem.
276:31285–31295. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zick Y, Eisenstein M, Goren RA, Hadari YR,
Levy Y and Ronen D: Role of galectin-8 as a modulator of cell
adhesion and cell growth. Glycoconj J. 19:517–526. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yamamoto H, Nishi N, Shoji H, Itoh A, Lu
L, Hirashima M and Nakamura T: Induction of cell adhesion by
galectin-8 and its target molecules in Jurkat T-cells. J Biochem.
143:311–324. 2008. View Article : Google Scholar
|
|
8
|
Diskin S, Chen WS, Cao Z, Gyawali S, Gong
H, Soza A, González A and Panjwani N: Galectin-8 promotes
cytoskeletal rearrangement in trabecular meshwork cells through
activation of Rho signaling. PLoS One. 7:e444002012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Landemarre L, Cancellieri P and Duverger
E: Cell surface lectin array: parameters affecting cell glycan
signature. Glycoconj J. 30:195–203. 2013. View Article : Google Scholar
|
|
10
|
Suzuki O, Tasaki K, Kusakabe T and Abe M:
UDP-GlcNAc2-epimerase regulates cell surface sialylation and
ceramide-induced cell death in human malignant lymphoma. Int J Mol
Med. 22:339–348. 2008.PubMed/NCBI
|
|
11
|
Suzuki O, Nozawa Y, Kawaguchi T and Abe M:
Phaseolus vulgaris leukoagglutinationg lectin-binding reactivity in
human diffuse large B cell lymphoma and its relevance to the
patient’s clinical outcome: lectin histochemistry and lectin blot
analysis. Pathol Int. 49:874–880. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Suzuki O, Nozawa Y and Abe M: The
regulatory roles of cell surface sialylation and N-glycans in human
B cell lymphoma cell adhesion to galectin-1. Int J Oncol.
28:155–160. 2006.
|
|
13
|
Suzuki O, Nozawa Y, Kawaguchi T and Abe M:
UDP-GlcNAc2-epimerase regulates cell surface sialylation and cell
adhesion to extracellular matrix in Burkitt’s lymphoma. Int J
Oncol. 20:1005–1011. 2002.PubMed/NCBI
|
|
14
|
Amano M, Galvan M, He J and Baum LG: The
ST6Gal I sialyltransferase selectively modifies N-glycans on CD45
to negatively regulate galectin-1-induced CD45 clustering,
phosphatase modulation, and T cell death. J Biol Chem.
278:7469–7475. 2003. View Article : Google Scholar
|
|
15
|
Suzuki O, Nozawa Y and Abe M: Regulatory
roles of altered N- and O-glycosylation of CD45 in
galectin-1-induced cell death in human diffuse large B cell
lymphoma. Int J Oncol. 26:1063–1068. 2005.PubMed/NCBI
|
|
16
|
Ideo H, Matsuzaka T, Nonaka T, Seko A and
Yamashita K: Galectin-8-N-domain recognition mechanism for
sialylated and sulfated glycans. J Biol Chem. 286:11346–11355.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Perillo NL, Pace KE, Seilhamer JJ and Baum
LG: Apoptosis of T cells mediated by galectin-1. Nature.
378:736–739. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rodig SJ, Ouyang J, Juszczynski P, Currie
T, Law K, Neuberg DS, Rabinovich GA, Shipp MA and Kutok JL:
AP1-dependent galectin-1 expression delineates classical Hodgkin
and anaplastic large cell lymphomas from other lymphoid
malignancies with shared molecular features. Clin Cancer Res.
14:3338–3344. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Juszczynski P, Ouyang J, Monti S, Rodig
SJ, Takeyama K, Abramson J, Chen W, Kutok JL, Rabinovich GA and
Shipp MA: The AP1-dependent secretion of galectin-1 by Reed
Sternberg cells fosters immune privilege in classical Hodgkin
lymphoma. Proc Natl Acad Sci USA. 104:13134–13139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fouillit M, Joubert-Caron R, Poirier F,
Bourin P, Monostori E, Levi-Strauss M, Raphael M, Bladier D and
Caron M: Regulation of CD45-induced signaling by galectin-1 in
Burkitt lymphoma B cells. Glycobiology. 10:413–419. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Suzuki O and Abe M: Cell surface
N-glycosylation and sialylation regulates galectin-3-induced
apoptosis in human diffuse large B cell lymphoma. Oncol Rep.
19:743–748. 2008.PubMed/NCBI
|
|
22
|
Hadari YR, Arbel-Goren R, Levy Y,
Amsterdam A, Alon R, Zakut R and Zick Y: Galectin-8 binding to
integrins inhibits cell adhesion and induces apoptosis. J Cell Sci.
113:2385–2397. 2000.PubMed/NCBI
|